Introduction
The brain is prone to oxidative stress-induced cell damage due to its high oxygen demand, the abundance of redox-active metals and relatively high levels of oxidizable polyunsaturated fatty acids (1). Neuronal cells are especially vulnerable to oxidative stress (1). Excessive oxidative stress leads to neuronal cell death, a key physiological process in a number of neurological disorders, including neurodegenerative disease (2,3), stroke (4) and traumatic brain injury (5). Therefore, neuroprotective antioxidants have long been considered promising for the treatment of neuronal disorders (6). As a crucial mediator of oxidative stress, hydrogen peroxide (H2O2) leads to lipid peroxidation, mitochondrial dysfunction and DNA damage, which ultimately causes neuronal cell death (7). Indeed, H2O2 treatment of cultured neuronal cells can be used to mimic oxidative stress in vitro (8).
Polydatin (PD) is a single compound isolated from the Chinese medicine Polygonum cuspidatum (Japanese knotweed) (9). This compound has a number of biological properties, including anti-inflammatory (10) and antioxidative (11) properties. It has also been reported that PD serves a neuroprotective role against neurodegenerative diseases, including Alzheimer's disease (12), Parkinson's disease (13), cerebral ischemia (14–16), traumatic brain injury (17) and spinal cord injury (18,19). In the brain, PD can upregulate the antioxidant Nrf2 to reduce oxidative stress (16,20) and alleviate Parkinsonism in MPTP-model mice by enhancing glycolysis in dopaminergic neurons (21). In cultured cells, PD has been reported to protect SH-SY5Y neuronal cells by promoting Atg5-mediated autophagy (22) and prevent Aβ-induced neuronal cytotoxicity by enhancing autophagy and decreasing oxidative stress (23). Although PD has been reported to protect neuronal cells and decrease oxidative stress, the neuroprotective roles of PD in primary neurons and the underlying neuroprotective mechanisms remain to be elucidated.
The present study further investigated the Nrf2-independent mechanisms underlying the role of PD in serving anti-oxidative and neuroprotective functions. Through a cell-based Ngb gene promoter-reporter assay, it was identified that PD could significantly induce Ngb expression (24). Ngb was originally found as a vertebrate globin that is predominantly expressed in the brain (25). Accumulating data has suggested that Ngb is a crucial endogenous neuroprotectant for a number of neurological disorders, including stroke, traumatic brain injury and neurodegenerative diseases (26). Ngb can directly interact with mitochondria and maintain mitochondrial function, as Ngb suppresses mitochondrial permeability transition pore opening by binding to voltage-dependent anion-selective channel 1 (27) and reduces mitochondrial reactive oxygen species (ROS) production by inhibiting mitochondrial complex III (28,29). In addition, Ngb has been reported to protect against oxidative stress-induced neuronal cell death (30). Notably, Ngb is reported to be regulated by cAMP response element-binding protein (CREB) (31). However, whether CREB/Ngb signaling axis mediates the neuroprotective effects of PD has not been investigated. The present study, therefore, hypothesized that PD might protect against H2O2-induced mitochondrial dysfunction and neurotoxicity by upregulating CREB/Ngb signaling.
The present study examined the effects of PD on H2O2-induced mitochondrial dysfunction and neurotoxicity and explored whether CREB/Ngb signaling is involved in the beneficial effects of PD on mitochondrial function and neuronal survival. The results showed that PD protects neuronal cells from H2O2-induced mitochondrial dysfunction and neurotoxicity via activation of CREB/Ngb signaling.
Materials and methods
Materials
Dulbecco's modified Eagle's medium (DMEM), Neurobasal medium, L-Glutamine, B-27 Supplement, geneticin, penicillin, streptomycin, Lipofectamine® 2000, Lipofectamine® 3000 and MitoSOX Red Mitochondrial Superoxide Indicator were purchased from Thermo Fisher Scientific, Inc. Fetal bovine serum (FBS) and 0.05% trypsin-EDTA were purchased from Gibco (Thermo Fisher Scientific, Inc.). The N2a cell line was purchased from the American Type Culture Collection. The chicken anti-Ngb primary antibody was purchased from BioVendor. AKT and phosphorylated CREB (Ser133; p-CREB) antibodies were purchased from Cell Signaling Technology, Inc. The MAP2 antibody was purchased from Novus Biologicals, LLC. β-actin antibody was purchased from Sigma-Aldrich (Merck KGaA). Mouse Ngb short interfering (si)RNA1, siRNA2 and siRNA3 were purchased from OriGene Technologies, Inc. Negative control (NC) siRNA was purchased from Santa Cruz Biotechnology, Inc. The pCMV-HA vector and CREB Dominant-Negative Vector Set were purchased from Takara Bio USA, Inc. The Dual-Luciferase Reporter Assay System was purchased from Promega Corporation. The pGL3-Basic vector, the pGEM-T Easy Vector System II and the Dual Luciferase Reporter Assay System were purchased from Promega Corporation. Reporter plasmids including the P2033(−2027/+6) and P2033(CRE2del) plasmids (mutated Ngb promoter plasmid with deletion of the specific CRE site at −854) were constructed as described previously (31). PD, with a purity of 98.95%, was purchased from MedChemExpress, LLC and DMSO was used as the solvent.
Cell culture and transfection
The animal protocol (approval number 2016#0101) was approved by the ethical committee of Hunan Normal University. The 10 week-old male and female C57BL/6 mice (weight, 25–30 g) were purchased from Hunan SJA Laboratory Animal Co., LTD, and bred in animal cages in the controlled environment with 12/12-h light/dark cycle, room temperature of 21–23°C and humidity of 55±10%. A total of four pregnant female mice (E16) were used for the present study. They were be anesthetized with isoflurane (5%) for ~3 min. Anesthetic adequacy was checked continuously by observing the respiration rate and checking tailpinches. Then they were sacrificed via immediate decapitation. Once it was confirmed that the mouse was dead, the fetuses were surgically remove. The fetal mouse pups were immediately decapitated, as immediate decapitation is the fastest, most humane and standard procedure to sacrifice fetal mice for purposes of extracting brains and growing primary neuron culture. Animals were removed from the study and sacrificed if any of the following clinical signs/conditions were found: Persistent recumbence; inability to rise; loss of righting reflex; pain or distress that cannot be alleviated by analgesics; difficulty with ambulation (paralysis, fractures and trauma); severe central nervous system signs (including circling, rolling, persistent seizures or convulsions); abnormal breathing (dyspnea) and cyanosis; body condition score (32) of ≥2 (out of 5); excessive weight loss; vomiting/diarrhea resulting in severe dehydration; and tumor production specific endpoints (maximum tumor diameter, ≤20 mm) (33). As described previously (29), primary mouse cortical neuronal cultures were prepared from the cortices of E16 C57/BL6 female mouse embryos and plated onto 12-, 24-, or 48-well plates precoated with poly-D-lysine (Sigma-Aldrich; Merck KGaA) at a density of 3.5×105 cells/ml in neurobasal medium (NBM) plus 2% B27, 0.8 mM L-glutamine and 100 µg/ml penicillin/streptomycin (Thermo Fisher Scientific, Inc.). Half of the medium was changed every 3 days. The purity of primary mouse cortical neurons on day 8 was determined by calculating the ratio of MAP2-positive cells to the total number of viable cells (Fig. S1). These data indicated that >95% of the cells in the cultures were neurons.
Mouse N2a neuronal cells were cultured in complete DMEM (DMEM containing 10% FBS and 100 µg/ml penicillin/streptomycin) and maintained in a humidified (5% CO2, 37°C) incubator. For transfection, N2a cells at ~80% confluence in 12 wells plates were transfected with 30 pmol NC siRNA or Ngb siRNAs (siNgb1 Target sequence: GCCAGGCTCTTCGCCCTGGAA, siNgb2 Target sequence: GCCAAGCTGTTTACTTTCCAA, siNgb3 Target sequence: GGTGATGCTAGTGATTGAT) with 3 µl Lipofectamine® 3000 or 3 µg plasmids [mouse Ngb promoter plasmid [P2033(−2027/+6)], pCMV-CREB or pCMV-CREBs133a or pCMV-KCREB] with 3 µl Lipofectamine® 2000 at 37°C according to the manufacturer's instructions for 6 h, followed by replacement of medium with fresh complete DMEM and culture for another 18 or 42 h. The transfected cells were treated with 200 µM H2O2 in the presence or absence of 10 µM PD at 37°C for 16 h for MTT assays, or for 4 h for JC-1 mitochondrial membrane potential and ROS production assay.
