Introduction
Myocardial infarction (MI) is a common disease of
the cardiovascular system (1).
Apoptosis is a distinctive mode of programmed cell death (2). There is a causal connection between
cardiac myocyte apoptosis and MI (1). MI can cause a progressive decline in
cardiac function that can develop into heart failure (3). Pyroptosis is an important form of
programmed cell death and is characterized by cellular swelling,
membrane disruption and the release of pro-inflammatory cytoplasmic
contents (4). The induction of
cysteine aspartic proteolytic enzyme (caspase) is known to induce
pyroptosis and inflammatory responses (4). Previous studies have focused on the
process of pyroptosis in a range of different heart diseases
(5,6). However, the investigation of
pyroptosis in MI remains to be elucidated.
Ventricular remodeling is an important pathological
process underlying MI. The severity of ventricular remodeling is
known to determine the prognosis of patients with MI (7). The death of myocardial cells is an
important factor in the process of ventricular remodeling and
represents the primary cause of cardiac dysfunction (8). The progress and development of
ventricular modelling may lead to mortality in patients with MI
(7). Since myocardial cells mainly
die during the early stages of ventricular remodeling (9), preventing the death of myocardial
cells during the early stages of ventricular remodeling following
MI is a key target if MI-induced ventricular remodeling is to
improve.
TGF-β serves an important role in the process of
ventricular remodeling and can target myocardial cells by
activating TGF-β-activated kinase 1 (TAK1) and its downstream
factor, JNK (10–12). TAK1 is involved in the
differentiation of cardiomyocytes and the growth and development of
the heart; consequently, TAK1 is important in cardiac disease
(13). The study demonstrates that
TAK1 regulates cell viability and inflammatory responses by
activating its downstream effectors: NF-κB and MAPK (14). Okada et al (15) firstly proposed that the TAK1/JNK
pathway acts as an essential regulator for the activation of the
NLRP3 inflammasome.
It has been confirmed that expression levels of
NLRP3 are increased in the early stages of ventricular remodeling
following MI and that the combination of NLRP3 with
apoptosis-associated speck-like protein containing a
caspase-recruitment domain (ASC) can recruit and activate caspase-1
thus promoting the maturation and release of IL-1β. IL-1β, an
important inflammatory molecule, recruits nearby
inflammation-related cells following MI, thus amplifying the
inflammatory response and participating in post-MI ventricular
remodeling, eventually leading to the death of myocardial cells
(16–19). Another study demonstrated that the
specific inhibition of IL-1β can significantly improve the
contractile function of a mouse model of MI mice and can also
reduce enlargement of the heart; these data suggested that early
ventricular remodeling might be related to cellular pyroptosis
(20).
Pyroptosis is an inflammasome-activated process.
Caspase-1-mediated pyroptosis refers to a process that takes place
in inflammasomes but is activated by caspase-1 (21). The specific blocking of the NLRP3
inflammasome with ASC markedly reduces cardiomyocyte death and
improves cardiac function, thus indicating that NLRP3 inflammasomes
are involved in the early stages of ventricular remodeling
following MI (22–24).
Sacubitril/valsartan (LCZ696) is a new form of
neuroendocrine inhibitor that can selectively and efficiently
inhibit the angiotensin II (Ang II) receptor and enkephalinase
(25). Ang II is not only a strong
vasoconstriction and growth-promoting peptide; research has shown
that this peptide is closely associated with cardiac remodeling and
can promote hypertrophy and fibrosis in cardiomyocytes (26–28).
In a previous study, Kompa et al (29) established a rat model of MI and
treated these animals with LCZ696 and perindopril. The study
demonstrated that the left ventricular ejection fraction and
shortened fraction of rats under LCZ696 treatment are enhanced
compared with those rats treated with perindopril; furthermore,
LCZ696 exhibits a more powerful effect in terms of myocardial
protection (29). Perindopril, an
ACE inhibitor and amlodipine, a dihydropyridine calcium channel
blocker, is known to be able to treat cardiovascular events
(30,31). In a previous clinical trial, LCZ696
significantly reduced the risk of cardiovascular death and
hospitalization of patients experiencing heart failure when
compared with enalapril; these effects were due to the inhibition
of TGF-β and the consequential reduction in ejection fraction and
improvement in cardiac function. Thus, LCZ696 represents a new
alternative treatment option for chronic heart failure compared
with the traditional treatment of renin-angiotensin-aldosterone
system (RAAS) blockade (32).
The present study established a rat model of MI and
the animals were treated with LCZ696 to explore its protective
effects on the heart and the inflammasome-mediated inflammatory
response. The present study also aimed to investigate the
regulatory mechanisms associated with LCZ696 that link the TAK1/JNK
pathway and NLRP3 inflammasomes.
Materials and methods
Experimental animals
A total of 72 male 3-month-old SPF-grade
Sprague-Dawley (SD) rats, weighing 260–300 g, were purchased from
VitalRiver Experimental Animal Techniques [Production License: SCXK
(Beijing) 20190001]. All animal were kept in a barrier room with
free access to food and water under 12-h light/dark cycle and
40–70% humidity at 20–25°C at Department of Laboratory Animal
Science of China Medical University (User License: SYXK (Liao)
2013001). Rats were randomly divided into the following groups:
Sham operation group (Sham group, n=24), myocardial infarction
model group (MI group, n=24) and a MI + LCZ696 treatment group
(LCZ696 group, n=24). The experiments were approved by the
Laboratory Animal Welfare and Ethics Committee of China Medical
University (approval. number IACUC NO. 2019109).
Establishment of a rat model of
myocardial infraction
Rats were anaesthetized with intraperitoneal
injection of 1% pentobarbital sodium (50 mg/kg). Electrocardiogram
parameters were recorded using BL-420 biological and functional
experimental system (TECHMAN). Respiratory support was provided by
a small animal ventilator. Next, the skin of each rat was cut in a
longitudinal manner from the left margin of the 3rd to 4th
intercostal space to expose the heart. The left anterior descending
coronary artery was then ligated ~3–4 mm below the intersection of
the left atrial appendage root and the pulmonary artery conical
clip. When ligated in this manner, the myocardial tissue began to
turn white and the ST segment of the electrocardiogram demonstrated
different levels of elevation. Collectively, these indicators
demonstrated that the rat model had been successfully established.
Rats in the Sham group were treated by thoracotomy only while rats
in the LCZ696 group were administered with 68 mg/kg LCZ696
(SML1380; Sigma-Aldrich; Merck KGaA) by oral gavage daily for a
total period of 7 days after the rat model of MI had been
established (33).
