Introduction
Amyotrophic lateral sclerosis (ALS) is a
neurodegenerative disease of a yet undetermined etiology, and it is
estimated to be the most common type of motor neuron disease during
adulthood (1). A great variation
has been observed in terms of the frequency of ALS between
different countries, with males exhibiting a slightly elevated risk
of developing ALS compared with females, particularly as regards
the form of ALS with limb onset (2–4).
On the other hand, there is a slight female predominance in bulbar
onset ALS (2–4).
ALS can be phenotypically manifested with
symptomatology from the upper (e.g., spasticity, weakness,
hyperreflexia and pseudobulbar palsy) and lower (e.g. atrophy of
muscles, fasciculations, hyporeflexia and muscle cramps) motor
neurons (5). The consequent
progressive and irreversible muscular weakness and atrophy, leads
to the paralysis of respiratory muscles and respiratory failure
(6). Apart from motor deficits,
non-motor symptoms [e.g., frontotemporal dementia (FTD)] can also
develop with ALS (7). The
association between ALS and FTD is evident through the crosstalk
that exists between them clinically, genetically and
neuropathologically (8).
From a neuropathological aspect, ALS presents as a
degeneration of motor neurons, while intraneuronal inclusions in
glial cells and neurons can also be observed (9). While the etiology and the exact
mechanisms that possible lead to the development of ALS remain to
be determined, a notable amount of research suggests that complex
mechanisms, that interact with each other, are possibly implicated
in the development of ALS (9,10).
Mitochondrial dysfunction, inflammatory abnormalities, pathological
protein aggregation, defective microtubule function, inadequate RNA
activity, oxidative stress and glutamate excitotoxicity, to mention
a few, are among the pathological processes that have been found to
confer susceptibility to ALS (11,12).
The molecular pathways through which the
aforementioned mechanisms can lead to ALS are far from being
completely elucidated (13).
However, there are a number of indications that genetic,
environmental and epigenetic factors all contribute to ALS
susceptibility (14). Among the
exogenous factors, head trauma, smoking, viral infections, exposure
to pesticides and heavy metals, antioxidants, physical exercise,
exposure to electromagnetic fields and body mass index have been
previously reported as possible triggers for the development of ALS
(15,16).
The contribution of genetic factors to ALS is
supported from robust data (17).
Firstly, there is familial ALS (fALS; accounting for almost 5-10%
of all ALS cases), where mutations >30 genes are considered as
ALS causative ones (10,18). Namely, mutations in the chromosome
9 open reading frame 72 (C9orf72), superoxide dismutase type 1
(SOD1), TAR DNA-binding protein 43 (TDP-43) and fused in
sarcoma/translocated in liposarcoma (FUS/TLS) genes are estimated
to be the commonest ones (10,18). Mutations in these genes can lead
to ALS; however, there are some genotype-phenotype associations,
where patients with ALS carrying specific mutations may exhibit
specific sub-phenotypes (19).
For example, C9orf72 can lead to ALS, behavioral variant FTD, or
ALS-FTD, while mutations in the TDP43 gene can lead to ALS with
either bulbar or limb onset (19). Moreover, the remaining 85-90% of
all ALS cases are considered as sporadic ALS (sALS) (5), where a large amount of genetic loci
have been reported to modify the risk of developing ALS (20). At this point, it is worth
mentioning that the conferred risk from rare genetic variants
suggests an oligogenic disease pattern concerning the ALS
architecture (21,22).
The Sec1 family domain containing 1 (SCFD1) protein,
belongs to the Senc1/Munk 18 (SM) family of proteins (23). These proteins are
vesicle-trafficking proteins, working closely with a particular
type of SNARE proteins, the syntaxins (24,25). SCFD1 is implicated in a number of
functions, such as cellular membrane fusion, oxidative-stress,
intra-Golgi-retrograde transport and apoptosis (23–26).
In 2016, a genome-wide association study (GWAS)
identified the SCFD1 rs10139154 variant at 14q12 as a genetic locus
associated with ALS in the discovery phase; however, statistical
significance was not found in the replication phase (27). Moreover, recently, Chen et
al (28) examined the effect
of the SCFD1 rs10139154 on a large Chinese population with ALS.
Based on the results of that study, the SCFD1 rs10139154
polymorphism was shown to be associated with the age at onset (AAO)
of patients with ALS.
Taking into consideration that SCFD1 is involved in
mechanisms (23–26) that are also possibly implicated in
ALS pathogenesis (e.g., oxidative stress and apoptosis) (11,12), and that the studies examining the
role of the SCFD1 rs10139154 variant regarding ALS have yielded
inconsistent results (27,28),
the present case-control study was conducted with the aim of
determining any possible association between the SCFD1 rs10139154
variant and ALS. A Southeastern European Caucasian (SEC) cohort
(Greek) was analyzed, primary aiming at detecting any association
between SCFD1 rs10139154 and ALS. The present study also wished to
examine the effect of this polymorphism on several other ALS
endophenotypes.