3-(4,5-Dimethylthiazol-2-yl)-2,5-dimethyltetrazoliumbromide (MTT) assay
An MTT assay was carried out as described previously (29). After different treatments, N2a cells were incubated with fresh DMEM containing 0.5 mg/ml MTT for 4 h in a 37°C cell culture incubator. Next, the medium was aspirated and DMSO was added to each well to dissolve the MTT formazan crystals. The absorbance of each sample at 570 nm was measured with a Molecualr Device Spectramax M5 plater reader. Percentages of live cell counts were used for assay normalization.
Lactate dehydrogenase (LDH) assay
LDH assays were used to measure neurotoxicity as previously described (27). Following treatment, the medium was removed and the cells were lysed with PBS containing 0.1% Triton at room temperature for 5 min. Subsequently, the lysate was collected and centrifuged at 380 × g for 10 min at 4°C. The supernatant was used for LDH activity measurement using a cytotoxicity detection kit (LDH) following the manufacturer's instructions (Roche Applied Science).
Mitochondrial membrane potential (∆Ψm) measurements
∆Ψm measurements were performed as described previously (29). Following treatment, primary mouse cortical neurons or N2a cells were stained with 2.0 µg/ml 5,5′,6,6′-tetrachloro-1,1′,3,3′-tetraethylbenzimidazolyl-carbocyanine iodide (JC-1) at 37°C for 20 min, followed by washing with Hanks' Balanced Salt Solution (HBSS) three times. Then, the cellular fluorescence was detected with a microplate reader (green fluorescence, excitation 489 nm and emission 529 nm; red fluorescence, excitation 570 nm and emission 600 nm) and imaged with a fluorescence microscope with 200× magnifications. Four random microscopic fields in each group were selected. The ∆Ψm was calculated as the fluorescence ratio of red (JC-1 aggregates) to green (JC-1 monomers).
Mitochondrial ROS measurements
Mitochondrial ROS production was assessed with MitoSOX Red Mitochondrial Superoxide Indicator according to the manufacturer's instructions. Briefly, following treatment, the medium was aspirated and HBSS was added to primary mouse cortical neurons or N2a cells, which were diluted with MitoSOX Red dye (10 µM) and incubated in a cell incubator at 37°C in the dark for 30 min. After loading the dye, the cells were washed twice with HBSS and the cellular fluorescence was imaged with a fluorescence microscope with 200× magnifications. Four random fields in each group were selected. The mitochondrial ROS was also measured at 510 nm excitation and 580 nm emission with a microplate reader.
Luciferase assay
N2a cells were seeded into 24-well plates and transfected with the indicated plasmids, including the mouse Ngb promoter plasmid P2033(−2027/+6) or mutated Ngb promoter plasmid P2033(CRE2del) plasmid (200 ng/well) and pRL-TK (10 ng/well). The wild-type and mutated mouse Ngb promoter were cloned into the pGL3-Basic vector (Promega Corporation) as previously described (31). After 24 h, cells were treated with H2O2 or PD as described. Reporter assays were performed with Dual Luciferase Reporter Assay System (Promega Corporation). All assays were carried out in a Veritas Microplate Luminometer (Turner BioSystems). The activity was normalized and defined as the Firefly/Renilla luciferase ratio. Each experiment was performed at least three times.
Western blotting analysis
Cells were lysed in RIPA buffer and centrifuged at 13,500 g for 10 min at 4°C. Supernatants were measured with a Bradford assay (Bio-Rad Laboratories, Inc.) and 30 µg of total protein were separated using a 10% SDS-PAGE gel and then transferred to a polyvinylidene difluoride membrane. The membrane was blocked in 5% nonfat dried milk in TBST (TBS containing 0.1% Tween-20) for 1 h at room temperature, followed by immunoblotting and incubation overnight at 4°C with the primary antibodies as follows: CREB (1:1,000, Cell Signaling Technology, cat. no. #4820), phospho-CREB antibody (1:1,000; Cell Signaling Technology, #9198), Ngb (1:500; Abnova, 2B5-A7), β-actin antibody (1:3,000; Sigma, A5441). Subsequently, the membrane was incubated with the appropriate horseradish peroxidase-conjugated secondary antibody (both 1:5,000; cat. nos. #31430 and #31460; Thermo Fisher Scientific, Inc.) for 1 h at room temperature. After washing with TBST, immunoreactive proteins were determined by an enhanced chemiluminescence substrate (Thermo Fisher Scientific, Inc.) according to the manufacturer's protocol. Images were captured with ChemiDoc Imaging Systems (Bio-Rad Laboratories). Quantification of protein band intensity obtained by Western blotting analysis was analyzed with ImageJ 1.52a (http://rsb.info.nih.gov/ij/) and normalized to the actin band intensity.
Electrophoretic mobility shift assay (EMSA)
An EMSA was performed as described previously (29,34). N2a cell nuclear extracts were prepared with a NE-PER Nuclear and Cytoplasmic Extraction kit (Thermo Fisher Scientific, Inc.). Oligonucleotides containing wild-type CRE2 (5′-TGGGGGCTGAGGTGATCCAAC-3′) were synthesized and labeled with biotin using a biotin 3-end DNA-labeling kit (Thermo Fisher Scientific, Inc.) and annealed to obtain double-stranded DNA probes. DNA/nuclear protein/antibody binding reactions for the supershift assay were performed using the LightShift EMSA Optimization and Control kit (Thermo Fisher Scientific, Inc.). The reaction mixtures were loaded onto a 6% nondenaturing polyacrylamide gel and electrophoresed in 0.5× TBE buffer at 100 V at 4°C. Following electrophoresis, the DNA/nuclear protein/antibody complexes were transferred to a nylon membrane (Ambion; Thermo Fisher Scientific, Inc.), followed by cross-linking of the membrane at 120 mJ/cm2 for 60 sec using a UV cross-linker (Stratagene; Agilent Sumitomo Dainippon Pharma Co., Ltd.). Biotin-labeled DNA was further detected using the Chemiluminescent Nucleic Acid Detection Module (Thermo Fisher Scientific, Inc.) and exposed to film (X-Omat; Kodak).
Statistical analysis
All data are expressed as the mean ± standard deviation of ≥3 independently repeated experiments. Differences between groups were analyzed by one-way ANOVA followed by Tukey-Kramer's test using GraphPad Prism 7 software (GraphPad Software, Inc.). P<0.05 was considered to indicate a statistically significant difference.
Results
PD protects primary mouse cortical neurons and N2a neuronal cells against H2O2-induced neurotoxicity
Primary cultured mouse cortical neurons and mouse N2a cells were treated with H2O2 at different doses and neuronal cell viability was measured by MTT assay. The data showed that treatment with H2O2 for 16 h significantly reduced cell viability of primary mouse cortical neurons and N2a cells in a dose-dependent manner (Fig. 1A and B). It was found the two cells have a different sensitivity to PD. Primary mouse cortical neurons are more sensitive to PD than N2a cells. It was found that ~30–40% neuronal cell death was induced by 50 and 200 µM H2O2 in primary mouse cortical neurons and N2a cells, respectively (Fig. 1A and 1B) and these H2O2 concentrations were selected for further experiments. To investigate whether PD can protect neuronal cells from oxidative stress, MTT assay and LDH assay were used for measurement of cytotoxicity. The primary mouse cortical neurons are non-dividing cells, but N2a cells are proliferated cells. If PD has a promoting effect on the proliferation of N2a cells, there would be more intracellular LDH in N2a cells, which may be accompanied by more LDH release. The present study thus performed an LDH assay with primary mouse cortical neurons and MTT assay for N2a cells, respectively. Primary mouse cortical neurons were treated with H2O2 (50 µM) plus PD (0, 5, 10 and 20 µM) for 16 h, followed by the MTT assay. The results showed that PD dose-dependently inhibited H2O2 (50 µM)-induced neurotoxicity in primary mouse cortical neurons. Compared with the control, 10 and 20 µM PD could inhibit H2O2-induced neurotoxicity. However, the cell viability of these two concentrations was significantly different compared with the control. Considering that the primary mouse neurons are more sensitive to PD treatment, there may be more reverse effects with a high dose of PD in primary mouse neurons; 10 µM PD exhibited the maximal protective effects among the tested doses (Fig. 1C). Moreover, LDH assays showed that 10 µM PD significantly prevented H2O2 (50 µM)-induced LDH release in primary mouse cortical neurons (Fig. 1D). Similarly, treatment with PD (10 µM) significantly attenuated neurotoxicity in N2a cells, which was induced by H2O2 at a dose of ≤400 µM (Fig. 1E). These results indicated the potent neuroprotective effects of PD.