Examining left ventricular
function
A total of eight rats from each group underwent
cardiac ultrasonic examination (EPIQ7C; Philips Medical Systems
B.V.) to analyze left ventricular (LV) function at each detection
time point (1, 3 and 7 days). Rats were first anaesthetized with 1%
sodium pentobarbital (50 mg/kg); the fur on the left side of the
chest was then removed using hair removal cream. M-mode ultrasound
was then used to detect the LV ejection fraction (LVEF), LV
fractional shortening (LVFS), LV internal diameter at systole
(LVIDs) and LV diastolic diameter (LVIDd).
Sample collection
At each detection time point (1, 3 and 7 days),
eight rats from each experimental group were randomly selected for
blood and tissue sample collection. First, rats were placed on an
operating table for anesthetized with 1% sodium pentobarbital (50
mg/kg). The abdominal cavity was then opened and the abdominal
aorta was cut to acquire 2 ml samples of blood. Blood samples were
stored in −80°C freezer for subsequent ELISA. Following euthanasia
through overdose of 1% sodium pentobarbital (200 mg/kg), the chest
cavity was opened to collect samples of heart tissue. Half of each
heart tissue sample was immersed in 10% formalin and fixed for 24 h
at room temperature for subsequent staining to investigate
pathological changes. The other half of each heart tissue sample
was stored in a −80°C freezer to await protein detection.
Determining the size of the
infarction
2,3,5-Triphenyltetrazolium chloride (TTC) staining
was used to determine the size of the myocardial infarct. Thin
sections of heart tissue were prepared from each sample collected
on the day 1, 3 and 7. Briefly, the rat heart was frozen quickly in
liquid nitrogen and cut into 2 mm sections. The sections were
incubated with 2% TTC staining solution at 37°C for 30 min.
Post-staining, the tissue slices were fixed overnight in 10%
formalin solution at 4°C. Representative images were then captured
from each tissue section using a digital camera. The area of
infarction within the sections of heart tissue (which remained
unstained following incubation with TTC staining solution) was then
determined by Sigma Scan Pro 5.0 software (Systat Software,
Inc.).
Hematoxylin and eosin (H&E)
staining
Post-perfused heart tissues were collected from
sacrificed rats on days 1, 3 and 7 and fixed in neutral formalin;
these tissues were then dehydrated with ethanol using a gradient
series of concentrations (75, 80, 95 and 100%). The tissues were
treated with xylene to clear them, embedded in paraffin and then
cut into 4 µm sections. For H&E staining, sections were
deparaffinized, stained first with hematoxylin and then stained
with eosin at room temperature. The experiment procedure was
according to the manufacturer's instructions (G1120, Solaibio,
China) Histopathological changes in the myocardium were then
evaluated by light microscopy.
Temperature and duration of staining
Masson's staining
A Masson's trichrome staining kit (Solarbio, G1345)
was used according to the manufacturer's instructions at room
temperature. Deparaffinized myocardium sections were stained in
hematoxylin for 3 min, differentiated in picric acid ethanol for 10
sec and were then rinsed with distilled water. Sections were then
stained blue by applying Masson's blueing solution for 3 min,
followed by 60 sec rinse in distilled water. The sections were then
stained in ponceau magenta solution for 5 min and rinsed with 2%
glacial acetic acid aqueous solution. Following this, sections were
washed with phosphomolybdic acid solution for 3 min, stained in
aniline blue for 3 min and differentiated in 1% glacial acetic
acid. Finally, sections were dehydrated three times with pure
ethanol (5 min/incubation), cleared with xylene for 5 min and then
mounted in neutral resin. Slides were then evaluated by light
microscopy. Myocardial collagen volume fraction (CVF) was analyzed
by Image-Pro Plus 6.0 (Media Cybernetics, Inc.). CVF was calculated
using the formula shown in Equation (1).
Equation (1):
CVF=(Area of myocardial collagen/Total area)
×100%
ELISA
The serum levels of IL-18 (cat. no. SEA064Ra; Wuhan
USCN Business Co., Ltd.) and IL-1β (cat. no. SEA563Ra; Wuhan USCN
Business Co., Ltd.) were detected on days 1, 3 and 7 by ELISA in
accordance with the manufacturer's guidelines. Blood samples were
collected from the abdominal aorta of euthanized rats. In brief,
100 µl of standard solutions and diluted samples were loaded into
each well of the ELISA plates and incubated at 37°C for 1 h.
Detection reagent A and B were added and then the plates rinsed
with the wash buffer provided in the ELISA kits. After thorough
rinsing, 90 µl of TMB substrate was added into each well and
incubated at 37°C for 20 min in the dark. Stop solution was then
applied to the plates and the optical density of each well was
detected on a microplate reader at 450 nm within 10 min of adding
the stop solution. The concentration of IL-18 and IL-1β in mice
sera were calculated by a formula obtained from the standard
curve.
Immunofluorescence
Heart tissue sections were prepared from days 1, 3
and 7 and then deparaffinized, rehydrated, incubated in 3%
H2O2 solution and then rinsed with PBS
buffer. Sodium citrate solution (0.1 M) was applied to the sections
for antigen retrieval and then the sections blocked with goat serum
at 37°C for 30 min. Following incubation, the residual goat serum
was decanted and then the sections incubated overnight at 4°C with
a far-red dye labelled reactive oxygen species (ROS) probe (cat.
no. C10422; Invitrogen; Thermo Fisher Scientific, Inc.) and primary
antibodies against TAK1 (1:1,000; cat. no. PA5-20083; Invitrogen;
Thermo Fisher Scientific, Inc.), phosphorylated (p)-JNK (1:1,000;
cat. no. MA5-15228; Invitrogen; Thermo Fisher Scientific, Inc.),
NLRP3 (1:1,000; cat. no. PA5-79740; Invitrogen; Thermo Fisher
Scientific, Inc.) and gasdermin D (1:500; GSDMD; cat. no.
NBP2-80427; Novus Biologicals, LLC). The sections were then rinsed
with PBS buffer, incubated with fluorescence-labelled secondary
antibody at 37°C for 30 min and rinsed thoroughly with PBS buffer.
Cell nuclei were then incubated with DAPI (cat. no. D3571;
Invitrogen; Thermo Fisher Scientific, Inc.) at room temperature for
10 min. Finally, the sections were rinsed in PBS and sealed with
anti-fade working solution. Expression levels were then evaluated
with a fluorescent microscope and quantified with ImageJ 1.52q
software (National Institutes of Health).
Western blotting
Frozen myocardial tissues were removed from the
−80°C freezer, thawed and crushed. After that, 1 ml of RIPA lysis
buffer (cat. no. 89901; Thermo Scientific, Inc.) containing 1% PMSF
was added and total protein extracted by homogenizing and
centrifuging the tissue for 15 min at 15,000 × g and 4°C. The
concentration of total protein extracts in the supernatants was
then determined with a BCA assay kit (cat. no. 23227; Thermo
Scientific, Inc.). Protein samples (30 mg) were then separated by
10% SDS-PAGE and transferred on to 0.45 µm PVDF membranes.
