Introduction
Hepatocellular carcinoma (HCC) is the third most
commonly diagnosed malignant tumor (1). The symptoms of patients are not
obvious during the initial stage, and consequently, most patients
with HCC are diagnosed at the middle and late stages of the
disease, often accompanied by intrahepatic and extrahepatic
metastasis (2). The prognosis is
poor, which brings a heavy burden to these patients. Approximately
110,000 people die of liver cancer in China every year, accounting
for 45% of the world's liver cancer deaths (3). The detection of serum α-fetoprotein
(AFP) combined with ultrasound imaging can provide a reliable
clinical basis for the early detection of liver cancer, which
allows for its successful resection (3). Combined with active comprehensive
treatment, resection can significantly improve the 5-year survival
rate of liver cancer patients. However, recurrence and metastasis
often occur in patients with liver cancer, resulting in low quality
of life among patients with liver cancer. At present, there is a
lack of effective prevention and treatment measures in the clinic.
Therefore, it is of great significance to clarify the molecular
mechanism of HCC and to find effective molecular therapeutic target
genes.
The main function of junctophilin is to participate
in the formation of coupled membrane complexes, maintain a very
short distance between the cell membrane and endoplasmic reticulum,
and provide a structural basis for signal transduction between the
cell membrane and endoplasmic reticulum, such as calcium-induced
aluminum release process of cardiomyocytes, voltage-dependent
release in skeletal muscle, and library controlled calcium influx
(4,5). As an important member of the
junctophilin family, junctophilin 3 (JPH3 is widely involved in the
pathophysiological process of cells, but its role in HCC is unknown
(6).
In recent years, it has been found that epigenetics
plays an important role in the malignant occurrence and development
of HCC (7). Epigenetic changes
refer to alterations in the functions and characteristics of genes
in various ways without changing the target gene sequence, and they
can be inherited through genomic modification during cell division
and proliferation cycle including DNA methylation, genomic
imprinting, maternal effects, and gene silencing (8,9).
These epigenetic modifications are widely involved in the
physiological and pathological processes, variability and
adaptability of organisms and can be affected by gene and
environmental factors (10).
JPH3 has been reported to play a tumor-suppressor
role in a variety of gastrointestinal tumors (11). Previous literature has reported
that DNA methylation regulates the expression of JPH3 and causes
its low expression in tumors, which plays a biological role in
promoting the malignant progression of tumors (6). However, it is still unclear whether
it also displays the above phenomenon in HCC. Our results showed
that JPH3 is expressed at a low level in HCC, thus DNA methylation
is suspected to cause the low expression of JPH3. In the present
study, the relationship between its DNA methylation level and
protein expression was analyzed. Furthermore, the relationship
between its expression level and the prognosis of HCC patients was
also analyzed. In cell function experiments, we used demethylation
drugs to observe the effect of JPH3 on the malignant biological
behavior of HCC cells after altering the DNA methylation level of
JPH3 and found that EMT regulates HCC.
Materials and methods
Tissue samples and related clinical
data
Seventy-three patients with primary hepatocellular
carcinoma were diagnosed at the Department of Hepatobiliary
Pancreatic Surgery, Affiliated Hospital of Zunyi Medical
University, and received surgical treatment (from May 2012 to
November 2015; age range, 25–71 years; sex distribution, 34 male,
39 female). The collected data included HCC tissues and paired
adjacent tissues. The present study was performed in accordance
with the principles outlined in the Declaration of Helsinki. The
Human Trial Ethics Committee of the Affiliated Hospital of Zunyi
Medical University approved this study (ethical approval no.
2011035). Written informed consent was obtained from the patients
who provided the specimens.
The detailed inclusion criteria were as follows: i)
HCC was confirmed by postoperative pathology, and the
histopathological diagnosis was clear; ii) patients were diagnosed
with HCC for the first time without distant metastasis; iii)
patients had not received any treatment prior to surgery; iv) no
other serious malignant disease had been diagnosed; v) the
clinical, pathological and surgical data were complete; vi) the
follow-up information was complete and available.
The exclusion criteria were as follows: i) those who
could not communicate normally; ii) patients with organic
dysfunction of the heart, brain, lung and kidney; iii) patients who
were diagnosed with HCC for the first time with distant metastasis;
iv) patients with incomplete clinical and pathological data; v)
patients with systemic diseases; vi) patients with autoimmune
dysfunction; and vii) follow-up information was not complete and
available.
Cell culture and 5-Aza
HCC cell lines Huh-7, MHCC-97H, MHCC-97L, Hep3B, and
human normal hepatocytes THLE2 were purchased from the Cell Bank of
the Chinese Academy of Sciences (Shanghai, China). All cells were
cultured in DMEM high glucose medium [containing 10% fetal bovine
serum (FBS)] and cultured in an incubator at constant temperature
of 37°C with 5% CO2. Huh-7 and Hep3B cells were treated
with 5-aza-2′-deoxycytidine (5-Aza) (Sigma-Aldrich; Merck KGaA) at
1 µM for 4 days as previously reported (12). The culture medium and 5-Aza were
replaced daily.
Cell transfection
Plasmid JPH3 and the plasmid control were
synthesized by Shanghai GenePharma Co., Ltd. Lipofectamine 2000
reagent (Thermo Fisher Scientific, Inc.) was used for transient
transfection, following the manufacturer's instructions. The cells
were harvested at 48 h following transfection.
Reverse transcription quantitative PCR
(RT-qPCR)
Total RNA was extracted from 100 mg HCC tissues,
paired adjacent tissue and HCC cell lines using TRIzol reagent
(Takara Bio, Inc.). Reverse transcription of cDNA was performed
using the PrimeScript RT Reagent Kit (Takara Bio, Inc.). The
temperature protocol for RT was as follows: 35°C for 5 min,
followed by 47°C for 30 min and 80°C for 5 min. In addition, qPCR
was performed with SYBR Premix Ex Taq II (Takara Bio, Inc.) and a
Light Cycler system (Roche Diagnostics GmbH). The thermocycling
conditions for qPCR were as follows: initial activation step at
45°C for 30 min followed by 40 cycles of denaturation at 92°C for
15 sec, annealing at 65°C for 30 sec and extension at 72°C for 30
sec. The results were analyzed using the 2−ΔΔCq method
(13). GAPDH was used as the
internal standard. The following primer sequences were used for the
qPCR: JPH3 forward, 5′-AATCCTTGCCTGTCGCTCTA-3′ and reverse,
5′-CCCAATCGTGTGGTTCTTCT-3′; and GAPDH forward,
5′-GGAGCGACATCCGTCCAAAAT −3′ and reverse,
5′-GGCTGTTGTCAATCTTCTCATGG-3′.