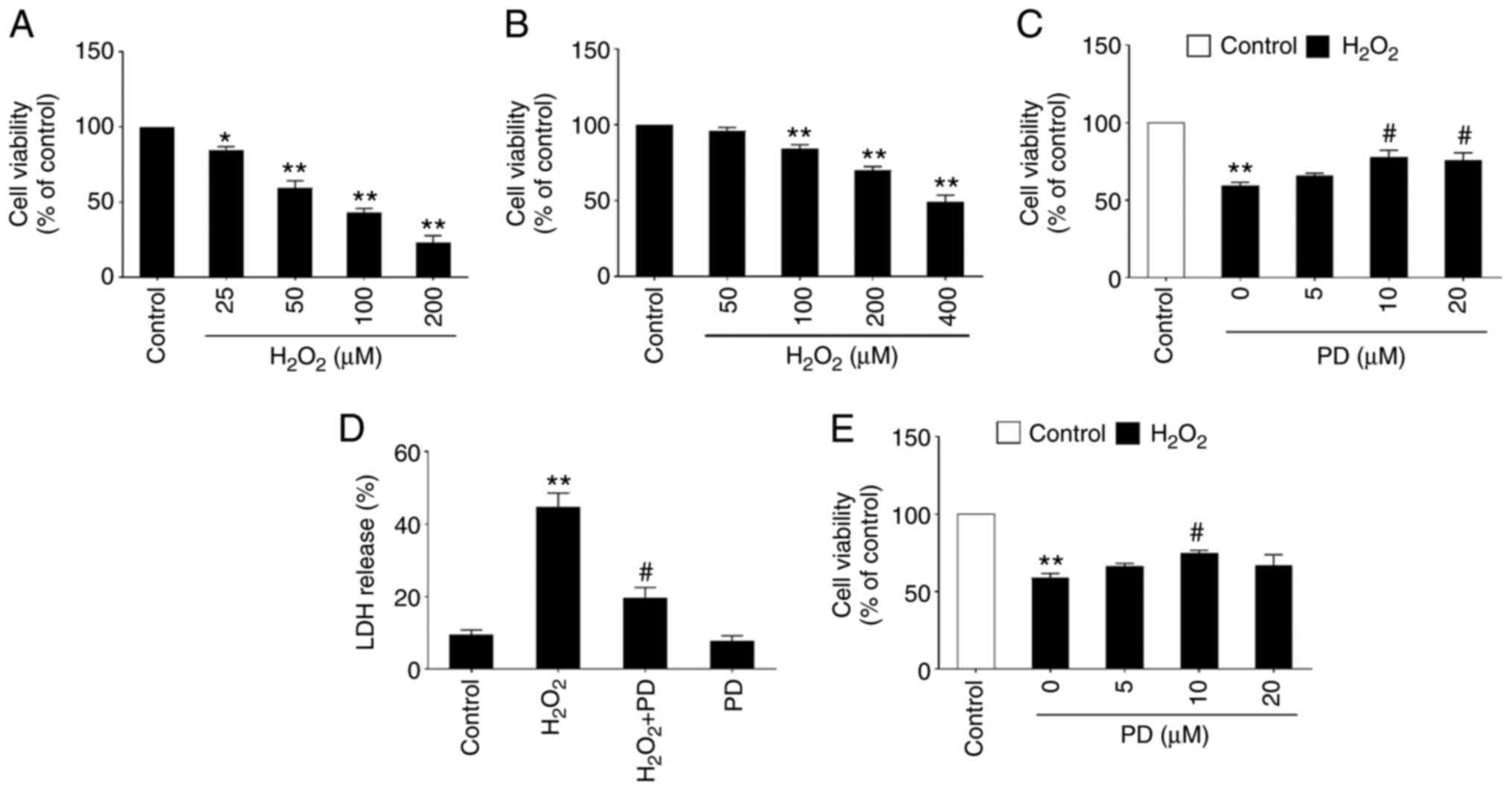 |
Figure 1.
PD protects primary mouse cortical neurons and N2a neuronal cells against H2O2-induced neurotoxicity. (A) Cell viability was measured by MTT assay in primary mouse cortical neurons cultured in 48-well plates treated with the indicated dose of H2O2 for 16 h. n=4, data are expressed as the mean ± SD, *P<0.05, **P<0.01 compared with the control (0 µM H2O2). (B) Cell viability was measured by MTT assay in N2a cells cultured in 48-well plates treated with the indicated dose of H2O2 (0, 50, 100, 200 and 400 µM) for 16 h. Data are expressed as the mean ± SD, *P<0.05, **P<0.01 compared with the control (0 µM H2O2). (C) Cell viability was measured by MTT assay in primary mouse cortical neurons cultured in 48-well plates treated with 50 µM H2O2 and the indicated dose of PD for 16 h. Data are expressed as the mean ± SD, **P<0.01 compared with the control (DMSO); n=4, #P<0.05 compared with PD (0 µM). (D) Ratio of LDH release was measured by LDH assay in primary mouse cortical neurons cultured in 48-well plates treated with DMSO (control) or 50 µM H2O2 with or without 10 µM PD. **P<0.01 compared with the control (DMSO); #P<0.05 compared with H2O2 (50 µM). (E) Cell viability was measured by MTT assay in N2a cells cultured in 48-well plates treated with 400 µM H2O2 and the indicated dose of PD (0, 5, 10 and 20 µM) for 16 h. n=5, data are expressed as the mean ± SD, **P<0.01 compared with the control (DMSO); #P<0.05 compared with H2O2 (50 µM). PD, polydatin; LDH, lactate dehydrogenase.
|
PD attenuates H2O2-induced Δψm and oxidative stress in neurons and N2a cells
Mitochondrial dysfunction has been proposed as a mediator of neurodegeneration (35,36). To investigate whether PD attenuates H2O2-induced Δψm, a JC-1 assay was performed to determine the Δψm in primary mouse cortical neurons treated with H2O2 (50 µM) or H2O2 (50 µM) plus PD (10 µM). It has been reported that the mitochondrial dysfunction that occurs as early as 4 h can affect neuronal cell viability (35). Δψm was thus detected at the early time point 4 h. The results showed that the JC-1 red/green fluorescence ratio in neurons significantly decreased after H2O2 (50 µM) treatment compared with the untreated control, while this decrease was significantly attenuated by cotreatment with PD (10 µM; Fig. 2A). Similar observations were also found in N2a cells, as PD (10 µM) treatment significantly preserved the Δψm, which was dramatically reduced by H2O2 (200 µM) treatment (Fig. 2C). To further investigate whether PD inhibits H2O2-induced mitochondrial oxidative stress, MitoSOX dye was used. The results showed that PD treatment significantly attenuated H2O2-induced mitochondrial ROS production in primary mouse cortical neurons (Fig. 2B) and N2a cells (Fig. 2D). Taken together, these results indicated that PD attenuates H2O2-induced mitochondrial dysfunction and oxidative stress in primary mouse cortical neurons and N2a cells.