Membranes were then blocked for 2 h with 5% skimmed milk at room
temperature. Next, membranes were incubated overnight at 4°C with
primary antibodies against TAK1 (1:1,000; cat. no. 4505; Cell
Signaling Technology, Inc.), p-JNK (1:1,000; cat. no. 9255; Cell
Signaling Technology, Inc.), JNK (1:1,000; cat. no. 9252; Cell
Signaling Technology, Inc.), NLRP3 (1:1,000; cat. no. ab263899;
Abcam), pro-caspase1 (1:1,000; cat. no. ab179515; Abcam), GSDMD
(1:500; cat no. 93709; Cell Signaling Technology, Inc.), N-terminal
cleavage product (GSDMD-NT) (1:1,000; cat. no. 93709; Cell
Signaling Technology, Inc.), pro-IL-lβ (1:1,000; cat. no. ab205924;
Abcam) and pro-IL-18 (1:1,000; cat. no. ab191860; Abcam); GAPDH
(1:2,000, cat. no. ab181602; Abcam) was used as an internal loading
control. Residual primary antibodies were removed by washing the
membranes three times in TBS-Tween 20 (0.1%) on a shaking
incubator. HRP-conjugated goat anti-rabbit secondary antibody (cat.
no. 7074; Cell Signaling Technology, Inc.) was applied to the
membranes and incubated for 1 h at room temperature. The membranes
were then washed three times in TBS-Tween 20 and imaged using an
imaging system and ECL chemiluminescent substrate (cat. no. 34580;
Thermo Scientific, Inc.). Finally, grey values were analyzed using
ImageJ 1.52q software (National Institutes of Health).
Reverse transcription-quantitative
(RT-q) PCR
The experiments of RNA extraction, cDNA synthesis
and qPCR were performed according to the manufacturer's protocols.
Myocardial tissues were recovered from −80°C and mixed with 1 ml of
TRIzol® (cat. no. 15596026; Thermo Fisher Scientific,
Inc.). Samples were then thoroughly homogenized at low temperature
to extract total RNA. cDNA was synthesized by reverse transcription
and RT-qPCR performed using primers designed against TAK1
and JNK; GAPDH was used as an internal control
(Table I). RT-qPCR was carried out
with a SYBR Primer-Script RT-PCR kit (Takara Biotechnology Co.,
Ltd.) using the following cycle parameters (for 40 cycles):
pre-denaturation (95°C, 30 sec), annealing (95°C, 5 sec), extension
(60°C, 20 sec). The relative expression levels of each gene were
then determined using the 2−ΔΔct method (34). All RT-qPCR experiments were repeated
at least three times.
 | Table I.Primers used for reverse
transcription-quantitative PCR. |
Table I.
Primers used for reverse
transcription-quantitative PCR.
| Gene | Primer |
|---|
| NLRP3 | F:
TGAAGAGTGTGATCTGCGGAA |
|
| R:
GAAAGTCATGTGGCTGAAGCT |
| GSDMD | F:
TGAATGTGTACTCGCTGAGTGT |
|
| R:
CAGCTGCTGCAGGACTTTGTG |
| Caspase-1 | F:
CAGAGCTGTGCAGATGAGT |
|
| R:
CTGCAGCCACTGGTTCTGT |
| IL-1β | F:
AGGAGAGACAAGCAACGACA |
|
| R:
CTTTTCCATCTTCTTCTTTGGGT |
| IL-18 | F:
GCCATGTCAGAAGACTCTTGCGTC |
|
| R:
GTACAGTGAAGTCGGCCAAAGTTGTC |
| TAK1 | F:
AGTAGGCCACATTACACTGCT |
|
| R:
GACCCACACCTCACAAATTGA |
| JNK | F:
TCATGTGGTCAAGACGAGAT |
|
| R:
GGAGGAAGGAAGGCATGA |
| GAPDH | F:
GCACACAGTACATCCGTCA |
|
| R:
TTCTCCGAACGTGTCACGT |
Cell culture
H9C2 cells were cultured in a hypoxic culture
chamber (37°C, 2.5% O2, 5% CO2, 92.5%
N2) to establish a cellular model of MI (35). Cells in the control group were
cultured in a normal culture chamber (37°C, 5% CO2).
Cells were cultured with DMEM medium (cat. no. 12491015; Gibco;
Thermo Fisher Scientific, Inc.) without serum. TAK1 overexpression
plasmids were obtained from Mingshanshang Medical Biotechnology
Co., Ltd. and validated by PCR, enzymic digestion and DNA
sequencing. Next, 500 ng of DNA was transfected into cells using
Lipofectamine® 3000 (cat. no. L3000001; Invitrogen;
Thermo Fisher Scientific, Inc.) and/or 20 µm of LCZ696 was added to
the cells transfected with the TAK1 overexpression plasmid, in
appropriate with the following experimental groupings: Control
group (negative control without plasmid transfection), hypoxia MI
model group, vector + LCZ696 + hypoxia MI group and a TAK1
overexpression group + LCZ696 + hypoxia MI group. Experiments were
replicated four times.
Statistical analysis
Data were represented as mean ± standard deviation
and analyzed using SPSS 21.0 (IBM Corp.). Multiple groups were
compared by one-way analysis of variance. Tukey's post hoc was used
to compare between groups at different time points (days 1, 3 and
7). The non-parametric Wilcoxon signed ranks test was used for
multiple comparisons with adjusted P-values. P<0.05 was
considered to indicate a statistically significant difference.
Results
LCZ696 improves MI-induced myocardial
damage and suppresses inflammatory responses
Cardiac ultrasound of LV function demonstrated that
MI could lead to a thicker diameter of LV in both diastole and
systole, thus resulting in a fraction reduction and a poorer
ejection fraction (Fig. 1A); this
effect was time-dependent. The size of the infarction was confirmed
by TTC staining; the area of infarction increased significantly
following MI (Fig. 1B).
Inflammatory cell infiltration was observed in myocardial tissues
following the occurrence of MI and cell degeneration was detected
on day 3 following MI; these indicators had increased significantly
by day 7 (Fig. 1C). There was no
obvious interstitial fibrosis on days 1 and 3 following MI.
However, obvious interstitial fibrosis was observed in the MI area
and the area surrounding the MI area, on day 7 following MI
(Fig. 1D). After seven days of
LCZ696 treatment, a neat arrangement of myocardial cells was
observed. The data demonstrated that LCZ696 reduced the level of
interstitial fibrosis, leading to a significant reduction in CVF
when compared to that in rats in the MI group on day 7 (Fig. 1E). It was found that inflammatory
responses began on day 1 after MI; this involved a significant
increase in the levels of IL-1 β and IL-18. However, LCZ696
effectively reduced the relative levels of these inflammatory
factors in the serum (Fig. 1F),
suggesting that LCZ696 can improve the myocardial damage caused by
MI and inhibit inflammation.