Patients and methods
Study participants
In the current protocol, a total of 310 participants
were drafted. In greater detail, 155 patients with sporadic ALS, as
well as 155 healthy controls were included. The participants were
recruited between March, 2017 and September, 2017 from the
University Hospital of Larissa (Department of Neurology), in
Larissa, Greece. Consultant neurologists made the diagnoses of ALS,
following the El Escorial criteria (29). The inclusion criteria for the
participants with ALS were as follows: i) An age >18 years; ii)
Greek ethnicity; and iii) a diagnosis of ALS based on the El
Escorial criteria. The respective inclusion criteria for healthy
controls were the following: i) An age >18 years; ii) of Greek
ethnic origin; and iii) no referred family or personal history of
ALS or other neurological disorders. The characteristics of the ALS
cohort have been previously described (30–33). The research study protocol of the
present study was approved by the Local Ethics Committee
(University Hospital of Larissa: 59295/23-01-2017) and it was
deemed in accordance to the Declaration of Helsinki. The
experimental protocol was explained and all the participants
provided their written informed consent in order to be included in
the study, with the freedom of withdrawing from the study at any
given time.
Molecular genetics
The salting out method was used to isolate genomic
DNA from leucocytes (blood was collected with peripheral
venipuncture from each participant), a method which has also been
previously described (34,35).
All the samples derived from the healthy volunteers and the
patients with ALS were genotyped for the SCFD1 rs10139154 variant.
The method which was applied for genotyping was the TaqMan allele
specific discrimination assay on an ABI PRISM 7900 Sequence
Detection System. The results were then analyzed using SDS software
(SDS 2.4). (Applied Biosystems; Thermo Fisher Scientific, Inc.). In
brief, this method consisted of an initial enzyme activation
occurring as the first step of PCR at a 50°C incubation for 2 min,
followed by enzyme activation at 95°C for 10 min (one cycle),
denaturation at 95°C for 15 sec and annealing/extension at 60°C for
1 min (40 cycles for the last two points). The personnel who
performed the genotyping was blinded to status of the samples.
Quality assessment
In order for the genotyping reproducibility be
evaluated, a random 10% of the sample was re-genotyped, with 100%
concordance. Moreover, the threshold for the genotype call rate
(percentage of successfully genotyped samples) was set as ≥95%.
Additionally, the deviation or not from the Hardy-Weinberg
equilibrium (HWE) was calculated with the Chi-squared test
(36), in both the patients with
ALS and the healthy controls.
Statistical analysis
The CaTS Power Calculator for Genetic Studies
(Center for Statistical Genetics, University of Michigan, Ann
Arbor, MI, USA) was used for the statistical power of the current
sample to be measured. With odds ratios (ORs) along with the
respective 95% confidence intervals (CIs), the study sample was
examined for associations between SCFD1 rs10139154 and ALS using
SNPStats software (https://www.snpstats.net/) (36). Towards this, five genetic models
were assumed: i) The co-dominant model, where a P-value with 2
degrees of freedom and 2 ORs were calculated [OR1: (mt/wt vs.
wt/wt) and OR2: (mt/mt vs. wt/wt)]; ii) the dominant model (mt/mt +
mt/wt vs. wt/wt); iii) the recessive model (mt/mt vs. mt/wt +
wt/wt); iv) the overdominant model (mt/wt vs. mt/mt + wt/wt) and v)
the log-additive model, where mt/mt carriers have a double risk of
disease compared with mt/wt, with wt/wt as a reference. In the
analyses, the ‘C’ was defined as the wild-type allele (wt) and the
‘T’ as the mutant allele (mt), for the SCFD1 rs10139154 variant. A
P-value <0.05 was considered to indicate a statistically
significant difference. Both univariate (unadjusted) and
multivariate (adjusted for age and sex) analyses were
performed.
The primary outcome of the protocol of the present
study was to examine the possible association between ALS and SCFD1
rs10139154 (ALS vs. healthy controls). Unadjusted and adjusted for
age and sex analyses were performed. In addition, the effects of
SCFD1 rs10139154 on the following parameters were examined: i) The
AAO of ALS (unadjusted and adjusted for sex); ii) the site of ALS
onset (patients with bulbar onset ALS vs. healthy controls; and
patients with limb onset ALS vs. healthy controls; unadjusted and
adjusted for age and sex); and iii) the AAO of ALS with subgroup
analyses on the basis of the site of onset (bulbar and limb, crude
and adjusted for sex). The analysis of all the outcomes was
performed on the basis of the aforementioned genetic models.
Results
A total of 310 individuals were recruited in the
present study; 155 patients with definite ALS [77 (49.7%) female,
age (mean ± standard deviation), 63.74±11.30 years], and 155
healthy volunteers (control group) matched for age and sex.
Approximately 7 out of 10 (67.1%) patients with ALS confirmed a
history of alcohol consumption, while 68.4% had a history of
smoking. The most common site of ALS onset was the lower limbs
(34.8%) and the bulbar type (32.3%). The characteristics of the
patients with ALS have been formerly reported (30–32).
The genotype call rate was 98.06% (304/310).
Furthermore, no deviation from the HWE was observed in both
patients with ALS and the healthy controls (P=0.37 and P=0.23,
respectively) (Table I). The
analysis for the power of the sample size of the study was 80.4 to
detect a significant association (P<0.05) between ALS and SCFD1
rs10139154, with the frequency of the minor T allele set at 37%, an
ALS prevalence of 5/100,000, and an approximate relative risk of
1.59 for the multiplicative mode of inheritance.
 | Table I.Results from the Chi-squared exact
test for HWE for the SCFD1 rs10139154 in healthy controls and in
ALS cases. |
Table I.