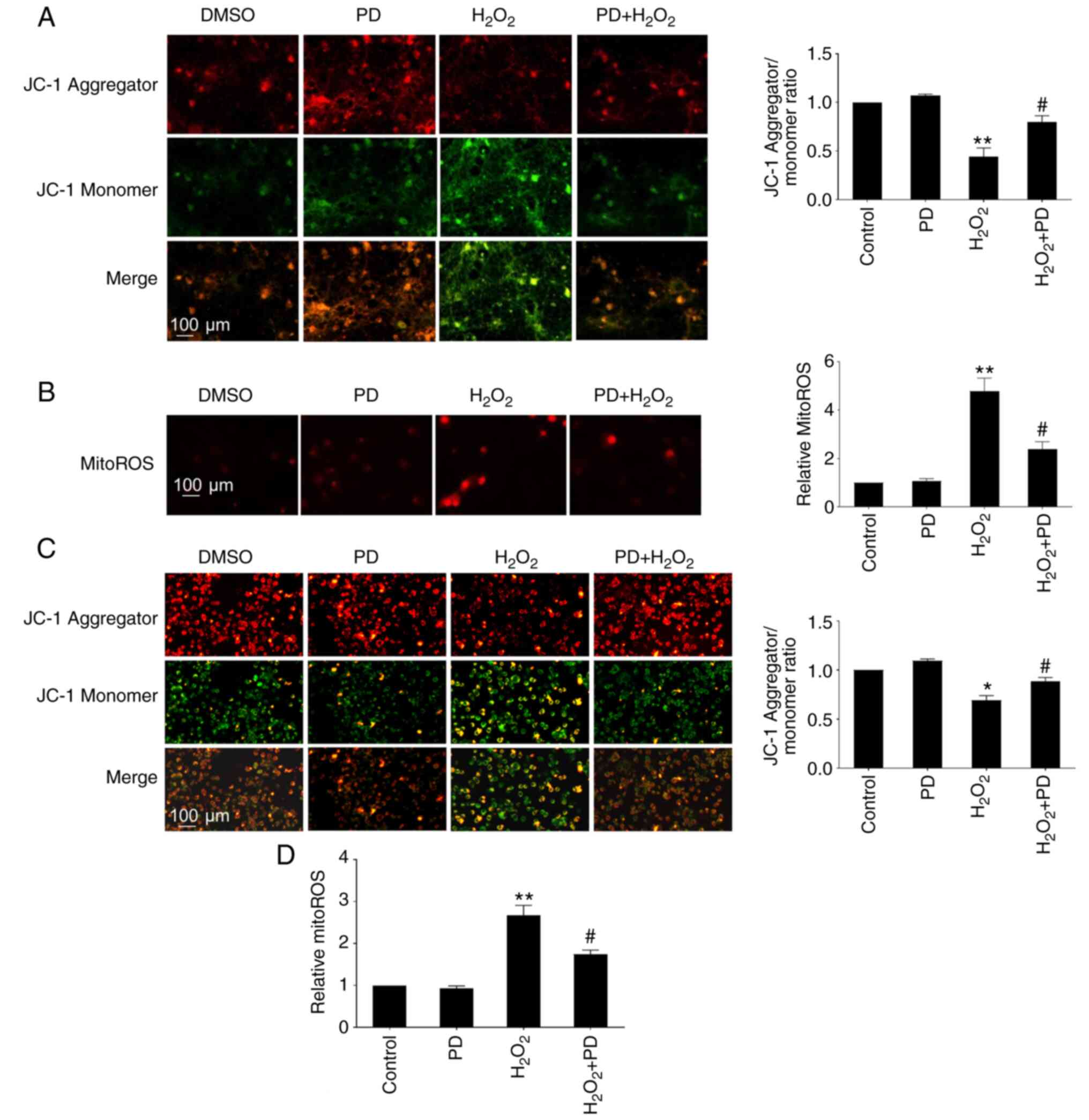 |
Figure 2.
PD attenuates H2O2-induced mitochondrial dysfunction (Δψm) and oxidative stress in neurons and N2a cells. (A) Δψm was measured by JC-1 dye in primary mouse cortical neurons cultured in 24-well plates treated with DMSO, 10 µM PD, 50 µM H2O2, or 10 µM PD + 50 µM H2O2 for 4 h. Fluorescence of the JC-1 aggregates (red) and monomers (green) in primary mouse cortical neurons was measured using fluorescence microscopy. The ratio of JC-1 aggregates/monomers was plotted. (B) Mitochondrial ROS were measured in primary mouse cortical neurons cultured in 24-well plates treated with DMSO, 10 µM PD, 50 µM H2O2, or 10 µM PD + 50 µM H2O2 for 4 h. Fluorescence was measured using fluorescence microscopy. The fluorescence intensity was plotted. (C) Δψm was measured by JC-1 dye in N2a cells cultured in 24-well plates treated with DMSO, 10 µM PD, 200 µM H2O2, or 10 µM PD + 200 µM H2O2 for 4 h. Fluorescence of the JC-1 aggregates (red) and monomers (green) in N2a cells was measured using fluorescence microscopy. The ratio of JC-1 aggregates/monomers was plotted. (D) Mitochondrial ROS contents were measured in N2a cells cultured in 24-well plates treated with DMSO, 10 µM PD, 200 µM H2O2, or 10 µM PD + 200 µM H2O2 for 4 h. n=4, data are expressed as the mean ± SD, *P<0.05 vs. control, **P<0.01 vs. control (DMSO); #P<0.05 vs. H2O2. JC-1, 5,5′,6,6′-tetrachloro-1,1′,3,3′-tetraethylbenzimidazolyl-carbocyanine iodide; PD, polydatin; ROS, reactive oxygen species.
|
PD attenuates H2O2-reduced p-AKT, p-CREB and Ngb expression in N2a cells
To investigate the molecular mechanism underlying the neuroprotective effects of PD, western blotting analysis was performed. The results showed that PD significantly induced the phosphorylation of AKT and CREB and Ngb protein expression in a dose-dependent manner and that there was no effect on the AKT and CREB protein expression (Fig. 3A). H2O2 induces neuronal cell death by reducing the levels of CREB and p-CREB (37). In agreement with this study, the results of the present study showed that H2O2 (200 µM) treatment led to a significant reduction in p-AKT, p-CREB and Ngb expression in N2a cells and that there was no effect on the AKT and CREB protein expression. Nevertheless, this decrease was rescued by cotreatment with PD (10 µM) (Fig. 3B). Taken together, these results suggested that PD attenuates H2O2-reduced p-AKT, p-CREB and Ngb expression in N2a cells.
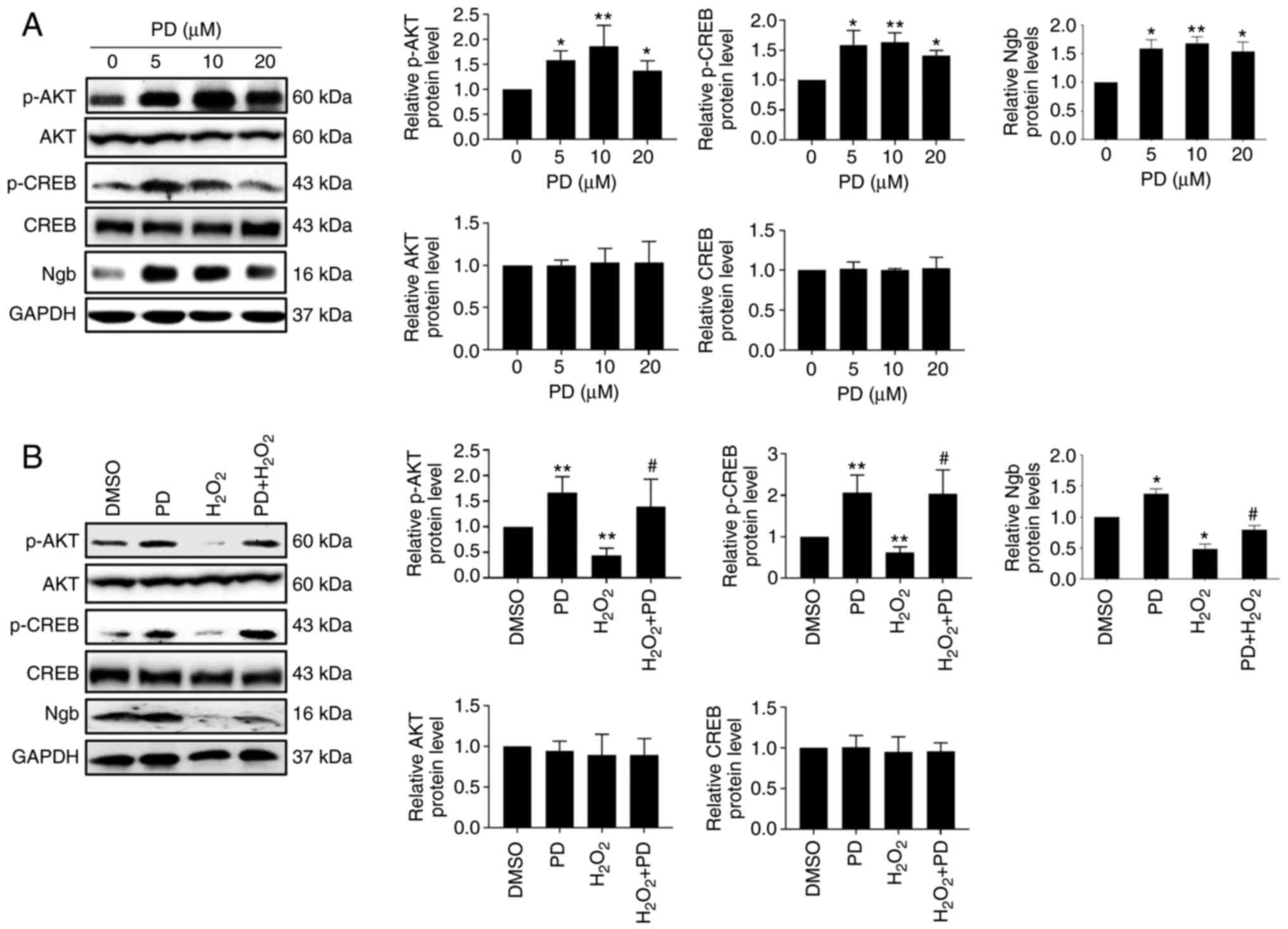 |
Figure 3.