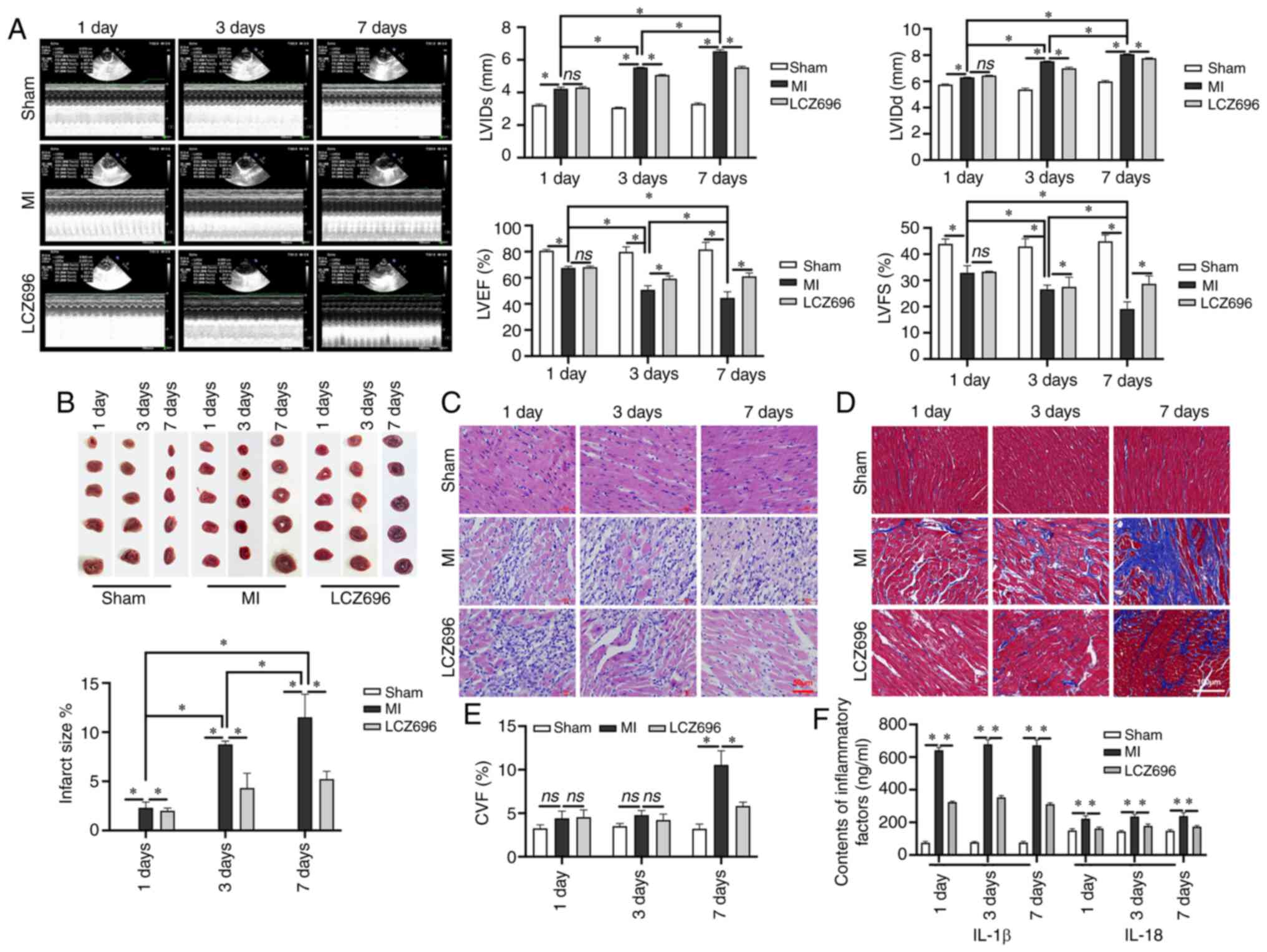 | Figure 1.LCZ696 rescues MI-induced
histopathological changes in myocardial cells and improves the
inflammatory response in rats. (A) M-mode ultrasonic examination
and LV function analysis (n=24). (B) Infarct size, as detected by
TTC staining. (C) H&E staining of myocardial tissues (scale bar
= 50 µm). (D) Interstitial fibrosis by Masson's staining (scale
bar=100 µm). (E) CVF (%) as determined by Masson's staining. (F)
Levels of IL-1 β and IL-18 in the serum of experimental rats, as
detected by ELISA. Data are given as mean ± standard deviation,
*P<0.05, ns; P>0.05. LVIDs: left ventricular systolic
internal dimension. LCZ696, sacubitril/valsartan; MI, myocardial
infarction; TTC, triphenyltetrazolium chloride; H&E,
hematoxylin and eosin; CVF, collagen volume fraction; LVEF, left
ventricular ejection fraction; LVFS, left ventricular fractional
shortening, as determined by the following formula:
(LVIDd-LVIDs)/LVIDd ×100%; CVF was calculated as follows:
CVF=collagen area/view area ×100%. |
LCZ696 inhibits the expression of the
NLRP3 inflammasome following MI
To investigate the inhibition of LCZ696 on NLRP3
inflammasome, it was found that the expression levels of the NLRP3
inflammasome increased steadily from day 1 following MI (Fig. 2A and B). A significant accumulation
of ROS following MI was also detected and the levels of ROS
remained high until day 7 following MI (Fig. 2C). When NLRP3 was activated, the
expression levels of pro-caspase-l were upregulated and the
expression levels of caspase-1 p10, pro-IL-lβ and pro-IL-18, were
also upregulated (Fig. 2D and E);
these proteins were subsequently transformed into biologically
activate IL-lβ and IL-18 (Fig. 2F).
LCZ696 was able to inhibit the accumulation of ROS and downregulate
the expression levels of NLRP3. LCZ696 was also able to
downregulate the expression levels of pro-caspase-l, pro-IL-lβ and
pro-IL-18 and suppress the release of IL-lβ and IL-18.
 | Figure 2.LCZ696 inhibits activation of the
NLRP3 inflammasome, downstream inflammatory responses and oxidative
damage. (A) Protein and (B) mRNA expression levels of the NLRP3
inflammasome, as detected by western blotting and reverse
transcription-quantitative PCR. (C) Expression levels of reactive
oxygen species, as detected by immunofluorescence (scale bar=50
µm). (D) Relative expression levels of pro-caspase-1, caspase-1
p10, pro-IL-1β and pro-IL-18, as determined by western blotting.
