Introduction
Ulcerative colitis (UC) is a chronic nonspecific
inflammatory bowel disease (1),
the typical clinical symptoms of which are abdominal pain and
diarrhea, which may contain mucus or blood (2). During the early stages, UC begins in
the rectum, and in the subsequent stages it can extend to the
proximal end of the colon or even the whole colon in a continuous
manner, becoming a lifelong recurrent disease with a high risk of
developing into colorectal cancer (3,4).
Worldwide, the incidence of UC is increasing annually (5). Therefore, UC is considered one of
the main diseases threatening human life and health, and the study
of its potential pathogenesis is of great importance for the
development of therapeutic drugs with new targets.
It has previously been reported that various genes
and proteins are involved in the regulation of the progression of
UC. For example, the UC-related gene RNF186 has been shown to
maintain intestinal homeostasis by controlling endoplasmic
reticulum stress in colonic epithelial cells (6). In addition, epithelial IL-18
equilibrium can control barrier function in colitis (7), and anti-TNF-α therapy could
effectively treat UC (8).
Therefore, the discovery of different molecular targeted therapies
may provide a basis for the development of personalized treatment
strategies to improve the treatment of patients with UC. As a
subfamily of the RING type E3 ubiquitin ligases, the tripartite
motif-containing (TRIM)22 protein is emerging as a key regulator in
the development of various diseases by modulating transcriptional
activity of the NF-κB pathway (9,10).
Previous studies have reported that knockdown of TRIM22 could
reduce cerebral ischemia/reperfusion-induced inflammation and
apoptosis by inhibiting the NF-κB/NLRP3 axis (11), and could regulate macrophage
autophagy via the NF-κB/Beclin 1 pathway (10). Notably, it has been demonstrated
that functional variation of TRIM22 is related to inflammatory
bowel disease (12) and may serve
a carcinogenic role in colon cancer (13). It should be noted that TRIM30 is
the murine ortholog of TRIM22 (14). Therefore, the present study
hypothesized that TRIM22 or TRIM30 may have a certain regulatory
role in UC. The present study focused on the role and potential
mechanism of TRIM30 and TRIM22 in DSS-induced UC mouse and HT-29
cell models, respectively.
Bioinformatics analysis has been widely used to
explore the molecular mechanisms of various diseases (15,16), and may contribute to the
identification of novel biomarkers to improve diagnostic and
prognostic strategies for UC. KLF2 has been reported to be
downregulated in UC, as determined by clinical information
analysis, and may be closely related to inflammatory factors
(17). KLF2 has also been shown
to serve a regulatory role in acute lung injury as a transcription
suppressor of HSPH1 (18).
Therefore, it was hypothesized that TRIM22 might be inhibited by
KLF2 transcription and may have a role in promoting disease
development in UC. The present study investigated the effects of
TRIM22 or TRIM30 on DSS-induced UC in vivo and in
vitro, as well as the underlying mechanisms.
Materials and methods
Cell culture and establishment of an
in vitro model of UC
The human colorectal cancer cell line HT-29, which
was verified by STR profiling, was obtained from Wuhan Procell Life
Science & Technology Co., Ltd. The cells were cultured in DMEM
(Gibco; Thermo Fisher Scientific, Inc.) supplemented with 10% fetal
bovine serum (Gibco; Thermo Fisher Scientific, Inc.). 100 U/ml
penicillin and 100 µg/ml streptomycin (Gibco; Thermo Fisher
Scientific, Inc.) at 37°C in 5% CO2. Subsequently, once
HT-29 cells reached the logarithmic phase, they were inoculated
into sterile 6-well plates at a density of 1×105
cells/well and DSS (MilliporeSigma) was dissolved in sterile water.
When cell confluence reached ~80%, cells were treated with 2% DSS
or sterile water for 24 h at 37°C to establish an in vitro
cell model of UC.
Bioinformatics analysis
The National Center for Biotechnology Information
Gene Expression Omnibus (https://www.ncbi.nlm.nih.gov/geo/) is a free public
database of microarray/gene profiles, which was used to obtain the
GSE59071 (19) and GSE107597
(20) in UC and normal tissue
gene expression profiles (21).