PD attenuates H2O2-reduced p-AKT, p-CREB and Ngb expression in N2a cells. (A) Protein levels of p-AKT, AKT, p-CREB, CREB and Ngb were measured by western blotting in N2a cells cultured in 60 mm dishes treated with the indicated dose of PD, n=3. Band intensities of p-AKT and p-CREB were normalized to AKT and CREB, respectively. Band intensities of AKT, CREB and Ngb were normalized to GAPDH. n=3, data are expressed as the mean ± SD, *P<0.05, **P<0.01 compared with the control (DMSO). (B) Protein levels of p-AKT, AKT, p-CREB, CREB and Ngb were measured by western blotting in N2a cells cultured in 6-well plates treated with DMSO, 10 µM PD, 200 µM H2O2, or 10 µM PD + 200 µM H2O2, n=3. Band intensities of p-AKT and p-CREB were normalized to AKT and CREB, respectively. Band intensities of AKT, CREB and Ngb were normalized to GAPDH. Data are expressed as the mean ± SD, *P<0.05, **P<0.01 compared with control (DMSO); #P<0.05 compared with H2O2. PD, polydatin; p-, phosphorylated; CREB, cAMP response element-binding protein; Ngb, neuroglobin.
|
PD upregulates Ngb via CREB in N2a cells
To investigate whether CREB is required for PD-induced Ngb upregulation, a mouse Ngb promoter plasmid [P2033(−2027/+6)] and wild-type CREB or mutant CREB (CREBs133a or KCREB) were cotransfected into N2a cells for 24 h, followed by PD (10 µM) treatment for another 16 h. The results showed that mutant CREB significantly attenuated PD-induced Ngb promoter activity (Fig. 4A). Moreover, it was observed that KG501, a potent inhibitor of CREB, significantly attenuated PD-induced Ngb promoter activity (Fig. 4B). These results indicated that CREB is responsible for PD-induced Ngb promoter activity. To determine whether PD promotes the binding of CREB to CRE in the mouse Ngb promoter, EMSA was performed. The results showed that compared with DMSO-treated N2a cell nuclear extracts, PD-treated N2a cell nuclear extracts produced a stronger supershift band after incubation with p-CREB antibody and specific Ngb CRE oligos, indicating that PD significantly induces the binding of p-CREB to the specific CRE site located at −854 in the promoter region of the mouse Ngb gene (Fig. 4C). To further investigate whether the specific CRE site in the Ngb promoter region is responsible for PD-induced Ngb promoter activity, P2033(−2027/+6) or P2033(CRE2del, mutated Ngb promoter plasmid with deletion of the specific CRE site at −854) plasmids were transfected into N2a cells for 24 h followed by PD treatment for another 16 h. The results showed that deletion of the specific CRE site significantly attenuated basal Ngb promoter activity and PD-induced Ngb promoter activity (Fig. 4D). Taken together, these results suggested that PD upregulates Ngb via CREB in N2a cells.
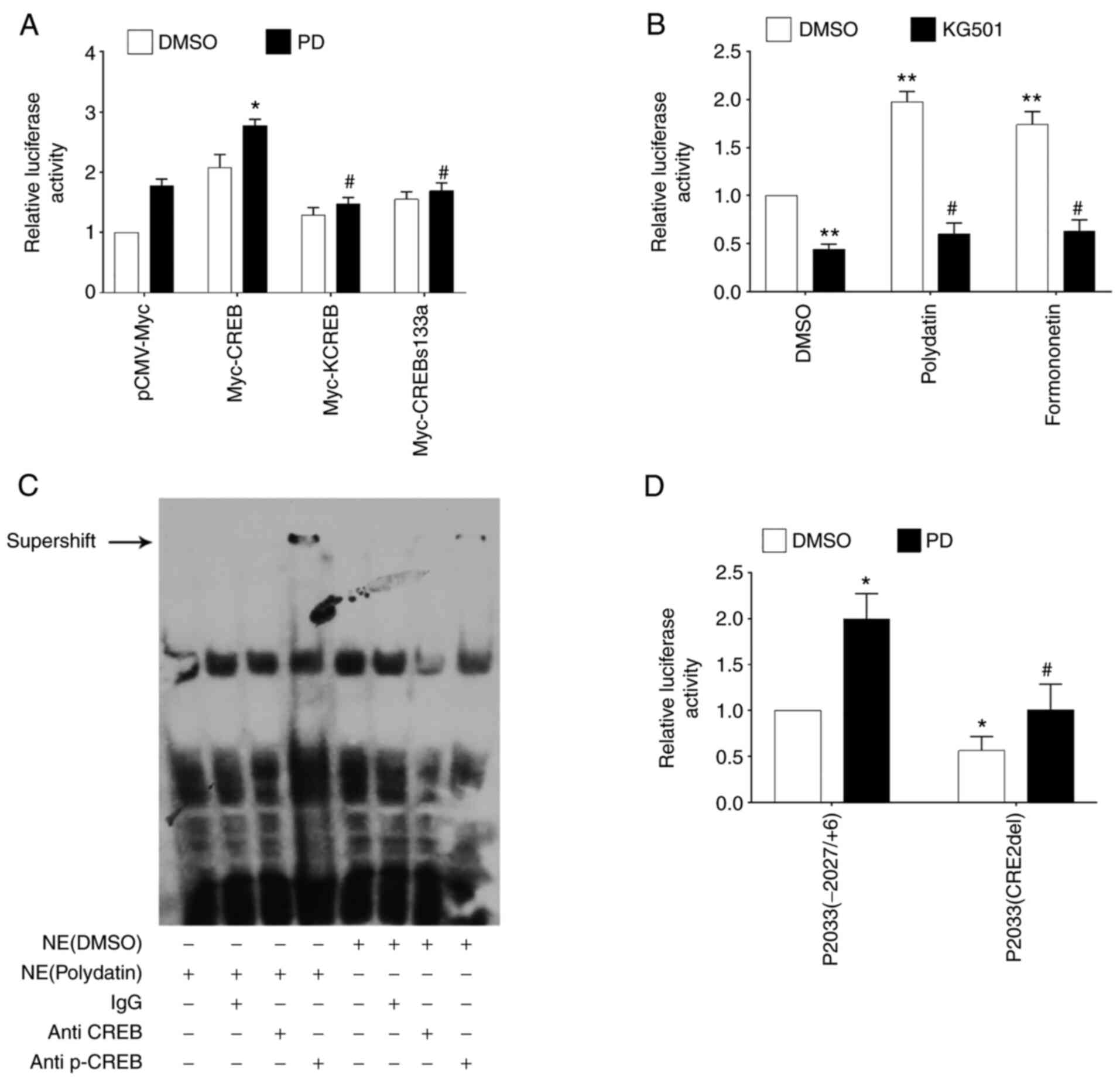 |
Figure 4.