Western blot analysis
Total protein from 100 mg HCC tissues, paired
adjacent tissue and HCC cell lines (1×106) was extracted
using RIPA lysis buffer (Beyotime Institute of Biotechnology Co.,
Ltd.). A Nanodrop spectrophotometer was used for quantification,
and an equal amount 30 µg was added to each well of 12% gels and
resolved by SDS-PAGE. Proteins were transferred to PVDF membranes
and blocked by incubation for 1 h at 37°C with 5% nonfat powdered
milk. The membranes were probed at 4°C overnight with antibodies
against JPH3 (dilution 1:3,000; ab79063; Abcam), N-cadherin
(dilution 1:5,000; ab76011; Abcam), vimentin (dilution 1:2,000;
ab92547; Abcam), Ki-67 (dilution 1:5,000; ab92742; Abcam),
E-cadherin (dilution 1:10,000; ab40772; Abcam), and GAPDH (dilution
1:5,000; ab9485; Abcam). The PVDF membranes were incubated with
horseradish peroxidase-conjugated secondary antibody (dilution
1:5,000; ab6721; Abcam) at room temperature for 1 h. Protein
expression levels were visualized using an enhanced
chemiluminescence detection system (Bio-Rad Laboratories, Inc.).
GAPDH was used as the internal loading control in the western blot
analysis. Protein expression was quantified using Quantity One
version 4.6.5 software (Bio-Rad Laboratories, Inc.).
Immunohistochemistry
The sample was dehydrated with xylene and alcohol.
Antigen retrieval was performed in 10 mmol/l sodium citrate
solution (pH 6.0) at 100°C for 15 min, and the samples were cooled
for 30 min. All slides were incubated with 5% goat serum (OriGene
Technologies, Inc.) for 15 min at room temperature to block
non-specific binding at room temperature. The slides were incubated
with antibodies against JPH3 (dilution 1:300; ab79063; Abcam) at
4°C overnight. The samples were incubated with biotinylated
secondary antibody at 37°C for 30 min then stained with DAB
(3,3-diaminobenzidine) and Mayer's hematoxylin.
JPH3 staining classifications were as follows: range
0–3: 0, negative; 1, weak; 2, moderate; and 3, strong; the
percentage of positive cells ranged from 0–4: 0, negative or
<5%; 1, 6–25%; 2, 26–50%; 3, 51–75%; and 4, 76–100%. The
percentage of positive cells and the intensity were used to
determine the final staining scores. Grades <4 were defined as
low JPH3 expression, while grades ≥4 were defined as high JPH3
expression.
Immunofluorescence
Huh-7 and Hep3B cells were fixed with 4%
paraformaldehyde and permeabilized with 0.25% Triton X-100 solution
for 30 min. Then, the cells were washed with PBS and blocked in 5%
bovine serum albumin for 1 h at room temperature. The coverslips
were incubated with antibodies against JPH3 (dilution 1:3,000;
ab79063; Abcam) overnight at 4°C. Then, the cells were washed with
PBS and incubated with appropriate secondary antibody and
4′,6-diamidino-2-phenylindole (DAPI). Slides were imaged using an
inverted fluorescence microscope. (magnification, ×200; Zeiss
AG).
DNA extraction and
methylation-specific PCR
DNA was isolated from HCC tissues (tumor and paired
normal adjacent tissues) and HCC cell lines by using a DNA
Isolation kit (Tiangen). Determination of bisulfite conversion was
performed using the EpiTect Bisulfite Kit (Qiagen Inc.).
Methylation-specific PCR (MSP) was performed with 2 µl of
bisulfite-modified DNA (100 ng/50 µl) and 48 µl of PCR mixture
consisting of 10X PCR Buffer (Mg2+ free), 25 mM
MgCl2, dNTP mixture (each 2.5 mM), sense primer (20 µM),
antisense primer (20 µM), and Takara EpiTaq HS (5 U/µl; Takara).
PCR amplification was conducted using 40 cycles (95°C for 20 sec,
41°C for 30 sec, and 75°C for 30 sec). For parallel quality
control, a plasmid containing a methylated JPH3 sequence and water
without DNA template were used as positive and negative controls,
respectively. The following primer sequences were used for the
methylation-specific PCR: JPH3 M forward,
5′-AGACGTTGGTTAGGTTTCGC-3′ and JPH3 U forward,
5′-GAGATGTTGGTTAGGTTTTGT −3′.
Bisulfite Sanger sequencing (BSP)
Five hundred nanograms of genomic DNA extracted from
HCC tissues (tumor tissues and paired normal adjacent tissues) was
bisulfite converted using a MethylCode™ Bisulfite
Conversion kit (Applied Biosystems; Thermo Fisher Scientific,
Inc.). The JPH3 promoter was amplified by PCR with Taq DNA
Polymerase (Invitrogen; Thermo Fisher Scientific, Inc.). The primer
sequence was designed using Methyl Primer Express™
Software v1.0 (Applied Biosystems; Thermo Fisher Scientific, Inc.).
The PCR products were electrophoresed, purified using Spin-X tubes,
and then cloned into the pUC-T vector (both from CWbiotech). Ten
single products were sequenced for each sample.
Transwell invasion assays
Cells (1×105 cells/well) were resuspended
in high-glucose DMEM containing 1% FBS and seeded into the upper
Transwell chamber at a density of 1×105 cells/well.
Tranwell inserts (Corning, Inc.) were precoated with Matrigel (1
mg/ml) at 37°C for 30 min. High-glucose DMEM (500 µl) of containing
10% FBS was added to the matched lower chamber. After 36 h
incubation, the Transwell chambers were fixed with 4%
paraformaldehyde at room temperature for 30 min, stained with 0.5%
crystal violet at room temperature for 20 mins, and finally counted
under an inverted optical microscope (×100 magnification; Olympus
Corp.).