Results from the Chi-squared exact
test for HWE for the SCFD1 rs10139154 in healthy controls and in
ALS cases.
|
| P-value |
|---|
|
|
|
|---|
| SNP | Healthy
controls | ALS |
|---|
| rs10139154 | 0.23 | 0.37 |
The frequency of the minor allele (T) was 35 and 38%
for the patients with ALS and healthy controls respectively. The
genotypic frequencies C/C, C/T and T/T for the patients with ALS,
were 44, 42 and 14%, respectively, while in the group of healthy
volunteers, the corresponding frequencies were 41, 42 and 17%,
respectively. The allelic and genotypic frequencies for SCFD1
rs10139154, for both patients with ALS and the healthy controls are
presented in Table II. The
allele and genotype numbers for SCFD1 rs10139154 in the healthy
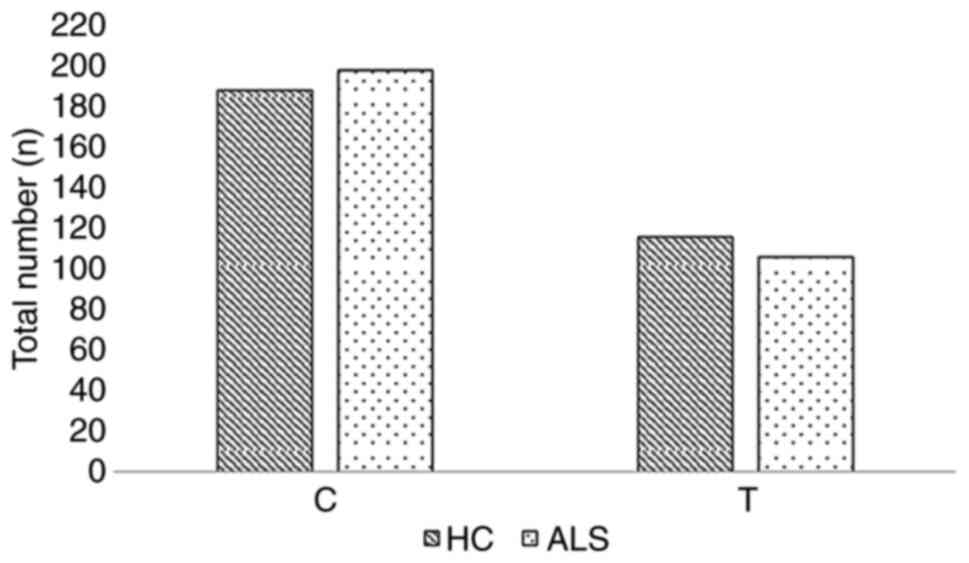
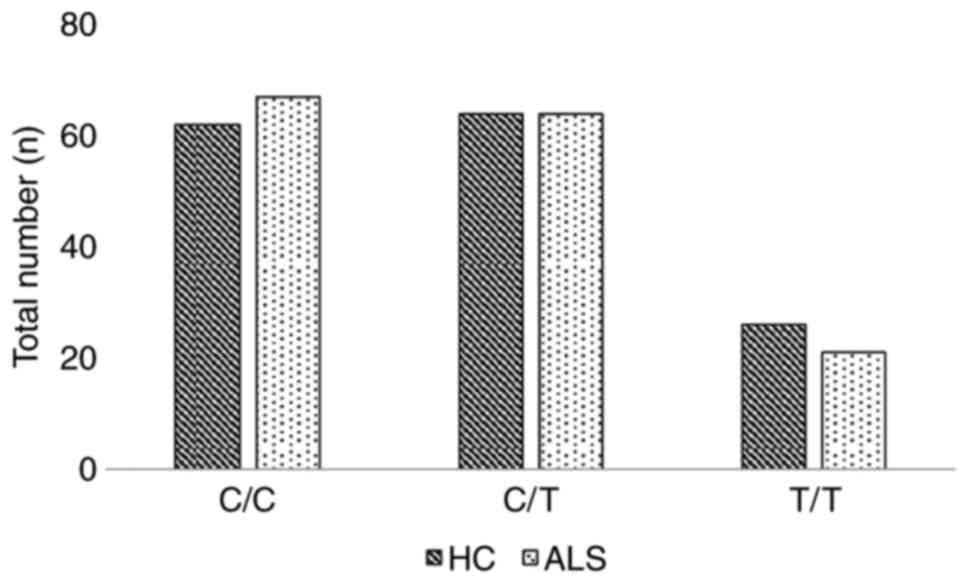
controls and ALS cases are graphically presented in Figs. 1 and 2, respectively.
| Table II.Allelic and genotype frequencies for
SCFD1 rs10139154 in the healthy controls, ALS cases and the whole
sample size. |
Table II.
Allelic and genotype frequencies for
SCFD1 rs10139154 in the healthy controls, ALS cases and the whole
sample size.
| rs10139154 SNP |
Genotypes/alleles | Healthy controls
(n=155), n (%) | ALS (n=155), n
(%) | Whole sample
(n=310), n (%) |
|---|
| Genotype | C/C | 62 (41) | 67 (44) | 129 (42) |
|
| C/T | 64 (42) | 64 (42) | 128 (42) |
|
| T/T | 26 (17) | 21 (14) | 47 (15) |
|
| Missing | 3 | 3 | 6 |
| Allele | C | 188 (62) | 198 (65) | 386 (63) |
|
| T | 116 (38) | 106 (35) | 222 (37) |
Based on the univariate (unadjusted) and
multivariate (adjusted for age and sex) analyses, no significant
association was found between the SCFD1 rs10139154 polymorphism and
ALS in any genetic model examined (Table III). Subgroup analysis
(unadjusted and adjusted for age and sex) according to the site of
ALS onset (limb, bulbar) also did not reveal any significant
association (Table IV).
| Table III.Single locus analysis (crude and
adjusted for age and sex) for association between SCFD1 rs10139154
and ALS, in co-dominant, dominant, recessive, overdominant and
log-additive mode. |
Table III.