PD upregulates Ngb via CREB in N2a cells. (A) Mouse Ngb promoter plasmid P2033(−2027/+6) and wild-type CREB or mutant CREB (KCREB or CREBs133a) were cotransfected into N2a cells for 24 h, followed by PD treatment for another 16 h. Relative Ngb promoter activity was measured with a luciferase assay. *P<0.05 compared with N2a cells transfected with pCMV and treated with PD (10 µM); #P<0.05 compared with N2a cells transfected with Myc-CREB and treated with PD (10 µM). (B) The mouse Ngb promoter plasmid P2033(−2027/+6) was transfected into N2a cells for 24 h. Cells were treated with or without KG501, PD and formononetin as described for another 16 h. Relative Ngb promoter activity was measured with a luciferase assay. **P<0.01 compared with N2a cells treated with DMSO; #P<0.05 compared with N2a cells treated with PD or formononetin. (C) Supershift assay of p-CREB binding to the CRE in the Ngb promoter. Ngb CRE biotin-labeled oligos were incubated with the nuclear extract of N2a cells treated with DMSO or polydatin, with or without the addition of antibodies against CREB and p-CREB. (D) The mouse Ngb promoter plasmid P2033(−2027/+6) or mutated Ngb promoter plasmid P2033(CRE2del) plasmid was transfected into N2a cells for 24 h. Then, the cells were treated with PD for another 16 h. Relative activities were measured with a luciferase assay. *P<0.05 compared with N2a cells treated with DMSO; #P<0.05 compared with N2a cells treated with PD. PD, polydatin; Ngb, neuroglobin; CREB, cAMP response element-binding protein; p-, phosphorylated; NE, nuclear extract.
|
PD inhibits neuronal cell death, maintains mitochondrial function and reduces ROS production via Ngb
Next, the role of Ngb in mediating PD protection against H2O2 (200 µM) was determined. Transfection of Ngb-specific siRNA (siNgb-2 and siNgb-3) into N2a cells for 48 h successfully led to the knockdown of endogenous Ngb (Fig. 5A) and siNgb-2 was chosen for further experiments. It was found that knockdown of Ngb significantly attenuated the protection of PD against H2O2 via an MTT assay (Fig. 5B). Next, the role of endogenous Ngb in mediating PD-preserved mitochondrial membrane potential in N2a cells was determined. It was found that the JC-1 ratio was significantly reduced in Ngb siRNA-transfected N2a cells compared with NC siRNA-transfected N2a cells under PD plus H2O2 treatment conditions (Fig. 5C). Furthermore, it was observed that there was a significant increase in ROS production in N2a cells treated with siNgb-2 transfection plus PD and H2O2 compared with NC siRNA transfection plus PD and H2O2 (Fig. 5D). These results indicate that PD inhibits neuronal cell death, maintains mitochondrial function and reduces ROS production via Ngb.
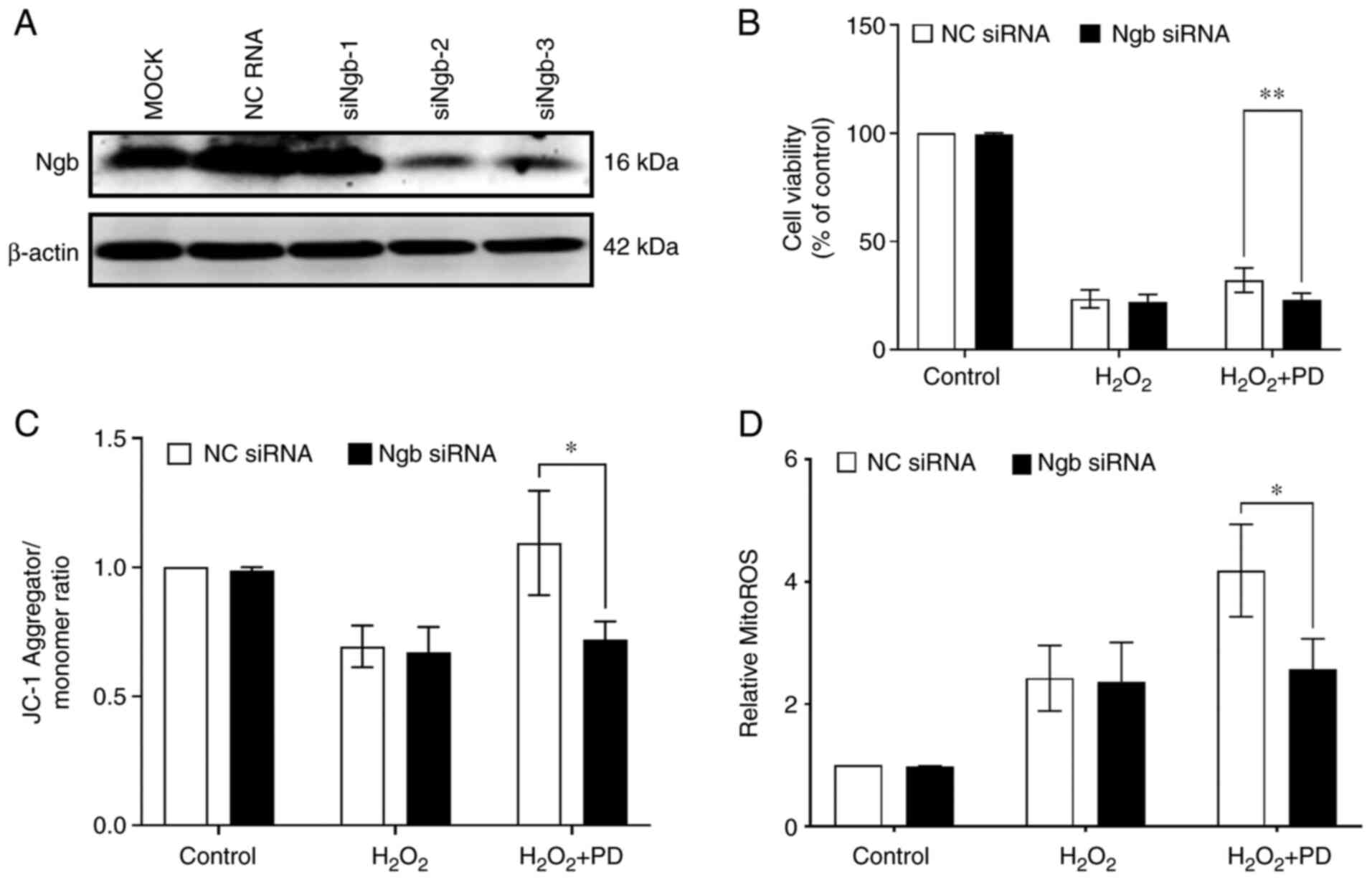 |
Figure 5.
PD inhibits neuronal cell death, maintains mitochondrial function and reduces ROS production by upregulating Ngb. (A) Ngb protein levels were measured by western blotting in N2a cells transfected with Ngb-specific siRNA (siNgb-1, siNgb-2 and siNgb-3) for 48 h. (B) N2a cells were transfected or not with Ngb-siRNA2 for 24 h and then treated with 200 µM H2O2 or 200 µM H2O2 plus 10 µM PD for 16 h. Cell viability was measured by MTT assay. n=3, data are expressed as the mean ± SD, **P<0.01. DMSO was used as a vehicle (control) and the final concentration of DMSO in cells was 0.1%. (C) N2a cells were transfected or not with Ngb-siRNA2 for 24 h and then treated with 200 µM H2O2 or with 200 µM H2O2 plus 10 µM PD for 4 h. The mitochondrial membrane potential was measured by a JC-1 mitochondrial membrane potential assay kit. n=4, data are expressed as the mean ± SD, *P<0.05. DMSO was used as a vehicle (control) and the final concentration of DMSO in cells was 0.1%. (D) N2a cells were transfected or not with Ngb-siRNA2 for 24 h and then treated with 200 µM H2O2 or with 200 µM H2O2 and 10 µM PD for 4 h. Mitochondrial ROS production was measured with a mitoROS assay kit. n=4, data are expressed as the mean ± SD, *P<0.05. DMSO was used as a vehicle (control) and the final concentration of DMSO in cells was 0.1%. Ngb, neuroglobin; ROS, reactive oxygen species; Ngb, neuroglobin; si, short interfering; JC-1, 5,5′,6,6′-tetrachloro-1,1′,3,3′-tetraethylbenzimidazolyl-carbocyanine iodide.
|
Discussion
Oxidative stress-induced neuronal cell death contributes significantly to the physiological processes of a number of neurological disorders, including neurodegenerative disease (2), stroke (4) and traumatic brain injury (5). The present study found that PD potently attenuated H2O2-induced mitochondrial dysfunction and mitochondrial ROS production. As a result, PD significantly attenuated H2O2-induced neuronal cell death and apoptosis. Moreover, it was found that activated AKT/CREB signaling and increased Ngb expression was crucial for mediating the neuroprotective role of PD against H2O2-induced neurotoxicity. These results suggested that PD protects neuronal cells against oxidative stress by upregulating AKT/CREB/Ngb signaling. Notably, PD can cross the blood-brain barrier (38). As oxidative stress participates in a number of physiological functions and pathologies of neuronal disorders, the present study further highlights the possible therapeutic potential of PD to treat various neuronal disorders. Future studies will be focused on testing the pharmacokinetics and efficacy of PD in animal models of Alzheimer's disease.