CCK-8
Hep3B and Huh-7 cells (1×103 cells/well)
were seeded into 96-well plates and incubated at 37°C for 24, 48 or
72 h. Then, CCK-8 reagent (Dojindo Molecular Technologies, Inc.)
(10 µl) was added to each well, and incubation at 37°C for 2 h. The
absorbance was measured at 450 nm by a spectrophotometer (Bio-Rad
Laboratories, Inc.).
Colony formation assay
Huh-7 and Hep3B cells were plated in 6-well plates
(500 cells/well) containing 2.5 ml of medium and cultured for 20
days. Colonies formed by cell proliferation were stained with
crystal violet, and colonies containing at least 50 cells were
counted. Colonies were fixed with 20% methanol for 30 min and
stained with 0.1% crystal violet for 5 min, and then counted. The
numbers of colonies were then counted manually using a light
microscope at ×20 magnification.
Wound healing assay
Huh-7 and Hep3B cells (5×105) were grown
in 6-well plates, and grown until they formed a confluent
monolayer. When the cells reached confluence, any nonadherent cells
were washed away with phosphate-buffered saline (PBS). The cell
monolayer was scratched with a pipette tip (10 µl) to generate 3
scratch wounds and then rinsed with PBS to remove nonadherent
cells, and the medium was replaced with fresh DMEM high glucose
medium without FBS. After 0 and 36 h, the distance between the
wound sides was measured.
Flow cytometry analysis of cell
apoptosis and cell cycle distribution
Huh-7 and Hep3B cells (1×105 cells) were
seeded into 6-well plates. Once they reached confluence, the cells
were collected and incubated with Annexin V-FITC (5 µl) and
propidium iodide (PI) solution (5 µl; Biogot Technology Co., Ltd.)
at room temperature for 15 min according to the manufacturer's
instructions. Cells were subsequently suspended in 400 µl binding
buffer. For cell cycle analysis, Huh-7 and Hep3B cells were
collected and fixed in 75% ethanol at −20°C overnight. The fixed
cells were washed with PBS and incubated with RNase A for 20 min at
room temperature. These cells were stained with PI and incubated in
the dark for 30 min at 4°C. Cell apoptosis and cell cycle
progression were analyzed using flow cytometry (BD Biosciences).
The percentages of cells within each phase of the cell cycle were
analyzed with ModFit version 4.0 (Verity Software House, Inc.) and
CellQuest version 5.1 (Thermo Fisher Scientific, Inc.).
Statistical analyses
Statistical data were analyzed using SPSS version
22.0 software (IBM Corp.) and GraphPad Prism version 5.0 software
(GraphPad Software Inc.). Paired Student's t-tests were used to
determine the expression of JPH3 in the HCC and the corresponding
peritumor tissues. If not specified, the unpaired Student's t-test
or analysis of variance was used, followed by Dunnett's multiple
comparison test to compare differences between groups. Associations
between clinicopathological parameters and JPH3 expression were
analyzed using Pearson's Chi-squared test. Kaplan-Meier survival
analysis and Cox regression assays were used to analyze the
prognostic significance of JPH3. P<0.05 was considered to
indicate a statistically significant difference. All data are
presented as the means ± SD.
Results
JPH3 is downregulated in HCC
We first analyzed the expression levels of JPH3 in
the HCC tissues. The mRNA expression of JPH3 was
significantly lower in HCC samples than that in the paired normal
adjacent tissues (Fig. 1A).
Immunohistochemical staining showed that JPH3 expression was lower
in the HCC samples than that in the paired normal adjacent tissues,
and JPH3 expression was primarily localized in the nucleus
(Fig. 1B). Western blot analysis
also confirmed that JPH3 expression was significantly lower in the
HCC samples than that in the paired normal adjacent tissues
(Fig. 1C). In addition, the
expression of JPH3 was also lower in HCC cell lines than in normal
hepatocytes THLE2 (Fig. 1D).
Hypermethylation of JPH3 promoter CpG
island contributes to low JPH3 expression in HCC
JPH3 has been reported to exhibit DNA methylation,
causing low protein expression. The methylation status of JPH3 was
assayed in randomly selected HCC tissues compared to paired normal
adjacent tissues. JPH3 promoter methylation in HCC tissues was
higher than that in the paired normal adjacent tissues (Fig. 2A). Analysis of TELE2, MHCC-97H,
MHCC-97L, Hep3B, and Huh-7 cell lines revealed a significantly high
degree of methylation in the HCC cell lines, but not in the TELE2
cells (Fig. 2B). Next, bisulfite
sequence-PCR (BSP) was used to analyze the specific methylation of
CpG island of JPH3, and the results showed that JPH3 promoter
methylation in HCC tissues was higher than that in the paired
normal adjacent tissues (Fig.
2C).
Low expression of JPH3 is
significantly associated with poor clinical prognosis in HCC
We further analyzed the association between JPH3
expression and the clinicopathological characteristics of the HCC
patients. JPH3 expression was significantly associated with TNM
stage (P=0.003) and tumor size (P=0.016) (Table I). The HCC patients with low JPH3
expression exhibited significantly shorter overall survival (OS)
than the HCC patients with high JPH3 expression (P=0.034, Fig. 3A). Cox proportional hazards
regression analysis indicated that TNM stage (P=0.014), tumor size
(P=0.030) and JPH3 (P=0.010) were significantly associated with OS
in HCC. In addition, multivariate analysis showed that TNM stage
(P=0.017), tumor size (P=0.027) and JPH3 (P=0.015) were independent
prognostic factors for OS in HCC (Table II). Moreover, HCC patients with
low JPH3 expression exhibited significantly shorter DSS than HCC
patients with high JPH3 expression (P=0.044, Fig. 3B). Cox proportional hazards
regression analysis indicated that TNM stage (P=0.009), tumor size
(P=0.024) and JPH3 (P=0.015) were significantly associated with DSS
in HCC. In addition, multivariate analysis showed that TNM stage
(P=0.007), tumor size (P=0.021) and JPH3 (P=0.019) were independent
prognostic factors for DSS in HCC (Table III).
 | Table I.Associations between JPH3 and
clinicopathological features of the HCC patients. |
Table I.
