Introduction
An abdominal aortic aneurysm (AAA) is diagnosed if
the diameter of an artery is >30 mm or if it has increased by
>50% compared with the adjacent normal aorta (1). The mortality rate of patients with an
AAA rupture is 65–85%; currently, surgery or minimally invasive
interventions are used for clinical treatment (2). However, randomized trials have
reported that, although selective repair of AAAs can prevent
rupture of the aneurysms and related deaths, it causes more
perioperative deaths than any other common types of surgery or
vascular surgery (3). Especially
for small aneurysms, the benefits of surgical repair are not
obvious, and an AAA rupture is likely to be prevented by aneurysm
therapy to inhibit the growth of small aneurysms (4). Therefore, examining effective drugs to
treat small AAAs is urgently required.
Chronic inflammation is the main pathological
feature in aneurysmal tissues, involving the infiltration of
inflammatory cells, elastic fiber degradation of the extracellular
matrix (ECM), neovascularization and apoptosis, dysfunction of
vascular smooth muscle cells and the release of a range of
proteolytic enzymes, such as MMPs, oxidation-derived free radicals,
cytokines and related products (5).
Macrophages are the dominant factor in the production of MMPs
during the inflammation of the media and adventitia. These MMPs
cause the degradation of the ECM (6).
Receptor for advanced glycosylation end products
(RAGE) is a cell-surface multiligand receptor of the immunoglobulin
superfamily (7). As an important
regulator of chronic and sustained inflammation, RAGE serves a key
role in the development and progression of pathological states,
such as cardiovascular disease, diabetic vascular complications,
Alzheimer's disease (AD), cancer and a range of inflammatory
diseases (8,9). RAGE exists as a full-length isoform
(fl-RAGE) and a soluble isoform (sRAGE). fl-RAGE consists of
extracellular domains, a short transmembrane helix and a cytosolic
C-terminal short tail. The cytosolic short tail domain is critical,
and is responsible for transducing downstream signaling from the
extracellular space to the intracellular space (10). sRAGE is one of the truncated forms
of RAGE and is found in the serum in humans. As an endogenous
competitor of RAGE, increased levels of sRAGE serve a critical role
within the modulatory network of the ligand/RAGE axis and acts to
alleviate the inflammatory response. As a biomarker of
RAGE-mediated pathogenesis, sRAGE can reflect RAGE activity in the
opposite direction (11).
Furthermore, ectodomain shedding by proteolysis of RAGE is a means
of sRAGE production.
High mobility group box 1 (HMGB1), one of the
ligands of RAGE and a proinflammatory factor, is an evolutionarily
highly conserved nuclear, nonhistone DNA-binding protein that
exerts a proinflammatory role in a series of pathologies by
regulating the release of inflammatory cytokines, activating
macrophages and increasing vascular permeability (12). HMGB1 is involved in the pathogenesis
of inflammatory conditions via its interactions with pivotal
transmembrane receptors, including RAGE and Toll-like receptor-4
(13,14). Extracellular hyperexpression of
HMGB1 has been documented in various diseases, such as
cardiovascular disease, septic shock, rheumatoid arthritis,
atherosclerosis, traumatic brain injury and ischemia/reperfusion
injury (15–17). After binding to membrane receptors,
HMGB1 activates downstream NF-κB and further stimulates the
expression of related inflammatory factors, such as oxidative
stress and proinflammatory cytokines, which has been observed in
AAAs (18).
ADAM metallopeptidase domain 10 (ADAM10) is one of
the most closely studied members of the ADAM family, which has been
shown to participate in the inflammatory response, and it is a
target for the treatment of inflammatory diseases (19). As a membrane-anchored protease,
ADAM10-mediated shedding is essential for numerous biological
processes, such as cell migration, proliferation and (20,21).
ADAM10 is also responsible for RAGE cleavage, and binding to its
ligand HMGB1 promotes RAGE shedding (22). Moreover, targeting of ADAM10 is a
therapeutic strategy for the treatment of AD and prion disease
(23), but its role in AAAs remains
unknown.
Therefore, in the present study, we hypothesized
that ADAM10 may attenuate the inflammatory progression of AAAs by
inhibiting HMGB1/RAGE/NF-κB signaling. The current study examined
the effect of ADAM10 on the expression levels of HMGB1 and RAGE,
and the activation of NF-κB in an experimental model of AAAs in
C57BL/6 mice.
Materials and methods
Animal groups
A total of 21 male C57BL/6 wild-type mice (age, 8–12
weeks; weight, 20–26 g) were purchased from the Experimental Animal
Center of Shandong University (Jinan, China). The mice were
randomly divided into the following groups (n=7 per group): i)
Sham-operated (Sham) group; ii) AAA model (AAA) group; and iii)
ADAM10-treated (ADAM10) group. Mice were housed at 20–25°C and a
humidity of 40–70% with a 12:12 h light-dark cycle. Water and food
were given ad libitum and were provided by the Experimental
Center of Shandong Provincial Hospital Affiliated with Shandong
University. This study was performed according to the Care and Use
of Laboratory Animals guidelines (24). The experimental protocols were
approved by the Ethical Review Board of Shandong University
(approval no. 2018-015). The animal experiments were performed
between 01/2019 and 12/2019.
Establishment of the AAA model and
administration of ADAM10
The AAA model was established as described
previously (25). Briefly, mice
were anesthetized via 2.5% isoflurane inhalation. A para-abdominal
median incision was made to expose the lower abdominal aorta to the
bifurcation of the abdominal aorta, and a 10-mm segment of the
infrarenal abdominal aorta was exposed. Subsequently, 30 µl porcine
pancreatic elastase (1.5 U/ml; cat. no. E1250; Sigma-Aldrich; Merck
KGaA) was perfused intraluminally for 5 min via a PPE-10 catheter
that had been inserted into the aorta. PBS was used as a control in
the Sham group. ADAM10 (6 mg/kg; cat. no. 936-AD; R&D Systems,
Inc.) was injected intraperitoneally after 3 days of porcine
pancreatic elastase perfusion in the ADAM10 group and the treatment
continued for 10 days. At the end of the study, the mice were
euthanized under CO2 exposure (flow rate of
CO2, 2 l/min; air displacement rate, 20%/min). Blood was
collected after mouse euthanasia via cardiac puncture. Blood
samples were separated via centrifugation at 3,000 × g at 4°C for
10 min and stored at −80°C. The aortas were embedded in Tissue-Tek
O.C.T. compound at −80°C or stored at −80°C for protein and reverse
transcription-quantitative (RT-q) PCR detection.