Single locus analysis (crude and
adjusted for age and sex) for association between SCFD1 rs10139154
and ALS, in co-dominant, dominant, recessive, overdominant and
log-additive mode.
|
|
| Univariate | Multivariate |
|---|
|
|
|
|
|
|---|
| Mode | Genotype | OR (95% CI) | P-value | OR (95% CI) | P-value |
|---|
| Co-dominant | C/C | 1.00 | 0.7 | 1.00 | 0.44 |
|
| C/T | 0.93
(0.57-1.51) |
| 0.77
(0.38-1.54) |
|
|
| T/T | 0.75
(0.38-1.46) |
| 0.57
(0.23-1.39) |
|
| Dominant | C/C | 1.00 | 0.56 | 1.00 | 0.28 |
|
| C/T-T/T | 0.87
(0.55-1.38) |
| 0.70
(0.37-1.33) |
|
| Recessive | C/C-C/T | 1.00 | 0.43 | 1.00 | 0.3 |
|
| T/T | 0.78
(0.42-1.45) |
| 0.64
(0.28-1.48) |
|
| Overdominant | C/C-T/T | 1.00 | 0.99 | 1.00 | 0.78 |
|
| C/T | 1.00
(0.63-1.58) |
| 0.91
(0.48-1.73) |
|
| Log-additive | - | 0.88
(0.64-1.21) | 0.42 | 0.76
(0.49-1.16) | 0.2 |
| Table IV.Single locus analysis (crude and
adjusted for age and sex) for association between SCFD1 rs10139154
and ALS, based on the site of disease onset (bulbar or limbs), in
co-dominant, dominant, recessive, overdominant and log-additive
modes. |
Table IV.
Single locus analysis (crude and
adjusted for age and sex) for association between SCFD1 rs10139154
and ALS, based on the site of disease onset (bulbar or limbs), in
co-dominant, dominant, recessive, overdominant and log-additive
modes.
|
|
| Bulbar onset | Limb onset |
|---|
|
|
|
|
|
|---|
|
|
| Univariate | Multivariate | Univariate | Multivariate |
|---|
| Mode | Genotype | OR (95% CI) | P-value | OR (95% CI) | P-value | OR (95% CI) | P-value | OR (95% CI) | P-value |
| Co-dominant | C/C | 1.00 | 0.39 | 1.00 | 0.16 | 1.00 | 0.35 | 1.00 | 0.35 |
|
| C/T | 1.37
(0.61-3.11) |
| 1.34
(0.40-4.46) |
| 0.63
(0.32-1.21) |
| 0.54
(0.23-1.26) |
|
|
| T/T | 0.60
(0.16-2.29) |
| 0.28
(0.04-1.72) |
| 0.92
(0.41-2.07) |
| 0.74
(0.27-2.05) |
|
| Dominant | C/C | 1.00 | 0.73 | 1.00 | 0.91 | 1.00 | 0.26 | 1.00 | 0.19 |
|
| C/T-T/T | 1.15
(0.52-2.52) |
| 0.94
(0.30-2.92) |
| 0.71
(0.39-1.28) |
| 0.60
(0.28-1.29) |
|
| Recessive | C/C-C/T | 1.00 | 0.25 | 1.00 | 0.065 | 1.00 | 0.74 | 1.00 | 0.98 |
|
| T/T | 0.50
(0.14-1.77) |
| 0.23
(0.04-1.23) |
| 1.14
(0.53-2.43) |
| 0.99
(0.39-2.52) |
|
| Overdominant | C/C-T/T | 1.00 | 0.26 | 1.00 | 0.21 | 1.00 | 0.15 | 1.00 | 0.18 |
|
| C/T | 1.56
(0.72-3.35) |
| 1.99
(0.67-5.90) |
| 0.64
(0.34-1.19) |
| 0.59
(0.27-1.30) |
|
| Log-additive | - | 0.91
(0.53-1.58) | 0.75 | 0.66
(0.29-1.46) | 0.29 | 0.89
(0.59-1.33) | 0.56 | 0.80
(0.49-1.32) | 0.38 |
Furthermore, the analyses based on the AAO of ALS
(unadjusted and adjusted for sex), failed to produce any
statistically significant differences. The analyses for the AAO of
ALS with regards to the site of onset (limb, bulbar), also revealed
non-statistically significant results. The respective results (ORs,
CIs, P-values, mean and standard error of the age for each
genotype) are presented in Table
V, for the ALS group overall. The results from the subgroup
analysis regarding the AAO, based on the site of the ALS onset are
presented in Table VI for the
bulbar onset and in Table VII
for the limb onset.
| Table V.Single locus analysis (crude and
adjusted for sex) for association between SCFD1 rs10139154 and AAO
of ALS, in co-dominant, dominant, recessive, overdominant and
log-additive mode. |
Table V.