Associations between JPH3 and
clinicopathological features of the HCC patients.
|
|
| JPH3
expression |
|
|---|
|
|
|
|
|
|---|
| Variables | Cases | High (n=21) | Low (n=52) | P-value |
|---|
| Age, years |
|
|
|
|
|
<50 | 40 | 12 | 28 | 0.798 |
|
≥50 | 33 | 9 | 24 |
|
| Sex |
|
|
| 0.527 |
|
Male | 34 | 11 | 23 |
|
|
Female | 39 | 10 | 29 |
|
| AFP (ng/ml) |
|
|
| 0.232 |
|
≤20 | 25 | 5 | 20 |
|
|
>20 | 48 | 16 | 32 |
|
| HBsAg |
|
|
| 0.478 |
|
Positive | 44 | 14 | 30 |
|
|
Negative | 29 | 7 | 22 |
|
| Tumor size
(cm) |
|
|
| 0.016 |
| ≤5 | 36 | 15 | 21 |
|
|
>5 | 57 | 6 | 31 |
|
| TNM stage |
|
|
| 0.003 |
|
I/II | 29 | 14 | 15 |
|
|
III/IV | 44 | 7 | 37 |
|
| Multiplicity |
|
|
| 0.438 |
|
Presence | 40 | 13 | 27 |
|
|
Absence | 33 | 8 | 25 |
|
| Intrahepatic
metastasis |
|
|
| 0.739 |
|
Presence | 36 | 11 | 25 |
|
|
Absence | 37 | 10 | 27 |
|
| Table II.Univariate and multivariate analysis
of different prognostic variables influencing overall survival (OS)
in the HCC patients. |
Table II.
Univariate and multivariate analysis
of different prognostic variables influencing overall survival (OS)
in the HCC patients.
|
|
| Univariate analysis
model | Multivariate
analysis model |
|---|
|
|
|
|
|
|---|
| Variables | n | HR (95% CI) | P-value | HR (95% CI) | P-value |
|---|
| Age (years) |
| 0.336
(0.276-1.084) | 0.536 |
|
|
|
<50 | 40 |
|
|
|
|
|
≥50 | 33 |
|
|
|
|
| Sex |
| 0.408
(0.339-1.127) | 0.506 |
|
|
|
Male | 34 |
|
|
|
|
|
Female | 39 |
|
|
|
|
| AFP (ng/ml) |
| 1.115
(0.869-2.064) | 0.726 |
|
|
|
≤20 | 25 |
|
|
|
|
|
>20 | 48 |
|
|
|
|
| HBsAg |
| 1.046
(0.479-1.069) | 0.551 |
|
|
|
Positive | 44 |
|
|
|
|
|
Negative | 29 |
|
|
|
|
| TNM stage |
| 1.148
(0.486-2.394) | 0.014 |
1.241(0.556-2.047) | 0.017 |
|
I/II | 29 |
|
|
|
|
|
III/IV | 44 |
|
|
|
|
| Tumor size, cm |
| 1.012
(0.784-1.840) | 0.030 | 1.047
(0.774-1.931) | 0.027 |
| ≤5 | 36 |
|
|
|
|
|
>5 | 57 |
|
|
|
|
| Intrahepatic
metastasis |
| 0.756
(0.841-1.894) | 0.447 |
|
|
|
Presence | 36 |
|
|
|
|
|
Absence | 37 |
|
|
|
|
| JPH3
expression |
| 1.427
(0.841-3.004) | 0.010 | 1.439
(0.715-3.046) | 0.015 |
|
Low | 52 |
|
|
|
|
|
High | 21 |
|
|
|
|
| Table III.Univariate and multivariate analysis
of different prognostic variables influencing disease-specific
survival (DSS) of the HCC patients. |
Table III.
Univariate and multivariate analysis
of different prognostic variables influencing disease-specific
survival (DSS) of the HCC patients.
|
|
| Univariate
analysis | Multivariate
analysis model |
|---|
|
|
|
|
|
|---|
| Variable | n | HR (95% CI) | P-value | HR (95% CI) | P-value |
|---|
| Age (years) |
| 0.304
(0.365-1.203) | 0.630 |
|
|
|
<50 | 40 |
|
|
|
|
|
≥50 | 33 |
|
|
|
|
| Sex |