GEO2R (ncbi.nlm.nih.gov/geo/geo2r/) was used for data preprocessing
and analyzing the expression of TRIM22 in the UC (n=6) and control
(n=6) groups (GSE59071: 97 UC and 11 control samples; GSE107597: 6
UC samples and 4 control samples). Furthermore, the UCSC database
(http://genome.ucsc.edu/) and JASPAR database
(http://jaspar.genereg.net/) were used to
predict transcriptional binding sites of KLF2 to the TRIM22
promoter.
Cell transfection
Cells (1×105 cells/well) were seeded into
6-well plates and cultured for 24 h at 37°C with 5% CO2.
The pGPH1 vector carrying short hairpin (sh)RNA plasmids
(sh-TRIM30-1; 5′-CAGTATAGAAGTTACAATA-3′ and sh-TRIM30-2;
5′-GGTGAATATCTGTGCACAA-3′), a sh-TRIM22 plasmid
(5′-GGAAGATGACATCAGACAA-3′), a sh-KLF2-1 plasmid
(5′-GCACCGACGACGACCTCAA-3′) and sh-KLF2-2 plasmid
(5′-GAAGCGCGGCCGCCGCTCTTG-3′), a negative control (NC) shRNA
plasmid (sh-NC), pCDNA3.1 vector targeting KLF2 (Oe-KLF2) and an
empty NC vector (Oe-NC) were all purchased from Shanghai GenePharma
Co., Ltd. and were dissolved in sterile phosphate-buffered saline
(PBS; Beyotime Institute of Biotechnology). Briefly, 70 µl of this
solution was gently mixed with 30 µl Lipofectamine® 3000
(Invitrogen; Thermo Fisher Scientific, Inc.) and incubated at room
temperature for 20 min to form the complex, according to the
manufacturer's protocol. Subsequently, HT-29 cells were seeded in
sterile 6-well plates (1×106 cells/well) at 37°C and
when cell confluence reached ~60%, 200 nM of the complex was
transfected into cells. Following transfection for 24 h, DSS was
added and cells were incubated for 24 h as aforementioned. Each
experiment was performed in triplicate.
TUNEL staining assay
Apoptosis was detected using a TUNEL kit
(MilliporeSigma) at 37°C for 60 min. Cells were washed with PBS and
fixed with 40 µl 4% paraformaldehyde for 15 min at room
temperature. The cells were then permeabilized in 0.3% Triton
X-100/PBS for 10 min, followed by two washes with PBS. After
washing, 3% H2O2 was added to inhibit
endogenous peroxide and 3,3-diaminobenzidine was added to induce
color generation at room temperature for 5 min, according to the
manufacturer's instructions. 0.5 µg/ml DAPI (Beyotime) was adopted
to stain the nuclear for 5 min at room temperature. Antifade
Mounting Medium was used to seal the cells. Images of the cells
randomly selected from 5 fields of view were captured under an
Olympus BX51 fluorescence microscope (Olympus Corporation;
magnification, ×200).
Animal model
A total of 20 C57BL/6 mice (male; age, 6 weeks;
weight, 20–22 g) were purchased from the Medical Comparative Center
of Yangzhou University (Yangzhou, China) and were maintained under
the following conditions: 55±15% humidity; 26±2°C; 12-h light/dark
cycle; with ad libitum access to food and water. The animal
experiments performed in the present study were approved by the
Ethics Committee of Suzhou Hospital Affiliated to Nanjing Medical
University (Suzhou, China). All operations and experimental
procedures complied with the Animal Management Regulations of the
Ministry of Health (22).