Measurements of the abdominal aortic
diameter
The maximum inner luminal diameters of the
infrarenal abdominal aortas were measured using an animal
ultrasound system (FUJIFILM VisualSonics, Inc.). Measurements and
analysis of dilation were carried out on days 3, 7 and 14 after AAA
induction by two experienced operators who did not know their group
assignment. AAA was defined as dilatation >50% of the average
diameter of the aorta.
ELISA examination
The levels of HMGB1 and sRAGE in the serum samples
were measured using ELISA kits (HMGB1, cat. no. CSB-E08225, Cusabio
Technology LLC; sRAGE, cat. no. MRG00, R&D Systems, Inc.)
according to the manufacturer's instructions. Sample diluent (100
µl) and serum (100 µl) were added to the wells and incubated at
37°C for 2 h. Subsequently, biotin-labeled antibody working fluid
(100 µl; 1X antibody) was added to each well and incubated at 37°C
for 1 h. Then, the plate was washed three times with 0.01 M PBS. A
total of 100 µl substrate solution was added and incubated at room
temperature for 1 h. TMB color developing agent (90 µl) was added
to each well and incubated at 37°C for 30 min in the dark, and then
50 µl termination solution was added. The optical density value was
measured using a ThermoMultiskan GO microplate reader (1510-01981;
Thermo Fisher Scientific, Inc.) at 450 nm.
H&E and Elastin van Gieson (EVG)
staining
The aortic specimens were fixed in 4%
paraformaldehyde for 24 h at room temperature, then cut into 5-µm
serial sections using a freezing microtome. H&E and EVG
staining were performed following a standard protocol. The sections
were stained with hematoxylin for 3 min and eosin solution for 15
sec at room temperature. The sections were stained with Weigerts
Resorcin Fuchsin solution for 45 min and Van Gieson solutions for 5
min at room temperature for EVG staining. The integrity of the
elastin layers and elastin degradation were observed by light
microscopy (BX3-CBH; OLYMPUS cellSens Standard 1.16; Olympus
Corporation; magnification, ×50 and ×200).
Immunohistochemical staining
The aortic specimens were fixed in 4%
paraformaldehyde for 24 h at room temperature, and then after
washing and dehydration they were embedded by optimal cutting
temperature compound and stored in −80°C. The sections were cut
into 5-µm serial sections by a freezing microtome as previously
described (26). The sections were
incubated with primary polyclonal antibodies against CD68 (1:400;
cat. no. ab125212; Abcam) at 4°C overnight and blocked with goat
serum (1:10; cat. no. ZLI-9021; ZSGB-BIO) for 30 min at room
temperature. The sections were incubated with secondary HRP-goat
anti-rabbit IgG antibodies for 30 min at room temperature by the
rabbit polymer detection system (1X; cat. no. PV-6001; ZSGB-BIO).
The sections were then stained with DAB reagent (1X) for 3 min at
room temperature. Stained sections were observed by light
microscopy (BX3-CBH; OLYMPUS cellSens Standard 1.16; Olympus
Corporation).
RT-qPCR analysis
Total RNA was extracted from the abdominal aorta
tissue using TRIzol® reagent (Invitrogen; Thermo Fisher
Scientific, Inc.) according to the manufacturer's protocol. cDNA
was synthesized from 1 µg total RNA according to the manufacturer's
protocol by PrimeScript RT reagent kit gDNAEraser (cat. no. RR047A;
Takara Bio, Inc.) in a 20 µl reaction volume. RT-qPCR was performed
with 2 µl cDNA and gene-specific primers in a final 20 µl reaction
system. The forward and reverse primers designed by Takara Bio,
Inc. are shown in Table I.
Amplification was performed using a SYBR Premix Ex Taq kit (Takara
Bio, Inc.), and the thermocycling conditions were as follows:
Initial denaturation at 95°C for 30 sec, followed by 45 cycles of
95°C for 3 sec and 60°C for 30 sec. The relative mRNA expression
was assessed using the 2−∆∆Cq method (27). The relative expression of the target
gene was normalized to that of β-actin.
 | Table I.Primers for reverse
transcription-quantitative PCR analysis. |
Table I.
Primers for reverse
transcription-quantitative PCR analysis.