| 0.348
(0.340-1.178) | 0.405 |
|
|
|
Male | 34 |
|
|
|
|
|
Female | 39 |
|
|
|
|
| AFP (ng/ml) |
| 0.842
(0.634-1.778) | 0.836 |
|
|
|
≤20 | 25 |
|
|
|
|
|
>20 | 48 |
|
|
|
|
| HBsAg |
| 1.135
(0.782-1.842) | 0.641 |
|
|
|
Positive | 44 |
|
|
|
|
|
Negative | 29 |
|
|
|
|
| TNM stage |
| 1.004
(0.447-1.236) | 0.009 | 1.047
(0.486-1.445) | 0.007 |
|
I/II | 29 |
|
|
|
|
|
III/IV | 44 |
|
|
|
|
| Tumor size
(cm) |
| 1.148
(0.556-2.014) | 0.024 | 1.214
(0.336-2.174) | 0.021 |
| ≤5 | 36 |
|
|
|
|
|
>5 | 57 |
|
|
|
|
| Intrahepatic
metastasis |
| 1.047
(0.660-1.930) | 0.475 |
|
|
|
Presence | 36 |
|
|
|
|
|
Absence | 37 |
|
|
|
|
| JPH3
expression |
| 1.410
(0.559-2.014) | 0.015 | 1.507
(0.663-2.474) | 0.019 |
|
Low | 52 |
|
|
|
|
|
High | 21 |
|
|
|
|
Restoration of JPH3 expression by
treatment with 5-Aza demethylation inhibits the proliferation,
migration and invasion abilities in HCC
To test whether methylation directly induces JPH3
silencing, the JPH3 gene was demethylated in the Huh-7 and Hep3B
cell lines with 5-Aza, an inhibitor of DNA methyltransferases. As
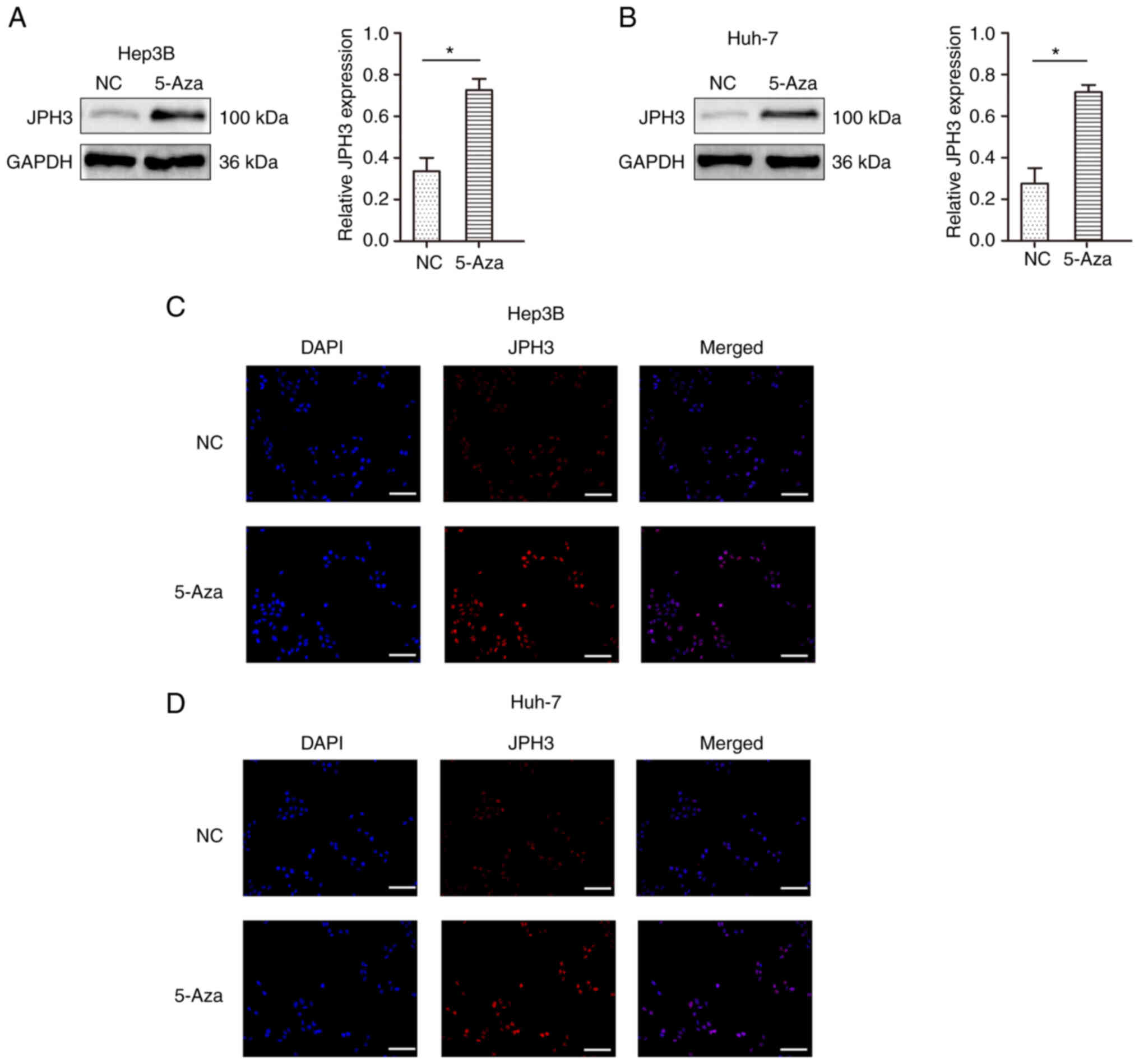
shown in Fig. 4A and B, after
treatment with 1 µM 5-Aza for 96 h, the expression of the JPH3
protein was restored. The immunofluorescence results confirmed the
above results (Fig. 4C and D). We
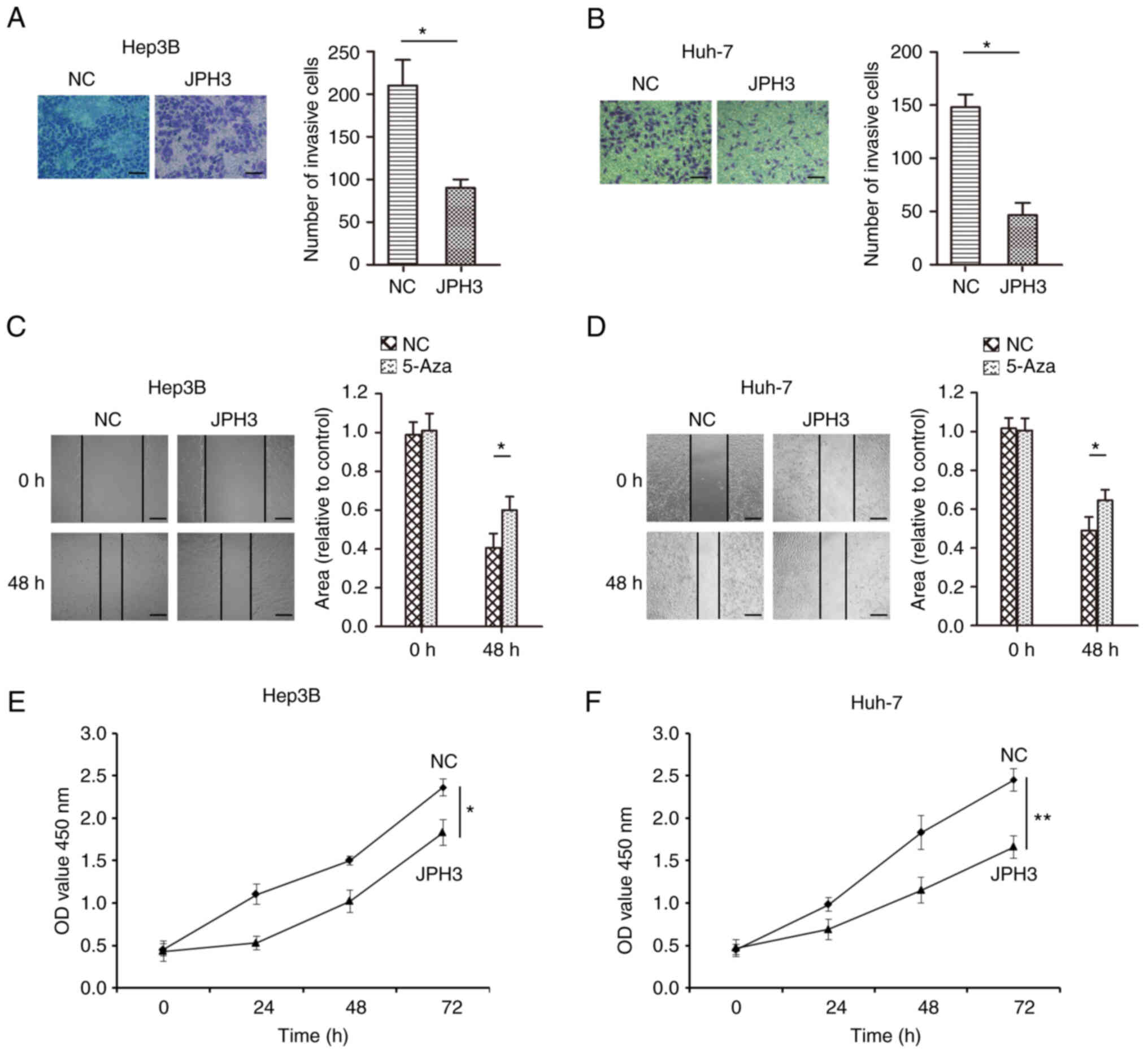
analyzed the effect of JPH3 on proliferation, migration and
invasion in HCC. The invasive ability of cells treated with 5-Aza
was less than that of the control Huh-7 and Hep3B cell lines
(Fig. 5A and B). In addition, the
migration ability of the cells treated with 5-Aza was less than
that of the control Huh-7 and Hep3B cell lines (Fig. 5C and D). The colony formation
experiment also showed that treatment with 5-Aza significantly
inhibited cell proliferation in the Huh-7 and Hep3B cell lines
(Fig. 5E and F). Moreover,
increasing the expression of JPH3 using JPH3 plasmid also inhibited
the proliferation, invasion and migration of HCC cells (Fig. 6A-F). The above results indicate
that demethylation of JPH3 can restore the protein expression of
JPH3 and inhibit proliferation, migration and invasion in HCC.
Restoration of JPH3 expression by
treatment with 5-Aza demethylation regulates the cell cycle and
apoptosis in HCC cells
We further analyzed the effect of JPH3 on cell cycle
distribution and apoptosis in the HCC cells. The results showed
that treatment with 5-Aza significantly promoted cell apoptosis in
the Huh-7 and Hep3B cell lines (Fig.
7A and B). In addition, the G1 stage of cells in the
5-Aza-treated group was higher than that noted in the control group
of Huh-7 and Hep3B cell lines (Fig.
7C and D). Moreover, increasing the expression of JPH3 promoted