The present study found that PD attenuates H2O2-induced mitochondrial dysfunction and mitochondrial ROS production, which can be ascribed to the activation of CREB signaling, at least in part. CREB, which belongs to the CREB/ATF family of transcription factors, is a critical component of the neuroprotective transcriptional network (39). In response to stimuli, CREB can be phosphorylated at the Ser133 site, enter cell nuclei and bind to CREB responsive elements to promote the transcription and expression of multiple antioxidant and detoxifying genes, thereby inhibiting neuronal oxidative injury (40). Among these genes, Ngb is a key candidate (31). In the present study, PD significantly elevated Ngb promoter activity and mRNA and protein levels. Notably, treatment with the CREB inhibitor KG-501 and ectopic overexpression of CREB s133a and the KCREB mutant in N2a cells largely attenuated PD-induced Ngb and neuroprotection. In addition, Ngb knockdown by specific Ngb siRNA largely attenuated PD-induced neuronal cell protection against H2O2. In agreement with the present study, it has been reported that Ngb is able to protect neuronal cells against oxidative stress (30,41). Notably, the neuroprotective roles of Ngb are associated with its ability to modulate mitochondria. Our previous data suggested that Ngb can directly interact with mitochondria and maintain mitochondrial function by binding to and suppressing the mitochondrial permeability transition pore (27) and reduce mitochondrial ROS production by inhibiting mitochondrial complex III (28,29). In agreement with our studies, accumulating studies suggest that PD protects the mitochondria of neuronal cells, myocardial cells, hepatocytes and arterial smooth muscle cells after acute severe hemorrhagic shock (42–45). Therefore, these studies indicate that PD protects against H2O2-induced mitochondrial dysfunction and neurotoxicity, in which Ngb might serve a key role.
Although the antioxidative and neuroprotective role of PD is mediated by the activation of CREB/Ngb signaling, the possibility that Nrf2 may also be involved cannot be ruled out. The antioxidative role of PD is mediated by the activation of Nrf2 (10,46,47). However, the direct physical interaction of PD with Nrf2 has not been demonstrated. As the members of the bZip family, CREB and Nrf2 may form a heterodimer (48). Notably, Nrf2 is a downstream target of CREB, as PKA-CREB signaling contributes to Exendin-4-induced Nrf2/ARE activation (49) and CREB is required for Nrf2 mediated induction of antioxidant genes heme oxygenase-1 (48). These studies suggest that CREB is upstream of Nrf2 and serves a central role in controlling Nrf2 signaling. Based on the studies of the authors and other studies, it is possible that PD activates CREB/Ngb and CREB/Nrf2 signaling pathways in parallel. This will be further investigated by employing Nrf2-deficient neurons in future studies. Considering that a number of Nrf2 activators are strong radical scavengers but also have some severe adverse effects and poor bioavailability (8), it is possible that compounds with multiple antioxidative effects may be more effective and have fewer adverse effects.
In conclusion, the results of the present study suggested that PD significantly induced AKT/CREB signaling and Ngb upregulation, which mediated the antioxidative effect of PD, at least in part, thereby protecting neuronal cells from H2O2-induced neurotoxicity. These results might provide novel molecular insights to explain the potent neuroprotective effects of PD in a variety of oxidative stress-associated neurological disorders.
Supplementary Material
Supporting Data
Acknowledgements
Not applicable.
Funding
The present study was funded in part by the National Natural Science Foundation of China (grant no. 81972642), the Natural Science Foundation of Hunan Province (grant no. 2018JJ3337), the Research Foundation of Education Bureau of Hunan Province (grant nos. 16C0018 and 17B162), the Hunan Provincial Health Commission Project (grant no. C2016045) and the Huxiang High‑Level Talent Innovation Team (grant no. 2018RS3072).
Availability of data and materials
All data generated or analyzed during this study are included in this published article.
Authors' contributions
YL and SX designed the experiments; HZ, YL, HL, CW, YX, HW and NL performed the experiments; and NL, YL, SY, XY and SX helped with data analysis and manuscript writing. HZ and YL confirm the authenticity of all the raw data. All authors have read and approved the manuscript.
Ethics approval and consent to participate
The animal protocol (approval number 2016#0101) was approved by the ethical committee of Hunan Normal University.
Patient consent for publication
Not applicable
Competing interests
The authors declare that they have no competing interests.
References
|
1
|
Wang X and Michaelis EK: Selective neuronal vulnerability to oxidative stress in the brain. Front Aging Neurosci. 2:122010.PubMed/NCBI
|
|
2
|
Singh A, Kukreti R, Saso L and Kukreti S: Oxidative stress: A key modulator in Neurodegenerative diseases. Molecules. 24:15832019. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Poddar MK, Banerjee S, Chakraborty A and Dutta D: Metabolic disorder in Alzheimer's disease. Metab Brain Dis. 36:781–813. 2021. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Chamorro A, Dirnagl U, Urra X and Planas AM: Neuroprotection in acute stroke: Targeting excitotoxicity, oxidative and nitrosative stress, and inflammation. Lancet Neurol. 15:869–881. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Khatri N, Thakur M, Pareek V, Kumar S, Sharma S and Datusalia AK: Oxidative stress: Major threat in traumatic brain injury. CNS Neurol Disord Drug Targets. 17:689–695. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Kamat CD, Gadal S, Mhatre M, Williamson KS, Pye QN and Hensley K: Antioxidants in central nervous system diseases: Preclinical promise and translational challenges. J Alzheimers Dis. 15:473–493. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Okuda S, Nishiyama N, Saito H and Katsuki H: Hydrogen peroxide-mediated neuronal cell death induced by an endogenous neurotoxin, 3-hydroxykynurenine. Proc Natl Acad Sci USA. 93:12553–12558. 1996. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Liu H, Feng Y, Xu M, Yang J, Wang Z and Di G: Four-octyl itaconate activates Keap1-Nrf2 signaling to protect neuronal cells from hydrogen peroxide. Cell Commun Signal. 16:812018. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Bonnefont-Rousselot D: Resveratrol and cardiovascular diseases. Nutrients. 8:2502016. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Zhao XJ, Yu HW, Yang YZ, Wu WY, Chen TY, Jia KK, Kang LL, Jiao RQ and Kong LD: Polydatin prevents fructose-induced liver inflammation and lipid deposition through increasing miR-200a to regulate Keap1/Nrf2 pathway. Redox Biol. 18:124–137. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Gu L, Liu J, Xu D and Lu Y: Polydatin prevents LPS-induced acute kidney injury through inhibiting inflammatory and oxidative responses. Microb Pathog. 137:1036882019. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Tang KS: Protective effects of polydatin against dementia-related disorders. Curr Neuropharmacol. 19:127–135. 2021. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Huang B, Liu J, Meng T, Li Y, He D, Ran X, Chen G, Guo W, Kan X, Fu S, et al: Polydatin prevents lipopolysaccharide (LPS)-induced Parkinson's disease via regulation of the AKT/GSK3beta-Nrf2/NF-κB signaling axis. Front Immunol. 9:25272018. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Cheng Y, Zhang HT, Sun L, Guo S, Ouyang S, Zhang Y and Xu J: Involvement of cell adhesion molecules in polydatin protection of brain tissues from ischemia-reperfusion injury. Brain Res. 1110:193–200. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Ji H, Zhang X, Du Y, Liu H, Li S and Li L: Polydatin modulates inflammation by decreasing NF-κB activation and oxidative stress by increasing Gli1, Ptch1, SOD1 expression and ameliorates blood-brain barrier permeability for its neuroprotective effect in pMCAO rat brain. Brain Res Bull. 87:50–59. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Shah FA, Kury LA, Li T, Zeb A, Koh PO, Liu F, Zhou Q, Hussain I, Khan AU, Jiang Y and Li S: Polydatin attenuates neuronal loss via reducing neuroinflammation and oxidative stress in rat MCAO models. Front Pharmacol. 10:6632019. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Li L, Tan HP, Liu CY, Yu LT, Wei DN, Zhang ZC, Lu K, Zhao KS, Maegele M, Cai DZ and Gu ZT: Polydatin prevents the induction of secondary brain injury after traumatic brain injury by protecting neuronal mitochondria. Neural Regen Res. 14:1573–1582. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Lv R, Du L, Zhang L and Zhang Z: Polydatin attenuates spinal cord injury in rats by inhibiting oxidative stress and microglia apoptosis via Nrf2/HO-1 pathway. Life Sci. 217:119–127. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Lv R, Du L, Liu X, Zhou F, Zhang Z and Zhang L: Polydatin alleviates traumatic spinal cord injury by reducing microglial inflammation via regulation of iNOS and NLRP3 inflammasome pathway. Int Immunopharmacol. 70:28–36. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Zhao X, Qin J, Li H, Feng X, Lv Y and Yang J: Effect of polydatin on neurological function and the Nrf2 pathway during intracerebral hemorrhage. J Mol Neurosci. 70:1332–1337. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Zhang S, Wang S, Shi X and Feng X: Polydatin alleviates parkinsonism in MPTP-model mice by enhancing glycolysis in dopaminergic neurons. Neurochem Int. 139:1048152020. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Bai H, Ding Y, Li X, Kong D, Xin C, Yang X, Zhang C, Rong Z, Yao C, Lu S, et al: Polydatin protects SH-SY5Y in models of Parkinson's disease by promoting Atg5-mediated but parkin-independent autophagy. Neurochem Int. 134:1046712020. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Wang X, Shas P, Liu A, Ma L, Lu M, Jiang W and Li N: Polydatin prevents Aβ-induced neuron cytotoxicity via enhancing autophagy and decreasing oxidative stress. Int J Clin Exp Med. 10:15250–15259. 2017.