(E) mRNAs expression levels of caspase-1, IL-1β and IL-18. (F)
Levels of inflammatory factors IL-1β and IL-18 in samples of rat
serum determined using ELISA. Data are given as mean ± standard
deviation. *P<0.05. LCZ696, sacubitril/valsartan; NLRP3, NLR
pyrin family domain containing 3; casp, caspase. |
MI mediates the extent of
pyroptosis
In view of the clear association between NLRP3
inflammasome activation and pyroptosis, whether pyroptosis occurred
after the establishment of MI model was next investigated.
Pyroptosis is known as GSDMD-dependent programmed cell death; thus,
GSDMD is a crucial executor of pyroptosis and is cleaved by
activated inflammatory caspases-1 into gasdermin N-terminal
(GSDMD-NT) and gasdermin C-terminal (36,37).
GSDMD-NT assembles membrane pores to promote cytolysis, thus
leading to programmed necrosis/cell death, which is a key step in
pyroptosis (36,37). The present study found that the
expression and lysis of GSDMD increased significantly from day 1 of
MI and that LCZ696 could reduce these effects (Fig. 3A and B). These results demonstrated
that pyroptosis might serve an important role in the progression of
myocardial injury following MI.
LCZ696 downregulates the expression of
the TAK1/JNK pathway-related proteins following MI
TAK1 is known to participate in the regulation of
cell pyroptosis (38,39) and may be activated by various
intracellular and extracellular stimuli in order to regulate cell
viability and inflammation. The present study investigated changes
in the expression levels of key proteins in the TAK1/JNK pathway
following MI and found that the protein and mRNA levels of TAK1,
and the phosphorylation of JNK, were upregulated from day 1
following MI (Fig. 4A and B). It
was also found that LCZ696 inhibited the expression of key proteins
in the TAK1/JNK pathway. The expression levels of proteins in the
TAK1/JNK pathway and NLRP3 in myocardial tissues was also
investigated; it was found that both TAK1/NLRP3 and JNK/NLRP3 were
co-expressed in myocardial tissues (Fig. 4C and D), suggesting that the
TAK1/JNK pathway may mediate the expression of the NLRP3
inflammasome.
The TAK1/JNK pathway activates the
NLRP3 inflammasome and pyroptosis
As the present study observed the co-expression of
TAK1/NLRP3 in the rat model of MI, the role of TAK1 in
NLRP3-induced myocardial cell pyroptosis was next investigated. A
hypoxia-simulated model of MI was created in H9C2 cardiomyocytes
in vitro. It was found that the protein expression levels of
TAK1 and the phosphorylation of JNK were both elevated in cell
model of MI (Fig. 5A and B).
However, LCZ696 was able to inhibit the effects on the NLRP3
inflammasome and pro-caspase-1 in the in vitro studies.
Notably, the overexpression of TAK1 reversed the inhibitory effects
of LCZ696 on hypoxia-induced inflammatory responses in myocardial
cells and this led to an increase in the in vitro expression
and lysis of GSDMD, the activation of NLRP3 and an increase in the
expression levels of pro-caspase-l (Fig. 5A). It was also found that the levels
of IL-1β and IL-18 were enhanced in cell supernatants (Fig. 5C), thus indicating that LCZ696
improved MI-induced inflammatory responses via the TAK1/JNK
signaling pathway.
 | Figure 5.Activation of the TAK1/JNK pathway
leads to accumulation of the NLRP3 inflammasome and the induction
of cellular pyroptosis, resulting in increased inflammatory
responses in H9c2 cells. (A) Relative expression levels of
proteins, as detected by western blotting. (B) mRNA expression
levels of TAK1, JNK, NLRP3, caspase-1 and GSDMD, as determined by
reverse transcription-quantitative PCR. (C) Levels of IL-1β and
IL-18, in samples of rat serum, as detected by ELISA. Data were
given as mean ± standard deviation, *P<0.05. TAK1, transforming
growth factor β-activated kinase-1; NLRP3, NLR pyrin family domain
containing 3; GSDMD, gasdermin D; p-, phosphorylated. |
Discussion
Ventricular remodeling is a key physiological
process following MI. Myocardial cells begin to die in the early
stages of ventricular remodeling (40,41).
Myocardial systolic and diastolic function depends on the integrity
of the cardiomyocytes (42).
However, due to regenerative limitations, the death of a
significant number of myocardial cells will inevitably lead to a
decline in cardiac function. The present study established a model
of MI model by ligating the left anterior descending coronary
artery in SD rats and then treated these animals with LCZ696. It
was found that LCZ696 improved MI-induced myocardial fibrosis,
inhibited inflammatory responses and improved cardiac functions.
Previous studies reported that the expression levels of NLRP3
increase during the early remodeling stage following MI and that
NLRP3 can recruit and activate caspase-1 by combining with ASC
(43–45). The activation of caspase-1 promotes
the maturation and release of IL-1β which ultimately leads to the
death of cardiomyocytes; caspase-1 regulates Ang II-induced
cardiomyocyte hypertrophy via the upregulation of IL-1 (46). TGF-β is known to serve a crucial
role in ventricular remodeling (47–50).
The present study explored the association between the
non-canonical TGF-β signaling pathway TAK1/JNK and cell pyroptosis.
It was found that over-activation of the NLRP3 inflammasome was
associated with the death of cardiomyocytes by pyroptosis and that
the TAK1/JNK pathway can promote the activation of NLRP3. The
inhibition of TAK1 and JNK led to a significant suppression of
myocardial damage. Therefore, the targeted inhibition of TAK1 can
block the process of pyroptosis in cardiomyocytes, thereby
improving myocardial injury. Thus, TAK1 represents a potential
novel therapeutic target.
LCZ696 is a novel drug that was designed for the
treatment of cardiovascular diseases. It combines valsartan (an
angiotensin receptor blocker) and sacubitril (an enkephalinase
inhibitor prodrug) at a ratio of 1:1 in a sodium supramolecular
complex (51). LCZ696 can
considerably increase the levels of natriuretic peptide, which can
exhibit diuretic and anti-proliferative effects and can exhibit
effects against cardiac hypertrophy (51). In addition, LCZ696 can also reduce
Ang II type 1 receptor activity, inhibit vasoconstriction and
sympathetic nerve excitement, reduce the secretion of aldosterone
and suppress inflammatory responses and oxidative stress by
inhibiting enkephalinase and Ang II receptors (52,53). A
previous study compared the efficacies of LCZ696 and enalapril in
the treatment of MI and illustrated that LCZ696 can significantly
increase the rate of survival following MI and improve the
imbalance of the RAAS and natriuretic peptide system, thus helping
to prevent the myocardial complication of heart rupture after
infarction (32). The in
vivo experiments of the present study found that LCZ696
exhibited significant inhibition of myocardial fibrosis and
improved ventricular remodeling. These data suggested that LCZ696
can inhibit pyroptosis and expression of the NLRP3 inflammasome in
myocardial cells.