cell apoptosis and prevented cell growth in the Huh-7 and Hep3B
cell lines (Fig. S1). These
results indicate that demethylation of JPH3 induced Huh-7 and Hep3B
cell cycle arrest in G1 stage and promoted apoptosis.
JPH3 affects the biological behavior
of HCC by regulating EMT
Based on the above research results, we further
analyzed whether JPH3 affects the malignant biological behavior of
HCC by regulating EMT. Western blot results revealed that
restoration of JPH3 expression by 5-Aza significantly suppressed
the expression of N-cadherin, vimentin and KI-67, which were
increased by downregulated expression of JPH3, and 5-Aza treatment
significantly increased the expression of E-cadherin, which was
inhibited by downregulated expression of JPH3 (Fig. 8A and B). Moreover, increasing the
expression of JPH3 significantly decreased the expression of
N-cadherin, vimentin and KI-67, which were increased by
downregulated expression of JPH3, and the expression of E-cadherin,
which was inhibited by downregulated expression of JPH3 was
increased (Fig. S2).
Discussion
Hepatocellular carcinoma (HCC) is a malignant tumor
with a high recurrence and mortality rate particularly in China
(14). Most patients are in the
middle and late stages of the disease at the time of diagnosis,
accompanied by extensive intrahepatic and extrahepatic metastasis,
and the patient prognosis is poor (15). Fundamentally speaking, tumors are
a disease of tissue growth regulation, and the malignant biological
behavior of a tumor is an important manifestation of abnormal
growth regulation (16). These
regulatory changes often involve gene mutations, deletion and
chromosome abnormalities, and epigenetic changes are among the most
common causes of gene function changes (16). The malignant growth of liver
cancer is a complex process involving many regulatory factors,
including gene mutations and epigenetic changes, changes in cell
surface signal transduction molecules and adhesion ability,
abnormal cell metabolism, and changes in tumor cells and the
surrounding microenvironment (17–20).
DNA methylation leads to the formation of
5-methylcytosine (5 mC) in DNA through the methyl donor S-adenosine
methionine (SAM) to the methyl of carbon 5 in the cytosine pyridine
ring. DNA methyltransferase family can catalyze this reaction
(21). In mammalian somatic
cells, this stable synthetic epigenetic marker exists only in the
dinucleotide of the CpG cytosine residue, while in embryonic stem
cells, DNA methylation occurs in both CpG and non-CpG sequences
(22). A large number of studies
have shown that DNA methylation plays an important role in the
proliferation and metastasis of HCC (7,23).