| Gene | Sequences |
|---|
| RAGE | Forward:
5′-GCTGTAGCTGGTGGTCAGAACA-3′ |
|
| Reverse:
5′-CCCCTTACAGCTTAGCACAAGTG-3′ |
| HMGB1 | Forward:
5′-ATGGGCAAAGGAGATCCTA-3′ |
|
| Reverse:
5′-ATTCATCATCATCATCTTCT-3′ |
| NF-κB | Forward:
5′-GCATTCTGACCTTGCCTATCT-3′ |
|
| Reverse:
5′-CTCCAGTCTCCGAGTGAAGC-3′ |
| MMP-2 | Forward:
5′-CCCTGGTGGCTGGAGGCTCT-3′ |
|
| Reverse:
5′-AACGGGGTCCCACGTCCCAA-3′ |
| MMP-9 | Forward:
5′-GCATCCGAGCAAGAAGACAAC-3′ |
|
| Reverse:
5′-CCCGACACACAGTAAGCATTC-3′ |
| IL-1β | Forward:
5′-TTGACGGACCCCAAAAGATG-3′ |
|
| Reverse:
5′-AGAAGGTGCTCATGTCCTCA-3′ |
| TNF-α | Forward:
5′-TCTCATCAGTTCTATGGCCC-3′ |
|
| Reverse:
5′-GGGAGTAGACAAGGTACAAC-3′ |
| MCP-1 | Forward:
5′-TTAAAAACCTGGATCGGAACCAA-3′ |
|
| Reverse:
5′-GCATTAGCTTCAGATTTACGGGT-3′ |
| β-actin | Forward:
5′-GTGGGCCGCTCTAGGCACCAA-3′ |
|
| Reverse:
5′-CTCTTTGATGTCACGCACGATTTC-3′ |
Western blot analysis
Mouse aortas were harvested for protein extraction
by RIPA buffer (cat. no. P0013B; Beyotime Institute of
Biotechnology) and a BCA protein concentration kit (cat. no. P0010;
Beyotime Institute of Biotechnology) was used for protein
determination. A total of 30 µg protein extract was loaded per lane
and separated by 10% SDS-PAGE and then electrotransferred to PVDF
membranes (MilliporeSigma), which were blocked with 5% skimmed milk
in PBS-0.05% Tween-20 (PBST) at room temperature for 1 h. Next,
primary antibodies against HMGB1 (1:1,000; cat. no. ab18256;
Abcam), RAGE (1:1,000; cat. no. ab3611; Abcam), phosphorylated
(p)-NF-κB p65 (Ser536; 93H1; 1:1,000; cat. no. 3033; Cell Signaling
Technology, Inc.), NF-κB p65 (1:1,000; cat. no. ab16502; Abcam) and
GAPDH (1:2,000; cat. no. ab181602; Abcam) were applied at 4°C
overnight. The membranes were washed three times, for 10 min each
time, in PBST (0.1% Tween-20). The appropriate horseradish
peroxidase-conjugated secondary antibodies were applied at room
temperature for 2 h including Goat Anti-Rabbit (1:3,000; cat. no.
ZB-5305; ZSGB-BIO) and Rabbit Anti-Mouse IgG H&L (1:2,000; cat.
no. ZB-2301; ZSGB-BIO). The bands were visualized using ECL assays
(Thermo Fisher Scientific, Inc.) and assessed via
semi-quantification of the optical density with Multi Gauge v3.2
software (FUJIFILM Wako Pure Chemical Corporation). GAPDH was used
as an internal reference.
Gelatin zymography analysis
Tissue extracts were isolated according to the
procedures described for the western blot analysis. Proteins were
extracted from mouse aorta using RIPA buffer (cat. no. P0013B;
Beyotime Institute of Biotechnology) and a BCA protein
concentration kit (cat. no. P0010; Beyotime Institute of
Biotechnology) was used for protein determination. The procedures
used were conducted as described previously (28). The gelatinolytic activities of MMP-2
and MMP-9 were quantified via densitometric analysis by Azure
Biosystems capture software v1.6 (29).
Statistical analysis
All the experiments were repeated three times. Data
were analyzed using SPSS 19.0 software (IBM Corp.) and are
presented as the mean ± SEM. The significant differences in mean
values among groups was examined via one-way ANOVA followed by
Tukey's post hoc test. P<0.05 was considered to indicate a
statistically significant difference.
Results
ADAM10 reduces pathological injury to
the abdominal aortic wall in a mouse AAA model
A total of 2 weeks after elastase perfusion, a
severe AAA developed with significant dilation of the infrarenal
aortic lumen in the AAA groups. Ultrasound examination revealed
that the largest diameters of the aorta were significantly
increased compared with those in the Sham groups, while they were
significantly decreased in the ADAM10 groups (Fig. 1A and B).
H&E staining identified that ADAM10 decreased
the injury to the abdominal aortic wall. A normal abdominal aortic
wall was shown in sham group (Fig. 1C-a
and d). The significant injury in elastic laminae presented
with flattening, fragmentation and degeneration in the medial
layer, accompanied with the thickened and remodeled aortic
adventitia in the AAA groups (Fig. 1C-b
and e). ADAM10 significantly reduced elastin degradation in the
aortic media, and the elastic lamellae partially recovered its wavy
structures, thereby maintaining the integrity of the aortic intima
in the ADAM10 groups (Fig. 1C-c and
f). EVA staining revealed the normal form of elastin (Fig. 1D-a and d). Moreover, elastin in the
aortic adventitia was thickened (Fig.
1D-b and e), which was reduced by ADAM10 (Fig. 1D-c and f).
ADAM10 reduces the inflammatory
response of the abdominal aortic wall in a mouse AAA model
The inflammatory responses in the experimental model
was examined via histopathologic analysis. Immunohistochemical
staining demonstrated that there was only a few CD68+
inflammatory cells in the normal aortic wall (Fig. 2A-a and d), while the inflammatory
cells accumulated and infiltrated into the aortic wall in the AAA
groups (Fig. 2A-b and e), and this
phenomenon was attenuated in the ADAM10 groups compared with the
AAA groups (Fig. 2A-c and f). The
increased number of CD68+ macrophages in the aortic wall
in the AAA groups increased significantly compared with sham
groups, which was significantly attenuated in the ADAM10 groups
(Fig. 2B). Furthermore, ADAM10
decreased the expression levels of the proinflammatory mediators,
including IL-1β, TNF-α and monocyte chemoattractant protein-1, in
the aortic wall of the AAA as shown via RT-qPCR analysis (Fig. 2C).
ADAM10 inhibits the expression of
MMPs
RT-qPCR analysis demonstrated that the mRNA
expression levels of MMP-2 and MMP-9 were significantly increased
in the AAA groups, but were significantly downregulated in the
ADAM10 groups on day 14 (Fig.
3A).
Gelatin zymography examination revealed that the
activities of MMP-2 and MMP-9 were notably increased in the AAA
groups and were significantly downregulated in the aneurysmal wall
in the ADAM10 groups on day 14 (Fig.
3B).