The death of cardiomyocytes is an essential
mechanism during ventricular remodeling (54). Cellular apoptosis is an important
mechanism underlying the death of cardiomyocytes (55). However, addressing apoptosis in
cardiomyocytes has failed to identify a specific means of
preventing cardiomyocyte death. As a newly discovered form of
programmed cell death, cellular pyroptosis has received widespread
attention because of its potential role in the occurrence and
development of a variety of diseases (56–59).
The present study investigated the expression levels of GSDMD and
GSDMD-NT and found that pyroptosis serves an important role in the
death of cardiomyocytes. Studies have also shown that inflammasomes
such as NLRP3 and NLRP1 can chemotactically activate caspase-1
following extracellular stimulation, thus triggering the classic
caspase-1-mediated cell pyroptosis pathway (60–62).
Subsequently, the activated caspase-1 can cleave GSDMD at specific
sites to release the GSDMD-N-terminal domain which can accumulate
inside the cell membrane and form non-selective pores, thus
resulting in an increase in the permeability of the cell membrane
and leading to cellular pyroptosis (23). The present study demonstrated the
dominant role of cell pyroptosis following MI and demonstrated that
LCZ696 can suppress the expression levels of proteins related to
the pyroptosis pathway. These data suggest that LCZ696 may improve
ventricular remodeling by inhibiting NLRP3 pathways and cellular
pyroptosis.
The NLRP3 inflammasome is an overall sensor of
infection and stress; the activation of this inflammasome has been
associated with a range of human diseases (63,64).
Research has demonstrated that TAK1 is the central regulator of
NLRP3 inflammasome activation and spontaneous cell death (65). The lack of TAK1 in macrophages has
been shown to induce the spontaneous activation of the NLRP3
inflammasome without TLR priming and subsequent activation signals,
thus suggesting the unique role of TAK1 in maintaining the steady
state of the NLRP3 inflammasome (65–67).
In a previous study, Malireddi et al (65). demonstrated that the loss of
macrophage-specific TAK1 can lead to the uncontrolled secretion of
TNF and abnormal signaling transmission, thereby leading to the
spontaneous activation of the NLRP3 inflammasome. The present study
first demonstrated that LCZ696 can inhibit the TAK1/JNK signaling
pathway, thus indicating a potential mechanism by which this new
drug exerts its effects. Furthermore, it confirmed that TAK1/JNK is
involved in the activation of the NLRP3 inflammasome. The
overexpression of TAK1 was shown to regulate the level of JNK
phosphorylation and simultaneously activated the NLRP3
inflammasome, thus promoting pyroptosis and the secretion of
inflammatory factors. Notably, it was found that LCZ696 inhibited
the expression of TAK1 and NLRP3. However, there are still some
limitations to the present study. It would be more useful if
whether LCZ696 could perform its effect on Myocardial injury after
myocardial infarction under the activation of TAK1/JNK signaling
pathway could be investigated.
In conclusion, the present study proposed that
LCZ696 downregulated the expression of the NLRP3 inflammasome,
reduced cell pyroptosis, improved ventricular remodeling and thus
reduced myocardial damage by inhibiting the TAK1/JNK signaling
pathway.
Acknowledgements
Not applicable.
Funding
The present study was supported by the National Key
Research and development program (grant. no. 2016YFC1301004).
Availability of data and materials
The datasets used and analyzed during the current
study are available from the corresponding author upon reasonable
request.
Authors' contributions
JS and GQ designed the research, analyzed the data
and wrote the manuscript. JS, ZF performed the experiments and
prepared the figures. GS prepared the figures and analyzed the
data. JS and GS confirmed the authenticity of all the raw data. GQ
performed critical revision of the manuscript and supervised the
study. All authors read and approved the final manuscript.
Ethics approval and consent to
participate
The experiments were approved by the Laboratory
Animal Welfare and Ethics Committee of China Medical University
(approval number IACUC no. 2019109).
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Konstantinidis K, Whelan RS and Kitsis RN:
Mechanisms of cell death in heart disease. Arterioscler Thromb Vasc
Biol. 32:1552–1562. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Elmore S: Apoptosis: A review of
programmed cell death. Toxicol Pathol. 35:495–516. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Cahill TJ and Kharbanda RK: Heart failure
after myocardial infarction in the era of primary percutaneous
coronary intervention: Mechanisms, incidence and identification of
patients at risk. World J Cardiol. 9:407–415. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Bergsbaken T, Fink SL and Cookson BT:
Pyroptosis: Host cell death and inflammation. Nat Rev Microbiol.
7:99–109. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Wree A, Eguchi A, McGeough MD, Pena CA,
Johnson CD, Canbay A, Hoffman HM and Feldstein AE: NLRP3
inflammasome activation results in hepatocyte pyroptosis, liver
inflammation, and fibrosis in mice. Hepatology. 59:898–910. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Man SM, Karki R and Kanneganti TD:
Molecular mechanisms and functions of pyroptosis, inflammatory
caspases and inflammasomes in infectious diseases. Immunol Rev.
277:61–75. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Azevedo PS, Polegato BF, Minicucci MF,
Paiva SAR and Zornoff LAM: Cardiac remodeling: Concepts, clinical
impact, pathophysiological mechanisms and pharmacologic treatment.
Arq Bras Cardiol. 106:62–69. 2016.PubMed/NCBI
|
|
8
|
Chen G, Li J, Song M, Wu Z, Zhang W, Wang
Z, Gao J, Yang Z and Ou C: A mixed component supramolecular
hydrogel to improve mice cardiac function and alleviate ventricular
remodeling after acute myocardial infarction. Adv Funct Materials.
27:17017982017.https://doi.org/10.1002/adfm.201701798 View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Kale P and Afzal A: Stage B heart failure:
To strain or not to strain. JACC Cardiovasc Imaging. 11:1401–1404.
2018. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Bujak M and Frangogiannis NG: The role of
TGF-beta signaling in myocardial infarction and cardiac remodeling.
Cardiovasc Res. 74:184–195. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Dobaczewski MW, Chen W and Frangogiannis
NG: Transforming growth factor (TGF)-β signaling in cardiac
remodeling. J Mol Cell Cardiol. 51:600–606. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Frangogiannis NG: The role of transforming
growth factor (TGF)-β in the infarcted myocardium. J Thorac Dis. 9
(Suppl 1):S52–S63. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Monzen K, Hiroi Y, Kudoh S, Akazawa H, Oka
T, Takimoto E, Hayashi D, Hosoda T, Kawabata M, Miyazono K, et al:
Smads, TAK1, and their common target ATF-2 play a critical role in
cardiomyocyte differentiation. J Cell Biol. 153:687–698. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Mihaly SR, Ninomiya-Tsuji J and Morioka S:
TAK1 control of cell death. Cell Death Differ. 21:1667–1676. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Okada M, Matsuzawa A, Yoshimura A and
Ichijo H: The lysosome rupture-activated TAK1-JNK pathway regulates
NLRP3 inflammasome activation. J Biol Chem. 289:32926–3236. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Toldo S, Mezzaroma E, Mauro AG, Salloum F,
Van Tassell BW and Abbate A: The inflammasome in myocardial injury
and cardiac remodeling. Antioxid Redox Signal. 22:1146–1161. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Zhaolin Z, Guohua L, Shiyuan W and Zuo W:
Role of pyroptosis in cardiovascular disease. Cell Prolif.