DNA methylation is a chemical modification process that transfers
S-adenosylmethionine as active methyl to a specific base in the DNA
chain under the catalysis of DNA methyltransferase (24). It causes changes in DNA
conformation, DNA stability and the mode of interaction between DNA
and protein, and controls gene expression (25). DNA methylation during the
occurrence and development of HCC is mainly manifested in the
hypomethylation activation of proto-oncogenes and the methylation
inactivation of tumor-suppressor genes (26). Studies have found that the
methylation level of proto-oncogenes Ras and myc in the promoter
region of HCC are significantly low, which enhances the expression
of c-myc and c-N-Rras and promotes the growth, survival and
metastasis of HCC cells (27). In
HCC, hypermethylation of tumor-suppressor gene promoter CpG island
interferes with the transcription of tumor-suppressor genes,
resulting in abnormal cell proliferation, which is closely related
to tumorigenesis (28). It has
been found that inactivation of DLL3 gene methylation in HCC can
promote the proliferation and metastasis of HCC cells through the
Notch signaling pathway (29). In
addition, Smad nuclear interacting protein-1 (snip1), whose
promoter is subjected to hypermethylation in HCC cells, can mediate
epithelial-mesenchymal transformation (EMT) and promote the
metastasis of HCC (30). In the
present study, we demonstrated that junctophilin 3 (JPH3) was also
modified by DNA methylation in HCC, resulting in its low expression
in HCC tissues. Its low expression in HCC was found to be
significantly related to the poor prognosis of patients.
When analyzing the role of DNA methylation of JPH3
in the malignant progression of HCC, we found that after treating
HCC cells with 5-Aza, the expression of JPH3 was significantly
increased, the proliferation, invasion and migration of HCC were
significantly inhibited, and the apoptosis level of HCC cells was
increased. These results suggest that DNA methylation and protein
expression of JPH3 may affect the signaling pathways regulating the
invasion and metastasis of HCC. Thus, we focused on EMT. EMT refers
to the process of epithelial cell phenotype transition to
mesenchymal cells under specific physiological or pathological
conditions (31). The main
morphological characteristics of EMT are when epithelial cells lose
their typical intercellular junction structure, reorganize the
cytoskeleton, and change from a polygonal to a spindle-shaped
fibroblast-like morphology (32).
After EMT, the cells become isolated, their movement capacity is
enhanced, and they are resistant to apoptosis (33). In HCC, TDP-43 inhibits GSK3β
protein translation and activates the Wnt/β-catenin pathway,
regulating EMT to induce proliferation and metastasis (34). In addition, miR-186 was found to
regulate EMT changes in HCC cells by targeting CDK6 and this
inhibits their proliferation, migration and invasion (35). MST4 inactivation was found to
induce EMT in HCC cells, promote their migration and invasion
potential in vitro, and promote intrahepatic metastasis
in vivo(36). MST4
inactivation was also found to increase the expression and nuclear
translocation of the key EMT transcription factor Snail1 through
the PI3K/Akt signaling pathway to induce the EMT phenotype of HCC
cells and enhance their invasion and metastasis potential (36). Our research confirmed that DNA
methylation regulates the protein expression of JPH3 and this
regulates the proliferation, migration, invasion and apoptosis of
HCC; the regulatory mechanism may be related to the changes in EMT
in tumor cells.
It has been reported that JPH3 is highly methylated
in gastrointestinal tumors, resulting in decreased expression of
its mRNA and protein levels. JPH3 can also upregulate the
cytoplasmic Ca2+ level and unfolded protein response due
to endoplasmic reticulum stress, induce calpain activation and
subsequent mitochondrial membrane depolarization and apoptosis, and
promote the malignant progression of gastrointestinal tumors
(6). Our results are consistent
with these findings. In the present study, the expression of JPH3
was analyzed in HCC for the first time and it was demonstrated that
DNA methylation may be associated with the low expression of JPH3
in HCC. Hypermethylated JPH3 in HCC, which caused a decrease in the
expression of JPH3, affected related signaling pathways and
regulated the malignant progression of tumors. Of course, our
research still has certain limitations. First, 5-Aza is a
broad-spectrum and comprehensive DNA methyltransferase inhibitor,
which can cause DNA demethylation (hypomethylation) and then
promote gene expression. However, the specific targeting sites of
5-Aza acting on JPH3 are largely unknown. In addition, evidence
verifying that the biological effects of JPH3 are related to 5-Aza,
is not solid enough. The limitations will be further investigated.
Understanding the above questions will better explain the important
role of JPH3 and its DNA methylation in HCC.
In conclusion, our research indicates that JPH3 is
expressed at low levels in HCC and its low expression is caused by
DNA methylation. The low expression of JPH3 in HCC was
significantly correlated with a poor prognosis of patients.
Mechanistically, JPH3 can inhibit proliferation, invasion and
migration and promote the apoptosis of HCC, and its mechanism of
action may be through the regulation of EMT. The present study
provides a scientific basis for further exploring the role of JPH3
in HCC and also provides novel ideas for the precise treatment of
HCC.
Supplementary Material
Supporting Data
Acknowledgements
Not applicable.
Funding
This study was supported by the National Natural Science
Foundation of China (grant no. 81960125).
Availability of data and materials
The datasets used and or/analyzed during the current
study are available from the corresponding author on reasonable
request.
Authors' contributions
YH and LZ conceived and designed the experiments.
ZY, MZ, XY and HH performed the experiments and analyzed the data.
YH and LZ drafted and revised the manuscript. All authors have read
and approved the final manuscript for publication. All authors read
and approved the manuscript and agree to be accountable for all
aspects of the research in ensuring that the accuracy or integrity
of any part of the work including the data are appropriately
investigated and resolved.
Ethics approval and consent to
participate
The study was approved by the Ethics Committee of
the Affiliated Hospital of Zunyi Medical University. Written
informed consent was obtained from all patients (ethical approval
no. 2011035).
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Couri T and Pillai A: Goals and targets
for personalized therapy for HCC. Hepatol Int. 13:125–137. 2019.
View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Yang JD, Hainaut P, Gores GJ, Amadou A,
Plymoth A and Roberts LR: A global view of hepatocellular
carcinoma: Trends, risk, prevention and management. Nat Rev
Gastroenterol Hepatol. 16:589–604. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Chen W, Zheng R, Baade PD, Zhang S, Zeng
H, Bray F, Jemal A, Yu XQ and He J: Cancer statistics in China,
2015. CA Cancer J Clin. 66:115–132. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Jiang J, Tang M, Huang Z and Chen L:
Junctophilins emerge as novel therapeutic targets. J Cell Physiol.