ADAM10 reduces serum HMGB1 levels,
while increasing serum sRAGE levels in a mouse AAA model
Serum HMGB1 and sRAGE levels changed in an opposite
manner in the AAA group. In the AAA group, serum HMGB1 was
significantly increased as detected via ELISA, and this effect was
reduced in the ADAM10 group (Fig.
4A). Furthermore, a lower level of serum sRAGE was detected in
the AAA group, while a significant increase in serum sRAGE levels
was observed in the ADAM10 group (Fig.
4B).
ADAM10 decreases aortic wall HMGB1,
RAGE and NF-κB expression in the AAA model
The mRNA and protein expression levels of HMGB1,
RAGE and NF-κB were detected in the aneurysm walls via RT-qPCR and
western blotting, respectively. The RT-qPCR results demonstrated
that the mRNA expression level of HMGB1 was higher in the AAA
group, which was significantly downregulated by ADAM10. Moreover,
the mRNA expression level of RAGE was higher in the AAA group,
which was significantly reduced by ADAM10. It was also found that
the mRNA expression level of NF-κB was higher in the AAA group,
which was decreased significantly by ADAM10 (Fig. 5A). The western blotting results were
consistent with those of the RT-qPCR. The protein expression levels
of HMGB1 and RAGE, and the ratio of the phosphorylation of NF-κB
p65/NF-κB p65 were higher in the AAA group, and were significantly
decreased in the ADAM10 group (Fig.
5B).
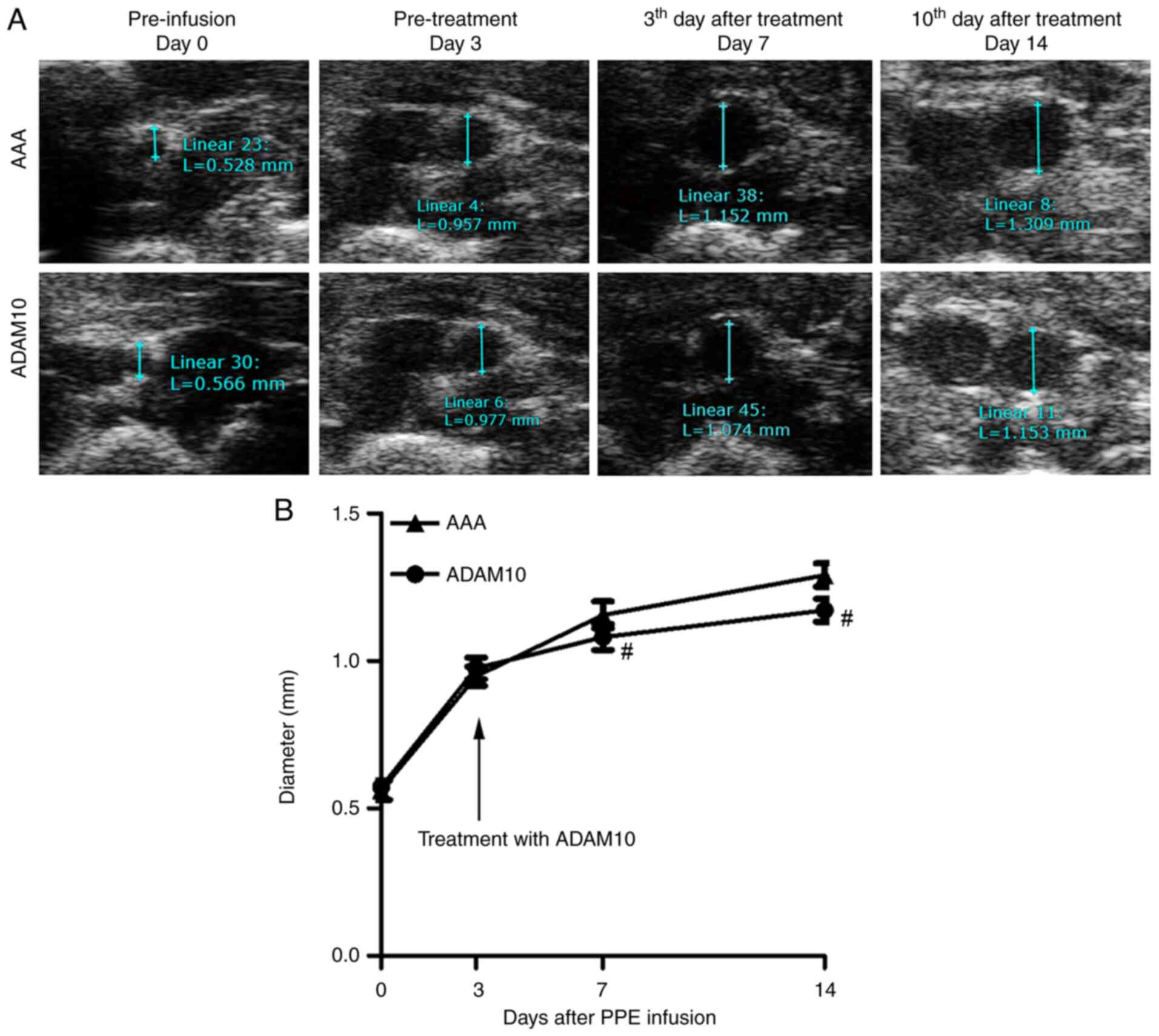
ADAM10 inhibits the progression rate
of small AAAs in a mouse model
Ultrasound examination showed the largest diameters
of the aorta on day 0, 3, 7 and 14 after infusion (Fig. 6A). In the ADAM10-treated group,
after 4 days, the growth curve of the AAA began to alter, showing a
downward trend, and the maximum aortic diameter was significantly
decreased after 3 and 10 days of continuous treatment (Fig. 6B). This indicated that ADAM10
treatment was able to significantly limit the progression rate of
small AAAs.