52:e125632019. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Mezzaroma E, Toldo S, Farkas D, Seropian
IM, Van Tassell BW, Salloum FN, Kannan HR, Menna AC, Voelkel NF and
Abbate A: The inflammasome promotes adverse cardiac remodeling
following acute myocardial infarction in the mouse. Proc Natl Acad
Sci USA. 108:19725–19730. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Liu D, Zeng X, Li X, Mehta JL and Wang X:
Role of NLRP3 inflammasome in the pathogenesis of cardiovascular
diseases. Basic Res Cardiol. 113:52018. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Ferrini A, Stevens MM, Sattler S and
Rosenthal N: Toward regeneration of the heart: Bioengineering
strategies for immunomodulation. Front Cardiovasc Med. 6:262019.
View Article : Google Scholar : PubMed/NCBI
|
|
21
|
McKenzie BA, Mamik MK, Saito LB, Boghozian
R, Monaco MC, Major EO, Lu JQ, Branton WG and Power C: Caspase-1
inhibition prevents glial inflammasome activation and pyroptosis in
models of multiple sclerosis. Proc Natl Acad Sci USA.
115:E6065–E6074. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Sharma B, McLeland CB, Potter TM, Stern ST
and Adiseshaiah PP: Assessing NLRP3 inflammasome activation by
nanoparticles. Methods Mol Biol. 1682:135–147. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Fann DY, Santro T, Manzanero S,
Widiapradja A, Cheng YL, Lee SY, Chunduri P, Jo DG, Stranahan AM,
Mattson MP and Arumugam TV: Intermittent fasting attenuates
inflammasome activity in ischemic stroke. Exp Neurol. 257:114–119.
2014. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Zhu ZD, Ye JY, Niu H, Ma YM, Fu XM, Xia ZH
and Zhang X: Effects of microRNA-292-5p on myocardial
ischemia-reperfusion injury through the peroxisome
proliferator-activated receptor-α/-γ signaling pathway. Gene Ther.
25:234–248. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Volpe M, Carnovali M and Mastromarino V:
The natriuretic peptides system in the pathophysiology of heart
failure: From molecular basis to treatment. Clin Sci (Lond).
130:57–77. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Zablocki D and Sadoshima J: Angiotensin II
and oxidative stress in the failing heart. Antioxid Redox Signal.
19:1095–1109. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Jones ES, Vinh A, McCarthy CA, Gaspari TA
and Widdop RE: AT2 receptors: Functional relevance in
cardiovascular disease. Pharmacol Ther. 120:292–316. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Michel MC, Brunner HR, Foster C and Huo Y:
Angiotensin II type 1 receptor antagonists in animal models of
vascular, cardiac, metabolic and renal disease. Pharmacol Ther.
164:1–81. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Kompa AR, Lu J, Weller TJ, Kelly DJ, Krum
H, von Lueder TG and Wang BH: Angiotensin receptor neprilysin
inhibition provides superior cardioprotection compared to
angiotensin converting enzyme inhibition after experimental
myocardial infarction. Int J Cardiol. 258:192–198. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Shirley M and McCormack PL:
Perindopril/amlodipine (Prestalia®): A review in
hypertension. Am J Cardiovasc Drugs. 15:363–370. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Campbell DJ: A review of perindopril in
the reduction of cardiovascular events. Vasc Health Risk Manag.
2:117–124. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Ishii M, Kaikita K, Sato K, Sueta D,
Fujisue K, Arima Y, Oimatsu Y, Mitsuse T, Onoue Y, Araki S, et al:
Cardioprotective effects of LCZ696 (Sacubitril/Valsartan) after
experimental acute myocardial infarction. JACC Basic Transl Sci.
2:655–668. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Chang PC, Lin SF, Chu Y, Wo HT, Lee HL,
Huang YC, Wen MS and Chou CC: LCZ696 therapy reduces ventricular
tachyarrhythmia inducibility in a myocardial infarction-induced
heart failure rat model. Cardiovasc Ther. 2019:60326312019.
View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Livak KJ and Schmittgen TD: Analysis of
relative gene expression data using real-time quantitative PCR and
the 2(-Delta Delta C(T)) method. Methods. 25:402–408. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Li W, Li Y, Chu Y, Wu W, Yu Q, Zhu X and
Wang Q: PLCE1 promotes myocardial ischemia-reperfusion injury in
H/R H9c2 cells and I/R rats by promoting inflammation. Biosci Rep.
39:BSR201816132019. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Shi J, Zhao Y, Wang K, Shi X, Wang Y,
Huang H, Zhuang Y, Cai T, Wang F and Shao F: Cleavage of GSDMD by
inflammatory caspases determines pyroptotic cell death. Nature.
526:660–665. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Liu X, Zhang Z, Ruan J, Pan Y, Magupalli
VG, Wu H and Lieberman J: Inflammasome-activated gasdermin D causes
pyroptosis by forming membrane pores. Nature. 535:153–158. 2016.
View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Malireddi R, Kesavardhana S and Kanneganti
TD: ZBP1 and TAK1: Master regulators of NLRP3
inflammasome/pyroptosis, apoptosis, and necroptosis (PAN-optosis).
Front Cell Infect Microbiol. 9:4062019. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Orning P, Weng D, Starheim K, Ratner D,
Best Z, Lee B, Brooks A, Xia S, Wu H, Kelliher MA, et al: Pathogen
blockade of TAK1 triggers caspase-8-dependent cleavage of gasdermin
D and cell death. Science. 362:1064–1069. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Bhatt AS, Ambrosy AP and Velazquez EJ:
Adverse remodeling and reverse remodeling after myocardial
infarction. Curr Cardiol Rep. 19:712017. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Li J, Cai SX, He Q, Zhang H, Friedberg D,
Wang F and Redington AN: Intravenous miR-144 reduces left
ventricular remodeling after myocardial infarction. Basic Res
Cardiol. 113:362018. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Caporizzo MA, Chen CY, Bedi K, Margulies
KB and Prosser BL: Microtubules increase diastolic stiffness in
failing human cardiomyocytes and myocardium. Circulation.