Single locus analysis (crude and
adjusted for sex) for association between SCFD1 rs10139154 and AAO
of ALS, in co-dominant, dominant, recessive, overdominant and
log-additive mode.
| Mode | Genotype | Mean (SE) | Difference (95%
CI) | Univariate
P-value | Multivariate
P-value |
|---|
| Co-dominant | C/C | 63.22 (1.5) | 0.00 | 0.7 | 0.68 |
|
| C/T | 64.75 (1.32) | 1.53 (−2.38 to
5.43) |
|
|
|
| T/T | 63.05 (2.44) | −0.18 (−5.77 to
5.42) |
|
|
| Dominant | C/C | 63.22 (1.5) | 0.00 | 0.55 | 0.58 |
|
| C/T-T/T | 64.33 (1.16) | 1.11 (−2.54 to
4.75) |
|
|
| Recessive | C/C-C/T | 63.97 (1.00) | 0.00 | 0.73 | 0.67 |
|
| T/T | 63.05 (2.44) | −0.92 (−6.17 to
4.33) |
|
|
| Overdominant | C/C-T/T | 63.18 (1.27) | 0.00 | 0.4 | 0.39 |
|
| C/T | 64.75 (1.32) | 1.57 (−2.09 to
5.23) |
|
|
| Log-additive | — | — | 0.33 (−2.26 to
2.93) | 0.8 | 0.85 |
| Table VI.Single locus analysis (crude and
adjusted for sex) for association between SCFD1 rs10139154 and the
AAO of ALS with bulbar onset, in co-dominant, dominant, recessive,
overdominant and log-additive mode. |
Table VI.
Single locus analysis (crude and
adjusted for sex) for association between SCFD1 rs10139154 and the
AAO of ALS with bulbar onset, in co-dominant, dominant, recessive,
overdominant and log-additive mode.
| Mode | Genotype | Mean (SE) | Difference (95%
CI) | Univariate
P-value | Multivariate
P-value |
|---|
| Co-dominant | C/C | 67.83 (2.28) | 0.00 | 0.46 | 0.51 |
|
| C/T | 63.12 (2.81) | −4.72 (−12.18 to
2.75) |
|
|
|
| T/T | 66.33 (4.67) | −1.50 (−14.29 to
11.29) |
|
|
| Dominant | C/C | 67.83 (2.28) | 0.00 | 0.25 | 0.28 |
|
| C/T-T/T | 63.6 (2.46) | −4.23 (−11.38 to
2.91) |
|
|
| Recessive | C/C-C/T | 65.07 (1.92) | 0.00 | 0.84 | 0.89 |
|
| T/T | 66.33 (4.67) | 1.26 (−10.86 to
13.38) |
|
|
| Overdominant | C/C-T/T | 67.53 (1.98) | 0.00 | 0.22 | 0.25 |
|
| C/T | 63.12 (2.81) | −4.42 (−11.32 to
2.49) |
|
|
| Log-additive | — | — | −2.27 (−7.88 to
3.33) | 0.43 | 0.44 |
| Table VII.Single locus analysis (crude and
adjusted for sex) for association between SCFD1 rs10139154 and the
AAO of ALS with limb onset, in co-dominant, dominant, recessive,
overdominant and log-additive mode. |
Table VII.
Single locus analysis (crude and
adjusted for sex) for association between SCFD1 rs10139154 and the
AAO of ALS with limb onset, in co-dominant, dominant, recessive,
overdominant and log-additive mode.
| Mode | Genotype | Mean (SE) | Difference (95%
CI) | Univariate
P-value | Multivariate
P-value |
|---|
| Co-dominant | C/C | 61.16 (2.5) | 0.00 | 0.92 | 0.93 |
|
| C/T | 62.6 (2.39) | 1.44 (−5.72 to
8.60) |
|
|
|
| T/T | 61.17 (3.61) | 0.01 (−8.48 to
8.49) |
|
|
| Dominant | C/C | 61.16 (2.5) | 0.00 | 0.78 | 0.8 |
|
| C/T-T/T | 62.06 (1.98) | 0.90 (−5.34 to
7.14) |
|
|
| Recessive | C/C-C/T | 61.73 (1.77) | 0.00 | 0.89 | 0.89 |
|
| T/T | 61.17 (3.61) | −0.56 (−8.51 to
7.39) |
|
|
| Overdominant | C/C-T/T | 61.16 (2.05) | 0.00 | 0.68 | 0.7 |
|
| C/T | 62.6 (2.39) | 1.44 (−5.26 to
8.14) |
|
|
| Log-additive | - | - | 0.24 (−3.82 to
4.29) | 0.91 | 0.92 |
Discussion
The present case-control study genotyped an ALS
cohort with SEC ancestry aiming to examine the role of the SCFD1
rs10139154 polymorphism in ALS. Based on the results obtained, the
SCFD1 rs10139154 polymorphism was not associated with ALS. More
precisely, the SCFD1 rs10139154 polymorphism neither conferred any
susceptibility to ALS nor influenced the AAO of ALS or its initial
manifestation. Therefore, based on this analysis, it appears rather
unlikely that the SCFD1 rs10139154 polymorphism is among the risk
factors for the development of ALS.
As aforementioned, the SCFD1 is a
vesicle-trafficking protein, functioning alongside with syntaxins
(24,25). The possible mechanisms via which
SCFD1 may be related to ALS pathophysiology are cellular membrane
fusion, oxidative-stress, intra-Golgi-retrograde transport and
apoptosis (23–26). The SCFD1 rs10139154 polymorphism
is an intronic variant located at chromosome 14: 30678292
(https://www.ensembl.org/index.html).
In a 2016 GWAS on patients with ALS, the SCFD1 rs10139154
polymorphism was initially reported as a possibly significant
genetic marker (27). More
precisely, in the discovery phase, a statistically significant
effect size (OR, 1.09) was reported; whereas the effect size
remained at the replication phase (OR, 1.06) statistical
significance was not achieved.