Discussion
In the present study, to test the hypothesis that
the inflammatory mediator ADAM10 is an important factor in the
pathophysiology of AAA, a mouse AAA model was established by
incubating the infrarenal aorta with pancreatic elastase. The
results demonstrated that ADAM10 inhibited HMGB1/RAGE/NF-κB
signaling and MMP activity in the pathogenesis of pancreatic
elastase-induced AAA.
AAA is described as a degenerative and dilated
disease of the abdominal aorta and small, asymptomatic AAAs
currently have no favorable therapeutic options, although it has
been demonstrated in animal and clinical models that inflammatory
processes have an important role in the pathophysiology of AAA
(5). Macrophages accumulating and
infiltrating the aneurysmal wall cause tissue injury, producing and
responding to inflammatory mediators, and they release proteases,
such as Cathepsin S protein and MMPs, resulting in ECM remodeling
during the formation of AAAs (30).
There are numerous drugs that have been tested in animals and
clinical trials with the goal of inhibiting the development of
AAAs, such as statins (31),
β-adrenoceptor antagonists (32),
renin-angiotensin system inhibitors (33) and doxycycline (34). However, at present, there is no
established drug therapy that can efficiently inhibit the
development of AAAs or reverse small AAAs (35).
The present study selected a mouse AAA model induced
by pancreatic elastase perfusion, the pathological changes of which
are considered to be the closest to the pathophysiological changes
of human AAAs (36). The aneurysm
wall has obvious inflammatory responses (37), which is consistent with the purpose
of the current study. This experimental mouse model has been widely
accepted and applied to study the pathophysiology of AAA (38). To examine its therapeutic effects,
ADAM10 was intraperitoneally injected into the mice on the first
day after the AAA was induced and the treatment continued for 10
days. The present results suggested that ADAM10 can significantly
alleviate the development of AAAs, which was reflected by the
observation that ADAM10 inhibited the dilatation of the infrarenal
aorta. After ADAM10 treatment, the flattening, fragmentation and
degeneration of the aortic intimal elastic laminae and remodeling
of the aortic adventitia were significantly relieved compared with
those of the AAA group. All of these results suggested that ADAM10
had an obvious preventive effect on the progression of AAA, which
provides a basis for its clinical transformation and its use for
the inhibition of AAAs. The mechanism may be mediated via
downregulation of HMGB/RAGE/NF-κB signaling.
HMGB-1, a typical damage-associated molecular
pattern protein released from various cells, such as macrophages,
smooth muscle cells, endothelial cells, cardiomyocytes, epithelial
cells and neurons (39), serves a
pivotal role in multiple cardiovascular diseases, inflammatory and
infectious disorders (40,41). It performs its biological function
by combining with receptors, including RAGE (42). In the present experiment, the
expression of HMGB1 was increased in the circulation at both the
transcriptional and translational levels in AAA model mice compared
with the Sham group, and HMGB1 has been shown to activate and
amplify inflammatory effector mechanisms (43). RAGE is the cellular target receptor
for HMGB1, and the binding of HMGB1 to the RAGE pathway is
associated with activating and modulating the inflammatory
pathological process (44);
therefore, the HMGB1/RAGE signaling axis may play a critical role
in the development of AAAs.
RAGE has been shown to be involved in the
pathological process of chronic inflammatory diseases, such as
atherosclerosis, AD, inflammatory bowel disease, rheumatoid
arthritis, amyloidosis and late diabetic vascular complications
(45). In general, the
extracellular domain is critical and is responsible for binding to
the specific ligand, and the cytosolic tail domain is essential for
transmitting RAGE signaling from the cell surface to the downstream
targets involved in the progression of inflammatory responses
(46). Ligand binding of fl-RAGE
can activate and amplify inflammatory states (47) as sRAGE lacks the transmembrane and
C-terminal cytoplasmic domain of fl-RAGE, and thus, is unable to
mediate intracellular signaling, acting as a ‘decoy’ receptor.
sRAGE competitively combines with the same ligand and prevents its
binding to fl-RAGE, without conducting signals to the intracellular
site to activate downstream signaling cascades (22,48).
Previous animal studies indicated that treatment of diabetic
RAGE−/−/apoE−/− mice suppressed inflammation
of the blood vessels and oxidative stress, as well as attenuated
atherosclerosis development (49).
ADAM10-mediated ectodomain shedding of the extracellular domain of
RAGE simultaneously generates sRAGE, and serves a role in
inhibiting the RAGE-mediated signaling pathway and alleviating
inflammation (50). In support of
this, in the current study, RAGE expression was increased in the
AAA model group and was decreased in the ADAM10 group. Increasing
serum sRAGE suggests that RAGE participates in the pathological
development of AAA. Additionally, ADAM10 inhibited RAGE activity
via ectodomain cleavage of functional fl-RAGE to produce more
inactive sRAGE, acting as a decoy, competitively binding ligands
and inhibiting its deleterious effects. The present finding was
also consistent with previous studies examining inflammatory,
autoimmune and infectious diseases (51–53).
NF-κB is a family implicated in numerous diseases,
ranging from cancer to inflammatory and immune disorders (54). Inflammation serves an important role
in contributing to the development of AAAs. Importantly, NF-κB also
regulates inflammation (55).
HMGB1/RAGE signaling stimulates a positive feedback loop, induces
multiple downstream signaling pathways, such as the
stress-activated protein kinase/JNK, cell division cycle 42/Rac,
p38 MAPK and ERK1/2, and activates the downstream proinflammatory
transcription factor NF-κB, which subsequently induces the
production of inflammatory factors (55,56).
Thus, the HMGB1/RAGE/NF-κB axis may be involved in the
pathophysiology of AAA.