234:16933–16943. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Margolis RL and Rudnicki DD: Pathogenic
insights from Huntington's disease-like 2 and other Huntington's
disease genocopies. Curr Opin Neurol. 29:743–748. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Hu X, Kuang Y, Li L, Tang H, Shi Q, Shu X,
Zhang Y, Chan FK, Tao Q and He C: Epigenomic and functional
characterization of junctophilin 3 (JPH3) as a novel tumor
suppressor being frequently inactivated by promoter CpG methylation
in digestive cancers. Theranostics. 7:2150–2163. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Han TS, Ban HS, Hur K and Cho HS: The
epigenetic regulation of HCC metastasis. Int J Mol Sci.
19:39782018. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Dawson MA and Kouzarides T: Cancer
epigenetics: From mechanism to therapy. Cell. 150:12–27. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Nebbioso A, Tambaro FP, Dell'Aversana C
and Altucci L: Cancer epigenetics: Movin forward. PLoS Genet.
14:e10073622018. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Zhang L, Lu Q and Chang C: Epigenetics in
health and disease. Adv Exp Med Biol. 1253:3–55. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Zhang D, Zhao J, Han C, Liu X, Liu J and
Yang H: Identification of hub genes related to prognosis in glioma.
Biosci Rep. 40:BSR201933772020. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Chen KH, He J, Wang DL, Cao JJ, Li MC,
Zhao XM, Sheng X, Li WB and Liu WJ: Methylation-associated
inactivation of LATS1 and its effect on demethylation or
overexpression on YAP and cell biological function in human renal
cell carcinoma. Int J Oncol. 45:2511–2521. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Livak KJ and Schmittgen TD: Analysis of
relative gene expression data using real-time quantitative PCR and
the 2(−Delta Delta C(T)) method. Methods. 25:402–408. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Huang A, Yang XR, Chung WY, Dennison AR
and Zhou J: Targeted therapy for hepatocellular carcinoma. Signal
Transduct Target Ther. 5:1462020. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Jiang Y, Han QJ and Zhang J:
Hepatocellular carcinoma: Mechanisms of progression and
immunotherapy. World J Gastroenterol. 25:3151–3167. 2019.
View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Berger MF and Mardis ER: The emerging
clinical relevance of genomics in cancer medicine. Nat Rev Clin
Oncol. 15:353–365. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Zahn LM: Effects of the tumor
microenvironment. Science. 355:1386–1388. 2017. View Article : Google Scholar
|
|
18
|
Villanueva A: Hepatocellular carcinoma. N
Engl J Med. 380:1450–1462. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Xie DY, Ren ZG, Zhou J, Fan J and Gao Q:
2019 Chinese clinical guidelines for the management of
hepatocellular carcinoma: Updates and insights. Hepatobiliary Surg
Nutr. 9:452–463. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Ganne-Carrié N and Nahon P: Hepatocellular
carcinoma in the setting of alcohol-related liver disease. J
Hepatol. 70:284–293. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Skvortsova K, Stirzaker C and Taberlay P:
The DNA methylation landscape in cancer. Essays Biochem.
63:797–811. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Edwards JR, Yarychkivska O, Boulard M and
Bestor TH: DNA methylation and DNA methyltransferases. Epigenetics
Chromatin. 10:232017. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Lee J, Kim Y, Friso S and Choi SW:
Epigenetics in non-alcoholic fatty liver disease. Mol Aspects Med.
54:78–88. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Pan Y, Liu G, Zhou F, Su B and Li Y: DNA
methylation profiles in cancer diagnosis and therapeutics. Clin Exp
Med. 18:1–14. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Dor Y and Cedar H: Principles of DNA
methylation and their implications for biology and medicine.
Lancet. 392:777–786. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Toh TB, Lim JJ and Chow EK: Epigenetics of
hepatocellular carcinoma. Clin Transl Med. 8:132019. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Raggi C and Invernizzi P: Methylation and
liver cancer. Clin Res Hepatol Gastroenterol. 37:564–571. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Nakamura M, Chiba T, Kanayama K, Kanzaki
H, Saito T, Kusakabe Y and Kato N: Epigenetic dysregulation in
hepatocellular carcinoma: An up-to-date review. Hepatol Res.
49:3–13. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Maemura K, Yoshikawa H, Yokoyama K, Ueno
T, Kurose H, Uchiyama K and Otsuki Y: Delta-like 3 is silenced by
methylation and induces apoptosis in human hepatocellular
carcinoma. Int J Oncol. 42:817–822. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Acun T, Oztas E, Yagci T and Yakicier MC:
SIP1 is downregulated in hepatocellular carcinoma by promoter
hypermethylation. BMC Cancer. 11:2232011. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Pastushenko I and Blanpain C: EMT
transition states during tumor progression and metastasis. Trends
Cell Biol. 29:212–226. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Saitoh M: Involvement of partial EMT in
cancer progression. J Biochem. 164:257–264. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Aiello NM and Kang Y: Context-dependent
EMT programs in cancer metastasis. J Exp Med. 216:1016–1026. 2019.
View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Guo F, Wang H, Jiang M, Yang Q, Xiang Q,
Zhou H, Hu X, Hao K, Yang J, Cao H and Shen Z: TDP-43 induces EMT
and promotes hepatocellular carcinoma metastasis via activating
Wnt/β-catenin signaling pathway. Am J Cancer Res. 10:3285–3301.
2020.PubMed/NCBI
|
|
35
|
Lu J, Zhao Z and Ma Y: miR-186 represses
proliferation, migration, invasion, and EMT of hepatocellular
carcinoma via directly targeting CDK6. Oncol Res. 28:509–518. 2020.
View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Dian MJ, Li J, Zhang XL, Li ZJ, Zhou Y,
Zhou W, Zhong QL, Pang WQ, Lin XL, Liu T, et al: MST4 negatively
regulates the EMT, invasion and metastasis of HCC cells by
inactivating PI3K/AKT/Snail1 axis. J Cancer. 12:4463–4477. 2021.
View Article : Google Scholar : PubMed/NCBI
|