Additionally, Chen et al (28) reported that the SCFD1 rs10139154
polymorphism was associated with the AAO of patients with ALS of
Chinese ancestry. More precisely, they found that carriers of the
C/C genotype had a lower AAO of ALS (49.53±10.75 years) compared
with the C/T carriers (53.75±11.84 years; P=0.002). Furthermore, in
the recessive model analysis, C/C carriers had a lower ALS AAO
(49.53±10.75 years) compared with (C/T + T/T; 53.37±11.60 years,
P=0.001) (28). Apart from ALS,
the SCFD1 rs10139154 ‘T’ allele has been shown to be associated
with a decreased risk of Alzheimer's disease (OR, 0.63; P=0.036),
in the recessive mode (24).
However, the aforementioned results could not be replicated in the
present study.
Of note, the minor allele frequency (MAF) at the
study of Chen et al (28)
was the ‘C’ allele, while this was found to be the ‘T’ allele in
the present study, suggesting that ancestry may be important for
the discrepancy between the studies. Based on 1,000 Genomes Project
Phase 3 allele frequencies, there is a great variability at the MAF
allele frequency between populations (https://www.ensembl.org/Homo_sapiens/Variation/Population?db=core;r=14:30677792-30678792;v=rs10139154;vdb=variation;vf=181642009).
In fact, while the ‘C’ allele appears to be the MAF at East Asian
and African population, the ‘T’ allele is considered the MAF at
Europeans, South Asians and Americans.
Apart from genetic variability between ethnicities,
there are other possible explanations for the fact that no
association was found between SCFD1 rs10139154 and ALS. For
instance, the effect of a polymorphism at expression quantitative
trait loci (eQTL); in fact there is evidence to suggest that
variants identified in GWASs may possibly alter the risk of disease
through gene regulation via eQTL (37). Concerning the rs10139154
polymorphism, it has emerged as the most significant SCFD1
polymorphism associated with ALS in a GWAS (27). In addition, for each SCFD1
rs10139154 ‘T’ allele, an increased SCFD1 expression has been found
in different tissue types obtained from genotype-tissue expression
and in post-mortem control data, as demonstrated in the study by
Iacoangeli et al (38).
However, no alteration in SCFD1 expression with the addition of the
SCFD1 rs10139154 ‘T’ alleles was found at the post-mortem ALS
cohort of that study (38). These
results suggested that between the post-mortem ALS cohort and
controls, different correlations existed between the SCFD1 genotype
and SCFD1 eQTLs expression, which may influence the risk of the
disease (38).
Currently, the available treatments for ALS are very
limited, while patients with ALS usually succumb to the disease
within 3-4 years from the time of diagnosis (39). Therefore, ongoing trials target
precision medicine approaches, and genetic targets that will
possibly influence the natural course of ALS (40). In this sense, research regarding
ALS (with genetic studies included) may reveal the molecular
mechanisms behind neurodegeneration, and may lead to the
development of novel therapeutic agents (41).
Certain limitations of the present study need to be
mentioned. Firstly, the participants with ALS were not screened for
the commonest fALS genes, namely the C9orf72, SOD1, TDP-43 and
FUS/TLS genes (10,18). The authors also acknowledge that
another constraint is that in the current statistical models,
several potential cofounders (genetic and non-genetic) were not
included (15,16). Finally, the present study had a
case-control design and thus included all the inherent limitations
of such a type of study (42). On
the contrary, it should be noted that a major strength of the
present case-control study is the homogeneity of the entire cohort,
as the data of both patients with ALS and the control group were
collected from a specific geographical area of central Greece.
In conclusion, the present study found that the
SCFD1 rs10139154 polymorphism was not associated with ALS. However,
whether this genetic locus is among the risk factors of ALS cannot
be established with absolute certitude at the moment. Bearing in
mind the latter considerations, further large-scale cooperative
studies examining the carriage of the SCFD1 rs10139154 polymorphism
in multiethnic cohorts are of great necessity, in order for the
attribute risk of this variant to ALS to be fully elucidated.
Acknowledgements
Not applicable.
Funding
The present study was supported in part by a research grant from
the Research Committee of the University of Thessaly, Greece (code:
5287).
Availability of data and materials
The datasets used and/or analyzed during the current
study are available from the corresponding author on reasonable
request.
Authors' contributions
VS and ED were involved in the conceptualization of
the study. VS, AMA, IL, CB, GN, KP and MS were involved in the
study methodology and validation, in data curation and software. VS
was involved in the formal analysis. VS, AMA, DPB, DAS, AT, PDM and
ED were involved in the investigative aspects and design of the
study. VS was involved in the writing and preparation of the
original draft. VS, AMA, IL, CB, GN, KP, MS, DPB, DAS, AT, PDM and
ED were involved in the writing, reviewing and editing of the
manuscript. ED supervised the study and was involved in project
administration and funding acquisition. All authors have read and
approved the final manuscript. VS and ED confirm the authenticity
of all the raw data.
Ethics approval and consent to
participate
The research study protocol of the present study was
approved by the Local Ethics Committee (University Hospital of
Larissa: 59295/23-01-2017) and it was deemed in accordance to the
Declaration of Helsinki. The experimental protocol was explained
and all the participants provided their written informed consent in
order to be included in the study, with the freedom of withdrawing
from the study at any given time.