In the present study, typical AAA formation was
observed in the AAA group, and an associated pathway
(HMGB1/RAGE/NF-κB) was activated, which was consistent with
previous studies (55–59). The diameter of the AAA was
significantly smaller and the HMGB1/RAGE/NF-κB pathway was
inactivated in the ADAM10 group. Therefore, it was suggested that
ADAM10 inhibited RAGE activity by cleaving fl-RAGE to
non-functional sRAGE as a decoy receptor that neutralizes
circulating ligands, thereby affecting HMGB1 binding with RAGE, and
ultimately inhibiting the activity of the HMGB1/RAGE/NF-κB
signaling axis and reducing inflammation in the AAA wall. ADAM10
decreased inflammatory factor induced aggregation by macrophages,
T-cells and B-cells, and further reduced MMP-2 and MMP-9
production, thus alleviating the development of AAA. However, the
results in the current study should be interpreted with caution
since they may not completely reflect changes that are occurring on
a functional level.
In conclusion, the present study demonstrated that
ADAM10 alleviated the development of AAA in an intraluminal
elastase-induced mouse AAA model and reversed small AAAs by
inhibiting aortic inflammation via the HMGB1/RAGE/NF-κB pathway to
further inhibit MMP activity. This study provides insight into the
molecular mechanism and a potential therapeutic strategy for
AAA.
Acknowledgements
Not applicable.
Funding
This research was funded by National Natural Science
Foundation of China (grant nos. 81800409 and 81670435) and the
Major Research and Development Program of Shandong (grant no.
2017GSF218074).
Availability of data and materials
The datasets used and/or analyzed during the current
study are available from the corresponding author on reasonable
request.
Authors' contributions
RQ, SC and GL were responsible for designing the
study, collecting the data, performing the statistical analysis and
drafting the manuscript. RQ, PG, KL, XF and HY collected the data,
performed the statistical analysis and conducted the literature
search. XW and GL supervised the project, helped to design the
study, analyzed the data and wrote the manuscript. All authors
confirm the authenticity of all the raw data. All authors have read
and approved the final manuscript.
Ethics approval and consent to
participate
This study was approved by the Animal Ethics
Committee of Shandong University (approval no. 2018-015).
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Johnston KW, Rutherford RB, Tilson MD,
Shah DM, Hollier L and Stanley JC: Suggested standards for
reporting on arterial aneurysms. Subcommittee on Reporting
Standards for Arterial Aneurysms, Ad Hoc Committee on Reporting
Standards, Society for Vascular Surgery and North American Chapter,
International Society for Cardiovascular Surgery. J Vasc Surg.
13:452–458. 1991. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Sampson UK, Norman PE, Fowkes FG, Aboyans
V, Yanna Song, Harrell FE Jr, Forouzanfar MH, Naghavi M, Denenberg
JO, McDermott MM, et al: Global and regional burden of aortic
dissection and aneurysms: Mortality trends in 21 world regions,
1990 to 2010. Glob Heart. 9:171–180.e10. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Ghaferi AA, Birkmeyer JD and Dimick JB:
Variation in hospital mortality associated with inpatient surgery.
N Engl J Med. 361:1368–1375. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Shang T, Ran F, Qiao Q, Liu Z and Liu CJ:
Tanshinone IIA attenuates elastase-induced AAA in rats via
inhibition of MyD88-dependent TLR-4 signaling. Vasa. 43:39–46.
2014. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Golledge J: Abdominal aortic aneurysm:
Update on pathogenesis and medical treatments. Nat Rev Cardiol.
16:225–242. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Raffort J, Lareyre F, Clément M,
Hassen-Khodja R, Chinetti G and Mallat Z: Monocytes and macrophages
in abdominal aortic aneurysm. Nat Rev Cardiol. 14:457–471. 2017.
View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Neeper M, Schmidt AM, Brett J, Yan SD,
Wang F, Pan YC, Elliston K, Stern D and Shaw A: Cloning and
expression of a cell surface receptor for advanced glycosylation
end products of proteins. J Biol Chem. 267:14998–15004. 1992.
View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Kierdorf K and Fritz G: RAGE regulation
and signaling in inflammation and beyond. J Leukoc Biol. 94:55–68.
2013. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Piperi C, Adamopoulos C, Dalagiorgou G,
Diamanti-Kandarakis E and Papavassiliou AG: Crosstalk between
advanced glycation and endoplasmic reticulum stress: Emerging
therapeutic targeting for metabolic diseases. J Clin Endocrinol
Metab. 97:2231–2242. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Huttunen HJ, Fages C and Rauvala H:
Receptor for advanced glycation end products (RAGE)-mediated
neurite outgrowth and activation of NF-kappaB require the
cytoplasmic domain of the receptor but different downstream
signaling pathways. J Biol Chem. 274:19919–19924. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Santilli F, Vazzana N, Bucciarelli LG and
Davì G: Soluble forms of RAGE in human diseases: Clinical and
therapeutical implications. Curr Med Chem. 16:940–952. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
12
|
van Zoelen MA, Yang H, Florquin S, Meijers
JC, Akira S, Arnold B, Nawroth PP, Bierhaus A, Tracey KJ and van
der Poll T: Role of toll-like receptors 2 and 4, and the receptor
for advanced glycation end products in high-mobility group box
1-induced inflammation in vivo. Shock. 31:280–284. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Paudel YN, Angelopoulou E, Piperi C,
Balasubramaniam VRMT, Othman I and Shaikh MF: Enlightening the role
of high mobility group box 1 (HMGB1) in inflammation: Updates on
receptor signalling. Eur J Pharmacol. 858:1724872019. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Andersson U, Yang H and Harris H:
High-mobility group box 1 protein (HMGB1) operates as an alarmin
outside as well as inside cells. Semin Immunol. 38:40–48. 2018.
View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Oozawa S, Mori S, Kanke T, Takahashi H,
Liu K, Tomono Y, Asanuma M, Miyazaki I, Nishibori M and Sano S:
Effects of HMGB1 on ischemia-reperfusion injury in the rat heart.
Circ J. 72:1178–1184. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Andrassy M, Volz HC, Igwe JC, Funke B,
Eichberger SN, Kaya Z, Buss S, Autschbach F, Pleger ST, Lukic IK,
et al: High-mobility group box-1 in ischemia-reperfusion injury of
the heart. Circulation. 117:3216–3226. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Hamada T, Torikai M, Kuwazuru A, Tanaka M,
Horai N, Fukuda T, Yamada S, Nagayama S, Hashiguchi K, Sunahara N,
et al: Extracellular high mobility group box chromosomal protein 1
is a coupling factor for hypoxia and inflammation in arthritis.