|
|
24
|
Liu N, Yu Z, Gao X, Song YS, Yuan J, Xun Y, Wang T, Yan F, Yuan S, Zhang J, et al: Establishment of cell-based neuroglobin promoter reporter assay for neuroprotective compounds screening. CNS Neurol Disord Drug Targets. 15:629–639. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Burmester T, Weich B, Reinhardt S and Hankeln T: A vertebrate globin expressed in the brain. Nature. 407:520–523. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Yu Z, Liu N, Liu J, Yang K and Wang X: Neuroglobin, a novel target for endogenous neuroprotection against stroke and neurodegenerative disorders. Int J Mol Sci. 13:6995–7014. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Yu Z, Liu N, Li Y, Xu J and Wang X: Neuroglobin overexpression inhibits oxygen-glucose deprivation-induced mitochondrial permeability transition pore opening in primary cultured mouse cortical neurons. Neurobiol Dis. 56:95–103. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Yu Z, Zhang Y, Liu N, Yuan J, Lin L, Zhuge Q, Xiao J and Wang X: Roles of neuroglobin binding to mitochondrial complex III subunit cytochrome c1 in oxygen-glucose deprivation-induced neurotoxicity in primary neurons. Mol Neurobiol. 53:3249–3257. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Liu N, Yu Z, Xun Y, Shu P, Yue Y, Yuan S, Jiang Y, Huang Z, Yang X, Feng X, et al: Amyloid-β25–35 upregulates endogenous neuroprotectant neuroglobin via NFκB Activation in vitro. J Alzheimers Dis. 64:1163–1174. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Amri F, Ghouili I, Amri M, Carrier A and Masmoudi-Kouki O: Neuroglobin protects astroglial cells from hydrogen peroxide-induced oxidative stress and apoptotic cell death. J Neurochem. 140:151–169. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Liu N, Yu Z, Li Y, Yuan J, Zhang J, Xiang S and Wang X: Transcriptional regulation of mouse neuroglobin gene by cyclic AMP responsive element binding protein (CREB) in N2a cells. Neurosci Lett. 534:333–337. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Ullman-Culleré MH and Foltz CJ: Body condition scoring: A rapid and accurate method for assessing health status in mice. Lab Anim Sci. 49:319–323. 1999.PubMed/NCBI
|
|
33
|
Eisenhauer EA, Therasse P, Bogaerts J, Schwartz LH, Sargent D, Ford R, Dancey J, Arbuck S, Gwyther S, Mooney M, et al: New response evaluation criteria in solid tumours: Revised RECIST guideline (version 1.1). Eur J Cancer. 45:228–247. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Liu N, Yu Z, Xiang S, Zhao S, Tjärnlund-Wolf A, Xing C, Zhang J and Wang X: Transcriptional regulation mechanisms of hypoxia-induced neuroglobin gene expression. Biochem J. 443:153–164. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Summers DW, DiAntonio A and Milbrandt J: Mitochondrial dysfunction induces Sarm1-dependent cell death in sensory neurons. J Neurosci. 34:9338–9350. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Huang TJ, Price SA, Chilton L, Calcutt NA, Tomlinson DR, Verkhratsky A and Fernyhough P: Insulin prevents depolarization of the mitochondrial inner membrane in sensory neurons of type 1 diabetic rats in the presence of sustained hyperglycemia. Diabetes. 52:2129–2136. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
See V and Loeffler JP: Oxidative stress induces neuronal death by recruiting a protease and phosphatase-gated mechanism. J Biol Chem. 276:35049–35059. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Chen Y, Zhang DQ, Liao Z, Wang B, Gong S, Wang C, Zhang MZ, Wang GH, Cai H, Liao FF and Xu JP: Anti-oxidant polydatin (piceid) protects against substantia nigral motor degeneration in multiple rodent models of Parkinson's disease. Mol Neurodegener. 10:42015. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Sakamoto K, Karelina K and Obrietan K: CREB: A multifaceted regulator of neuronal plasticity and protection. J Neurochem. 116:1–9. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Lee B, Cao R, Choi YS, Cho HY, Rhee AD, Hah CK, Hoyt KR and Obrietan K: The CREB/CRE transcriptional pathway: Protection against oxidative stress-mediated neuronal cell death. J Neurochem. 108:1251–1265. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Li RC, Morris MW, Lee SK, Pouranfar F, Wang Y and Gozal D: Neuroglobin protects PC12 cells against oxidative stress. Brain Res. 1190:159–166. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Li P, Wang X, Zhao M, Song R and Zhao KS: Polydatin protects hepatocytes against mitochondrial injury in acute severe hemorrhagic shock via SIRT1-SOD2 pathway. Expert Opin Ther Targets. 19:997–1010. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Ling Y, Chen G, Deng Y, Tang H, Ling L, Zhou X, Song X, Yang P, Liu Y, Li Z, et al: Polydatin post-treatment alleviates myocardial ischaemia/reperfusion injury by promoting autophagic flux. Clin Sci (Lond). 130:1641–1653. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Wang X, Song R, Bian HN, Brunk UT, Zhao M and Zhao KS: Polydatin, a natural polyphenol, protects arterial smooth muscle cells against mitochondrial dysfunction and lysosomal destabilization following hemorrhagic shock. Am J Physiol Regul Integr Comp Physiol. 302:R805–R814. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Wang X, Song R, Chen Y, Zhao M and Zhao KS: Polydatin-a new mitochondria protector for acute severe hemorrhagic shock treatment. Expert Opin Investig Drugs. 22:169–179. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
46
|
Ye J, Piao H, Jiang J, Jin G, Zheng M, Yang J, Jin X, Sun T, Choi YH, Li L and Yan G: Polydatin inhibits mast cell-mediated allergic inflammation by targeting PI3K/Akt, MAPK, NF-κB and Nrf2/HO-1 pathways. Sci Rep. 7:118952017. View Article : Google Scholar : PubMed/NCBI
|
|
47
|
Zhan J, Li X, Luo D, Hou Y, Hou Y, Chen S, Xiao Z, Luan J and Lin D: Polydatin promotes the neuronal differentiation of bone marrow mesenchymal stem cells in vitro and in vivo: Involvement of Nrf2 signalling pathway. J Cell Mol Med. 24:5317–5329. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
48
|
Mylroie H, Dumont O, Bauer A, Thornton CC, Mackey J, Calay D, Hamdulay SS, Choo JR, Boyle JJ, Samarel AM, et al: PKCε-CREB-Nrf2 signalling induces HO-1 in the vascular endothelium and enhances resistance to inflammation and apoptosis. Cardiovasc Res. 106:509–519. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
49
|
Zhou T, Zhang M, Zhao L, Li A and Qin X: Activation of Nrf2 contributes to the protective effect of Exendin-4 against angiotensin II-induced vascular smooth muscle cell senescence. Am J Physiol Cell Physiol. 311:C572–C582. 2016. View Article : Google Scholar : PubMed/NCBI
|