Patient consent for publication
Not applicable.
Competing interests
DAS is the Editor-in-Chief for the journal, but had
no personal involvement in the reviewing process, or any influence
in terms of adjudicating on the final decision, for this article.
The other authors declare that they have no competing
interests.
References
|
1
|
Kojima Y, Kasai T, Noto YI, Ohmichi T,
Tatebe H, Kitaoji T, Tsuji Y, Kitani-Morii F, Shinomoto M, Allsop
D, et al: Amyotrophic lateral sclerosis: Correlations between fluid
biomarkers of NfL, TDP-43, and tau, and clinical characteristics.
PLoS One. 16:e02603232021. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Manjaly ZR, Scott KM, Abhinav K,
Wijesekera L, Ganesalingam J, Goldstein LH, Janssen A, Dougherty A,
Willey E, Stanton BR, et al: The sex ratio in amyotrophic lateral
sclerosis: A population based study. Amyotroph Lateral Scler.
11:439–442. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Marin B, Boumédiene F, Logroscino G,
Couratier P, Babron MC, Leutenegger AL, Copetti M, Preux PM and
Beghi E: Variation in worldwide incidence of amyotrophic lateral
sclerosis: A meta-analysis. Int J Epidemiol. 46:57–74.
2017.PubMed/NCBI
|
|
4
|
Mitsumoto H, Przedborski S and Gordon PH:
Amyotrophic Lateral Sclerosis. CRC Press; pp. p8722005, PubMed/NCBI
|
|
5
|
Masrori P and Van Damme P: Amyotrophic
lateral sclerosis: A clinical review. Eur J Neurol. 27:1918–1929.
2020. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Niedermeyer S, Murn M and Choi PJ:
Respiratory failure in amyotrophic lateral sclerosis. Chest.
155:401–408. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Abramzon YA, Fratta P, Traynor BJ and Chia
R: The overlapping genetics of amyotrophic lateral sclerosis and
frontotemporal dementia. Front Neurosci. 14:422020. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Ahmed RM, Devenney EM, Strikwerda-Brown C,
Hodges JR, Piguet O and Kiernan MC: Phenotypic variability in
ALS-FTD and effect on survival. Neurology. 94:e2005–e2013. 2020.
View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Saberi S, Stauffer JE, Schulte DJ and
Ravits J: Neuropathology of amyotrophic lateral sclerosis and its
variants. Neurol Clin. 33:855–876. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Mejzini R, Flynn LL, Pitout IL, Fletcher
S, Wilton SD and Akkari PA: ALS genetics, mechanisms, and
therapeutics: Where are we now? Front Neurosci. 13:13102019.
View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Dardiotis E, Aloizou AM, Siokas V,
Patrinos GP, Deretzi G, Mitsias P, Aschner M and Tsatsakis A: The
role of MicroRNAs in patients with amyotrophic lateral sclerosis. J
Mol Neurosci. 66:617–628. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Dardiotis E, Siokas V, Sokratous M,
Tsouris Z, Aloizou AM, Florou D, Dastamani M, Mentis AA and Brotis
AG: Body mass index and survival from amyotrophic lateral
sclerosis: A meta-analysis. Neurol Clin Pract. 8:437–444. 2018.
View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Hardiman O, Al-Chalabi A, Chio A, Corr EM,
Logroscino G, Robberecht W, Shaw PJ, Simmons Z and van den Berg LH:
Amyotrophic lateral sclerosis. Nat Rev Dis Primers. 3:170712017.
View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Siokas V, Aloizou AM and Pateraki G:
Chapter 21 - Toxicology of neurodegenerative diseases.
Toxicological Risk Assessment and Multi-System Health Impacts from
Exposure. Tsatsakis AM: Academic Press; pp. 247–258. 2021,
View Article : Google Scholar
|
|
15
|
Dardiotis E, Siokas V, Sokratous M,
Tsouris Z, Michalopoulou A, Andravizou A, Dastamani M, Ralli S,
Vinceti M, Tsatsakis A and Hadjigeorgiou GM: Genetic polymorphisms
in amyotrophic lateral sclerosis: Evidence for implication in
detoxification pathways of environmental toxicants. Environ Int.
116:122–135. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Tsatsakis AM: Toxicological Risk
Assessment and Multi-System Health Impacts from Exposure. Elsevier;
2021, View Article : Google Scholar
|
|
17
|
Mathis S, Goizet C, Soulages A, Vallat JM
and Masson GL: Genetics of amyotrophic lateral sclerosis: A review.
J Neurol Sci. 399:217–226. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Oskarsson B, Gendron TF and Staff NP:
Amyotrophic lateral sclerosis: An update for 2018. Mayo Clin Proc.
93:1617–1628. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Li HF and Wu ZY: Genotype-phenotype
correlations of amyotrophic lateral sclerosis. Transl Neurodegener.
5:32016. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Brown RH and Al-Chalabi A: Amyotrophic
lateral sclerosis. N Engl J Med. 377:162–172. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Ghasemi M and Brown RH Jr: Genetics of
amyotrophic lateral sclerosis. Cold Spring Harb Perspect Med.