Arthritis Rheum. 58:2675–2685. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Golledge J, Muller J, Daugherty A and
Norman P: Abdominal aortic aneurysm: Pathogenesis and implications
for management. Arterioscler Thromb Vasc Biol. 26:2605–2613. 2006.
View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Saftig P and Reiss K: The ‘A Disintegrin
And Metalloproteases’ ADAM10 and ADAM17: Novel drug targets with
therapeutic potential? Eur J Cell Biol. 90:527–535. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Donners MM, Wolfs IM, Olieslagers S,
Mohammadi-Motahhari Z, Tchaikovski V, Heeneman S, van Buul JD,
Caolo V, Molin DG, Post MJ and Waltenberger J: A disintegrin and
metalloprotease 10 is a novel mediator of vascular endothelial
growth factor-induced endothelial cell function in angiogenesis and
is associated with atherosclerosis. Arterioscler Thromb Vasc Biol.
30:2188–2195. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Maretzky T, Reiss K, Ludwig A, Buchholz J,
Scholz F, Proksch E, de Strooper B, Hartmann D and Saftig P: ADAM10
mediates E-cadherin shedding and regulates epithelial cell-cell
adhesion, migration, and beta-catenin translocation. Proc Natl Acad
Sci USA. 102:9182–9187. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Raucci A, Cugusi S, Antonelli A, Barabino
SM, Monti L, Bierhaus A, Reiss K, Saftig P and Bianchi ME: A
soluble form of the receptor for advanced glycation endproducts
(RAGE) is produced by proteolytic cleavage of the membrane-bound
form by the sheddase a disintegrin and metalloprotease 10 (ADAM10).
FASEB J. 22:3716–3727. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Jiao T, Yao Y, Zhang B, Hao DC, Sun QF, Li
JB, Yuan C, Jing B, Wang YP and Wang HY: Role of MicroRNA-103a
targeting ADAM10 in abdominal aortic aneurysm. Biomed Res Int.
2017:96458742017. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
National Research Council (US) Institute
for Laboratory Animal Research, . Guide for the care and use of
laboratory animals. Washington (DC): National Academies Press;
1996
|
|
25
|
Anidjar S, Salzmann JL, Gentric D, Lagneau
P, Camilleri JP and Michel JB: Elastase-induced experimental
aneurysms in rats. Circulation. 82:973–981. 1990. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Zhang F, Kent KC, Yamanouchi D, Zhang Y,
Kato K, Tsai S, Nowygrod R, Schmidt AM and Liu B: Anti-receptor for
advanced glycation end products therapies as novel treatment for
abdominal aortic aneurysm. Ann Surg. 250:416–423. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Livak KJ and Schmittgen TD: Analysis of
relative gene expression data using real-time quantitative PCR and
the 2(-Delta Delta C(T)) method. Methods. 25:402–408. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Li G, Yang L, Yuan H, Liu Y, He Y, Wu X
and Jin X: Cold-inducible RNA-binding protein plays a central role
in the pathogenesis of abdominal aortic aneurysm in a murine
experimental model. Surgery. 159:1654–1667. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Snoek-van Beurden PA and Von den Hoff JW:
Zymographic techniques for the analysis of matrix
metalloproteinases and their inhibitors. Biotechniques. 38:73–83.
2005. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Murray PJ and Wynn TA: Protective and
pathogenic functions of macrophage subsets. Nat Rev Immunol.
11:723–737. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Shiraya S, Miyake T, Aoki M, Yoshikazu F,
Ohgi S, Nishimura M, Ogihara T and Morishita R: Inhibition of
development of experimental aortic abdominal aneurysm in rat model
by atorvastatin through inhibition of macrophage migration.
Atherosclerosis. 202:34–40. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Lindholt JS, Henneberg EW, Juul S and
Fasting H: Impaired results of a randomised double blinded clinical
trial of propranolol versus placebo on the expansion rate of small
abdominal aortic aneurysms. Int Angiol. 18:52–57. 1999.PubMed/NCBI
|
|
33
|
Morris DR, Cunningham MA, Ahimastos AA,
Kingwell BA, Pappas E, Bourke M, Reid CM, Stijnen T, Dalman RL,
Aalami OO, et al: TElmisartan in the management of abDominal aortic
aneurYsm (TEDY): The study protocol for a randomized controlled
trial. Trials. 16:2742015. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Meijer CA, Stijnen T, Wasser MN, Hamming
JF, van Bockel JH and Lindeman JH; Pharmaceutical Aneurysm
Stabilisation Trial Study Group, : Doxycycline for stabilization of
abdominal aortic aneurysms: A randomized trial. Ann Intern Med.
159:815–823. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Kokje VB, Hamming JF and Lindeman JH:
Editor's Choice-pharmaceutical management of small abdominal aortic
aneurysms: A systematic review of the clinical evidence. Eur J Vasc
Endovasc Surg. 50:702–713. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Busch A, Holm A, Wagner N, Ergün S,
Rosenfeld M, Otto C, Baur J, Kellersmann R and Lorenz U: Extra- and
intraluminal elastase induce morphologically distinct abdominal
aortic aneurysms in mice and thus represent specific subtypes of
human disease. J Vasc Res. 53:49–57. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Busch A, Chernogubova E, Jin H, Meurer F,
Eckstein HH, Kim M and Maegdefessel L: Four surgical modifications
to the classic elastase perfusion aneurysm model enable
haemodynamic alterations and extended elastase perfusion. Eur J
Vasc Endovasc Surg. 56:102–109. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Johnston WF, Salmon M, Su G, Lu G,
Ailawadi G and Upchurch GR Jr: Aromatase is required for female
abdominal aortic aneurysm protection. J Vasc Surg.