141:902–915. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Tang YS, Zhao YH, Zhong Y, Li XZ, Pu JX,
Luo YC and Zhou QL: Neferine inhibits LPS-ATP-induced endothelial
cell pyroptosis via regulation of ROS/NLRP3/caspase-1 signaling
pathway. Inflamm Res. 68:727–738. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Gao R, Shi H, Chang S, Gao Y, Li X, Lv C,
Yang H, Xiang H, Yang J, Xu L and Tang Y: The selective
NLRP3-inflammasome inhibitor MCC950 reduces myocardial fibrosis and
improves cardiac remodeling in a mouse model of myocardial
infarction. Int Immunopharmacol. 74:1055752019. View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Takahashi M: Role of NLRP3 inflammasome in
cardiac inflammation and remodeling after myocardial infarction.
Biol Pharm Bull. 42:518–523. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
46
|
Bai Y, Sun X, Chu Q, Li A, Qin Y, Li Y,
Yue E, Wang H, Li G, Zahra SM, et al: Caspase-1 regulates Ang
II-induced cardiomyocyte hypertrophy via up-regulation of IL-1β.
Biosci Rep. 38:BSR201714382018. View Article : Google Scholar : PubMed/NCBI
|
|
47
|
Cohn JN, Ferrari R and Sharpe N: Cardiac
remodeling-concepts and clinical implications: A consensus paper
from an international forum on cardiac remodeling. Behalf of an
international forum on cardiac remodeling. J Am Coll Cardiol.
35:569–582. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
48
|
Schnee JM and Hsueh WA: Angiotensin II,
adhesion, and cardiac fibrosis. Cardiovasc Res. 46:264–268. 2000.
View Article : Google Scholar : PubMed/NCBI
|
|
49
|
Yamazaki T, Komuro I, Shiojima I and
Yazaki Y: The renin-angiotensin system and cardiac hypertrophy.
Heart. 76 (Suppl 3):S33–S35. 1996. View Article : Google Scholar : PubMed/NCBI
|
|
50
|
Brand T and Schneider MD: Transforming
growth factor-beta signal transduction. Circ Res. 78:173–179. 1996.
View Article : Google Scholar : PubMed/NCBI
|
|
51
|
da Silva PM and Aguiar C:
Sacubitril/valsartan: An important piece in the therapeutic puzzle
of heart failure. Rev Port Cardiol. 36:655–668. 2017.(In English,
Portuguese). PubMed/NCBI
|
|
52
|
Forte M, Madonna M, Schiavon S, Valenti V,
Versaci F, Zoccai GB, Frati G and Sciarretta S: Cardiovascular
pleiotropic effects of natriuretic peptides. Int J Mol Sci.
20:38742019. View Article : Google Scholar : PubMed/NCBI
|
|
53
|
Lee NS and Daniels LB: Current
understanding of the compensatory actions of cardiac natriuretic
peptides in cardiac failure: A clinical perspective. Card Fail Rev.
2:14–19. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
54
|
Ni J, Li Y, Xu Y and Guo R: Salidroside
protects against cardiomyocyte apoptosis and ventricular remodeling
by AKT/HO-1 signaling pathways in a diabetic cardiomyopathy mouse
model. Phytomedicine. 82:1534062021. View Article : Google Scholar : PubMed/NCBI
|
|
55
|
Xia P, Liu Y and Cheng Z: Signaling
pathways in cardiac myocyte apoptosis. Biomed Res Int.
2016:95832682016. View Article : Google Scholar : PubMed/NCBI
|
|
56
|
Jia C, Chen H, Zhang J, Zhou K, Zhuge Y,
Niu C, Qiu J, Rong X, Shi Z, Xiao J, et al: Role of pyroptosis in
cardiovascular diseases. Int Immunopharmacol. 67:311–318. 2019.
View Article : Google Scholar : PubMed/NCBI
|
|
57
|
Wu J, Lin S, Wan B, Velani B and Zhu Y:
Pyroptosis in liver disease: New insights into disease mechanisms.
Aging Dis. 10:1094–1108. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
58
|
Guo H, Xie M, Zhou C and Zheng M: The
relevance of pyroptosis in the pathogenesis of liver diseases. Life
Sci. 223:69–73. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
59
|
Zheng Z and Li G: Mechanisms and
therapeutic regulation of pyroptosis in inflammatory diseases and
cancer. Int J Mol Sci. 21:14562020. View Article : Google Scholar : PubMed/NCBI
|
|
60
|
Teng JF, Mei QB, Zhou XG, Tang Y, Xiong R,
Qiu WQ, Pan R, Law BYK, Wong VKW, Yu CL, et al: Polyphyllin VI
induces caspase-1-mediated pyroptosis via the induction of
ROS/NF-κB/NLRP3/GSDMD signal axis in non-small cell lung cancer.
Cancers (Basel). 12:1932020. View Article : Google Scholar : PubMed/NCBI
|
|
61
|
Li A, Yu Y, Ding X, Qin Y, Jiang Y, Wang
X, Liu G, Chen X, Yue E, Sun X, et al: MiR-135b protects
cardiomyocytes from infarction through restraining the
NLRP3/caspase-1/IL-1β pathway. Int J Cardiol. 307:137–145. 2020.
View Article : Google Scholar : PubMed/NCBI
|
|
62
|
Tan M, Tan L, Jiang T, Zhu XC, Wang HF,
Jia CD and Yu JT: Amyloid-β induces NLRP1-dependent neuronal
pyroptosis in models of Alzheimer's disease. Cell Death Dis.
5:e1382. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
63
|
Burdette D, Haskett A, Presser L, McRae S,
Iqbal J and Waris G: Hepatitis C virus activates interleukin-1beta
via caspase-1-inflammasome complex. J Gen Virol. 93:235–246. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
64
|
Theofani E, Semitekolou M, Morianos I,
Samitas K and Xanthou G: Targeting NLRP3 inflammasome activation in
severe asthma. J Clin Med. 8:16152019. View Article : Google Scholar : PubMed/NCBI
|
|
65
|
Malireddi RS, Gurung P, Mavuluri J, Dasari
TK, Klco JM, Chi H and Kanneganti TD: TAK1 restricts spontaneous
NLRP3 activation and cell death to control myeloid proliferation. J
Exp Med. 215:1023–1034. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
66
|
Malireddi R, Gurung P, Kesavardhana S,
Samir P, Burton A, Mummareddy H, Vogel P, Pelletier S, Burgula S
and Kanneganti TD: Innate immune priming in the absence of TAK1
drives RIPK1 kinase activity-independent pyroptosis, apoptosis,
necroptosis, and inflammatory disease. J Exp Med. 217:201916442020.
View Article : Google Scholar
|
|
67
|
Yang Y, Wang, Kouadir M, Song H and Shi F:
Recent advances in the mechanisms of NLRP3 inflammasome activation
and its inhibitors. Cell Death Dis. 10:1282019. View Article : Google Scholar : PubMed/NCBI
|
