8:a0241252018. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
van Es MA, Hardiman O, Chio A, Al-Chalabi
A, Pasterkamp RJ, Veldink JH and van den Berg LH: Amyotrophic
lateral sclerosis. Lancet. 390:2084–2098. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Hou N, Yang Y, Scott IC and Lou X: The Sec
domain protein Scfd1 facilitates trafficking of ECM components
during chondrogenesis. Dev Biol. 421:8–15. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Stamati P, Siokas V, Aloizou AM,
Karampinis E, Arseniou S, Rakitskii VN, Tsatsakis A, Spandidos DA,
Gozes I, Mitsias PD, et al: Does SCFD1 rs10139154 polymorphism
decrease Alzheimer's disease risk? J Mol Neurosci. 69:343–350.
2019. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Bando Y, Katayama T, Taniguchi M,
Ishibashi T, Matsuo N, Ogawa S and Tohyama M: RA410/Sly1 suppresses
MPP+ and 6-hydroxydopamine-induced cell death in SH-SY5Y cells.
Neurobiol Dis. 18:143–151. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Carr CM and Rizo J: At the junction of
SNARE and SM protein function. Curr Opin Cell Biol. 22:488–495.
2010. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
van Rheenen W, Shatunov A, Dekker AM,
McLaughlin RL, Diekstra FP, Pulit SL, van der Spek RA, Võsa U, de
Jong S, Robinson MR, et al: Genome-wide association analyses
identify new risk variants and the genetic architecture of
amyotrophic lateral sclerosis. Nat Genet. 48:1043–1048. 2016.
View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Chen Y, Zhou Q, Gu X, Wei Q, Cao B, Liu H,
Hou Y and Shang H: An association study between SCFD1 rs10139154
variant and amyotrophic lateral sclerosis in a Chinese cohort.
Amyotroph Lateral Scler Frontotemporal Degener. 19:413–418. 2018.
View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Ludolph A, Drory V, Hardiman O, Nakano I,
Ravits J, Robberecht W and Shefner J; WFN Research Group On
ALS/MND, : A revision of the El Escorial criteria-2015. Amyotroph
Lateral Scler Frontotemporal Degener. 16:291–292. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Dardiotis E, Karampinis E, Siokas V,
Aloizou AM, Rikos D, Ralli S, Papadimitriou D, Bogdanos DP and
Hadjigeorgiou GM: ERCC6L2 rs591486 polymorphism and risk for
amyotrophic lateral sclerosis in Greek population. Neurol Sci.
40:1237–1244. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Siokas V, Aloizou AM, Liampas I, Tsouris
Z, Mentis AA, Nasios G, Papadimitriou D, Bogdanos DP, Hadjigeorgiou
GM and Dardiotis E: Lack of association between TREM2 rs75932628
variant and amyotrophic lateral sclerosis. Mol Biol Rep.
48:2601–2610. 2021. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Siokas V, Karampinis E, Aloizou AM, Mentis
AA, Liakos P, Papadimitriou D, Liampas I, Nasios G, Bogdanos DP,
Hadjigeorgiou GM and Dardiotis E: CYP1A2 rs762551 polymorphism and
risk for amyotrophic lateral sclerosis. Neurol Sci. 42:175–182.
2021. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Liampas I, Siokas V, Aloizou AM, Bakirtzis
C, Tsouris Z, Nousia A, Nasios G, Papadimitriou D, Liakos P,
Bogdanos DP, et al: MOBP rs616147 polymorphism and risk of
amyotrophic lateral sclerosis in a greek population: A case-control
study. Medicina (Kaunas). 57:13372021. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Siokas V, Kardaras D, Aloizou AM,
Asproudis I, Boboridis KG, Papageorgiou E, Hadjigeorgiou GM,
Tsironi EE and Dardiotis E: BDNF rs6265 (Val66Met) polymorphism as
a risk factor for blepharospasm. Neuromolecular Med. 21:68–74.
2019. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Siokas V, Aslanidou P, Aloizou AM,
Peristeri E, Stamati P, Liampas I, Arseniou S, Drakoulis N, Aschner
M, Tsatsakis A, et al: Does the CD33 rs3865444 polymorphism confer
susceptibility to Alzheimer's disease? J Mol Neurosci. 70:851–860.
2020. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Solé X, Guinó E, Valls J, Iniesta R and
Moreno V: SNPStats: A web tool for the analysis of association
studies. Bioinformatics. 22:1928–1929. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
GTEx Consortium: The genotype-tissue
expression (GTEx) project. Nat Genet. 45:580–585. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Iacoangeli A, Fogh I, Selvackadunco S,
Topp SD, Shatunov A, van Rheenen W, Al-Khleifat A, Opie-Martin S,
Ratti A, Calvo A, et al: SCFD1 expression quantitative trait loci
in amyotrophic lateral sclerosis are differentially expressed.
Brain Commun. 3:fcab2362021. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Keon M, Musrie B, Dinger M, Brennan SE,
Santos J and Saksena NK: Destination amyotrophic lateral sclerosis.
Front Neurol. 12:5960062021. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Kiernan MC, Vucic S, Talbot K, McDermott
CJ, Hardiman O, Shefner JM, Al-Chalabi A, Huynh W, Cudkowicz M,
Talman P, et al: Improving clinical trial outcomes in amyotrophic
lateral sclerosis. Nat Rev Neurol. 17:104–118. 2021. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Amado DA and Davidson BL: Gene therapy for
ALS: A review. Mol Ther. 29:3345–3358. 2021. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Yuan W, Beaulieu-Jones BK, Yu KH, Lipnick
SL, Palmer N, Loscalzo J, Cai T and Kohane IS: Temporal bias in
case-control design: Preventing reliable predictions of the future.
Nat Commun. 12:11072021. View Article : Google Scholar : PubMed/NCBI
|