61:1565–74.e1-e4. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Wu H, Li R, Pei LG, Wei ZH, Kang LN, Wang
L, Xie J and Xu B: Emerging role of high mobility group Box-1 in
thrombosis-related diseases. Cell Physiol Biochem. 47:1319–1337.
2018. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Bae JS: Role of high mobility group box 1
in inflammatory disease: Focus on sepsis. Arch Pharm Res.
35:1511–1523. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Ding HS and Yang J: High mobility group
box-1 and cardiovascular diseases. Saudi Med J. 31:486–489.
2010.PubMed/NCBI
|
|
42
|
van Beijnum JR, Buurman WA and Griffioen
AW: Convergence and amplification of toll-like receptor (TLR) and
receptor for advanced glycation end products (RAGE) signaling
pathways via high mobility group B1 (HMGB1). Angiogenesis.
11:91–99. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Hreggvidsdottir HS, Ostberg T, Wähämaa H,
Schierbeck H, Aveberger AC, Klevenvall L, Palmblad K, Ottosson L,
Andersson U and Harris HE: The alarmin HMGB1 acts in synergy with
endogenous and exogenous danger signals to promote inflammation. J
Leukoc Biol. 86:655–662. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Sims GP, Rowe DC, Rietdijk ST, Herbst R
and Coyle AJ: HMGB1 and RAGE in inflammation and cancer. Annu Rev
Immunol. 28:367–388. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Bierhaus A, Humpert PM, Morcos M, Wendt T,
Chavakis T, Arnold B, Stern DM and Nawroth PP: Understanding RAGE,
the receptor for advanced glycation end products. J Mol Med (Berl).
83:876–886. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
46
|
Dattilo BM, Fritz G, Leclerc E, Kooi CW,
Heizmann CW and Chazin WJ: The extracellular region of the receptor
for advanced glycation end products is composed of two independent
structural units. Biochemistry. 46:6957–6970. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
47
|
Basta G, Lazzerini G, Massaro M, Simoncini
T, Tanganelli P, Fu C, Kislinger T, Stern DM, Schmidt AM and De
Caterina R: Advanced glycation end products activate endothelium
through signal-transduction receptor RAGE: A mechanism for
amplification of inflammatory responses. Circulation. 105:816–822.
2002. View Article : Google Scholar : PubMed/NCBI
|
|
48
|
Sung JY, Chung W, Kim AJ, Kim HS, Ro H,
Chang JH, Lee HH and Jung JY: Calcitriol treatment increases serum
levels of the soluble receptor of advanced glycation end products
in hemodialysis patients with secondary hyperparathyroidism. Tohoku
J Exp Med. 230:59–66. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
49
|
Soro-Paavonen A, Watson AM, Li J, Paavonen
K, Koitka A, Calkin AC, Barit D, Coughlan MT, Drew BG, Lancaster
GI, et al: Receptor for advanced glycation end products (RAGE)
deficiency attenuates the development of atherosclerosis in
diabetes. Diabetes. 57:2461–2469. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
50
|
Zhang L, Bukulin M, Kojro E, Roth A, Metz
VV, Fahrenholz F, Nawroth PP, Bierhaus A and Postina R: Receptor
for advanced glycation end products is subjected to protein
ectodomain shedding by metalloproteinases. J Biol Chem.
283:35507–35516. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
51
|
Detzen L, Cheng B, Chen CY, Papapanou PN
and Lalla E: Soluble forms of the receptor for advanced glycation
endproducts (RAGE) in periodontitis. Sci Rep. 9:81702019.
View Article : Google Scholar : PubMed/NCBI
|
|
52
|
Ramasamy R and Schmidt AM: Schmidt,
Soluble RAGE: Therapy and biomarker in unraveling the RAGE axis in
chronic disease and aging. Biochem Pharmacol. 79:1379–1386. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
53
|
Lee EJ and Park JH: Receptor for advanced
glycation endproducts (RAGE), its ligands, and soluble RAGE:
Potential biomarkers for diagnosis and therapeutic targets for
human renal diseases. Genomics Inform. 11:224–229. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
54
|
Baud V and Karin M: Is NF-kappaB a good
target for cancer therapy? Hopes and pitfalls. Nat Rev Drug Discov.
8:33–40. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
55
|
Nakashima H, Aoki M, Miyake T, Kawasaki T,
Iwai M, Jo N, Oishi M, Kataoka K, Ohgi S, Ogihara T, et al:
Inhibition of experimental abdominal aortic aneurysm in the rat by
use of decoy oligodeoxynucleotides suppressing activity of nuclear
factor kappaB and ets transcription factors. Circulation.
109:132–138. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
56
|
Chen YJ, Chan DC, Chiang CK, Wang CC, Yang
TH, Lan KC, Chao SC, Tsai KS, Yang RS, Liu SH, et al: Advanced
glycation end-products induced VEGF production and inflammatory
responses in human synoviocytes via RAGE-NF-κB pathway activation.
J Orthop Res. 34:791–800. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
57
|
Medeiros MC, Frasnelli SC, Bastos Ade S,
Orrico SR and Rossa C Jr: Modulation of cell proliferation,
survival and gene expression by RAGE and TLR signaling in cells of
the innate and adaptive immune response: Role of p38 MAPK and
NF-KB. J Appl Oral Sci. 22:185–193. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
58
|
Lappas M, Permezel M and Rice GE: Advanced
glycation endproducts mediate pro-inflammatory actions in human
gestational tissues via nuclear factor-kappaB and extracellular
signal-regulated kinase 1/2. J Endocrinol. 193:269–277. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
59
|
Hofmann MA, Drury S, Fu C, Qu W, Taguchi
A, Lu Y, Avila C, Kambham N, Bierhaus A, Nawroth P, et al: RAGE
mediates a novel proinflammatory axis: A central cell surface
receptor for S100/calgranulin polypeptides. Cell. 97:889–901. 1999.
View Article : Google Scholar : PubMed/NCBI
|