sh-TRIM30 or sh-NC was dissolved in sterile PBS and
mixed with Lipofectamine 3000 to form the complex as
aforementioned. Mice were randomly assigned into the following four
groups (n=5 mice/group): Group I (control), mice were given normal
drinking water; Group II [dextran sulphate sodium (DSS)], mice were
administered drinking water containing 2% of DSS for 1 week; Group
III (DSS + sh-NC), mice were administered 200 µl sh-NC mixture by
tail vein injection twice per week for 1 week and were then given
drinking water containing 2% DSS for 1 week; Group IV (DSS +
sh-TRIM30), mice were administered 200 µl sh-TRIM30 mixture by tail
vein injection twice per week for 1 week and were then given
drinking water containing 2% DSS for 1 week. The body weights of
the mice were recorded daily and disease activity index (DAI) was
determined by an investigator blinded to the protocol by scoring
the extent of body weight loss, stool hemoccult positivity or gross
bleeding, and stool consistency in accordance with the method
described by Kihara et al (23). DAI was calculated as follows:
DAI=(weight loss score + stool characters score + bleeding score)/3
(24). Subsequently, mice were
administered 150 mg/kg sodium pentobarbital intraperitoneally to
halt their breathing and cardiac arrest was confirmed. The colon
was then dissected, photographed, measured for length. Then, colon
tissue was embedded and then cut into 4 µm thick slices, fixed in
4% paraformaldehyde for 15 min at room temperature, before being
stained with hematoxylin for 10 min and eosin for 2 min at room
temperature. The tissue was observed under an Olympus BX40 light
microscope (Olympus Corporation).
Cell Counting Kit-8 (CCK-8) assay
The CCK-8 assay was carried out to evaluate cell
viability. Briefly, the transfected cells were inoculated into
96-well plates at a density of 2×103 cells/well, and
when cell confluence reached 70–80%, 2% DSS or a moderate amount of
sterile water was added and incubated at 37°C for 24 h.
Subsequently, 10 µl CCK-8 solution (Phygene Scientific) was added
to each well and the cells were cultured for another 2 h at 37°C.
The absorbance in each well was measured at a wavelength of 450 nm
using a microplate reader (BioTek Instruments, Inc.).
Reverse transcription-quantitative PCR
(RT-qPCR) analysis
Total RNA (~860 ng/µl) was extracted using
TRIzol® reagent (Invitrogen; Thermo Fisher Scientific,
Inc.) with a purity of A260/A280 ~1.8, according to the
manufacturer's instructions. cDNA was synthesized using the
QuantiTect Reverse Transcription kit (Qiagen GmbH) according to the
kit's operating procedures. Subsequently, RT-qPCR was performed on
an ABI 7500 Real-Time PCR instrument (Applied Biosystems; Thermo
Fisher Scientific, Inc.) using the SYBR Green PCR kit (Takara Bio,
Inc.), according to the manufacturer's protocol. The reaction
system was as follows: 95°C for 10 min; followed by 40 cycles at
95°C for 10 sec and 60°C for 60 sec and final extension at 72°C for
60 sec. The following primers (GenScript) were used for qPCR:
TRIM22, forward 5′-CCGCCTGGAAGATCGAGAG-3′ and reverse
5′-CTGCCAGGTTATCCAGCACA-3′; TRIM30, forward
5′-CTCCCAAGAGGACCAGCAC-3′ and reverse 5′-ACTCCTCACACTGCAGAGCA-3′;
KLF2, forward 5′-ACTCACACCTGCAGCTACGC-3′ and reverse
5′-AGTGGTAGGGCTTCTCACCTGT-3′; GAPDH (applicable species: human),
forward 5′-GTAGAGCGGCCGCCATGT-3′ and reverse
5′-GCCCAATACGACCAAATCAGAGAA-3′; and GAPDH (applicable species:
mouse), forward 5′-ACCCTTAAGAGGGATGCTGC-3′ and reverse
5′-CCCAATACGGCCAAATCCGT-3′. mRNA expression levels were quantified
using the 2−ΔΔCq method (25) and normalized to the internal
reference gene GAPDH. Each sample was tested in triplicate and
average values were obtained.
Western blot analysis
Cells in each group were collected and RIPA lysis
buffer (cat. no. P0013C; Beyotime Institute of Biotechnology) was
used to extract total protein. The protein concentration was
determined using a BCA protein assay kit (cat. no. P0012S; Beyotime
Institute of Biotechnology). Protein samples (20 µg) were separated
by 10% SDS-PAGE and then transferred to PVDF membranes soaked in
methanol and blocked with 5% bovine serum albumin (Shanghai
Rebiosci Biotech Co., Ltd) at room temperature for 30 min. The
membranes were then incubated with primary antibodies against
TRIM22 (cat. no. ab68071; 1:1,000 dilution; Abcam), TRIM30 (cat.
no. ab76972; 1:1,000 dilution; Abcam), NF-κB (cat. no. ab32536;
1:1,000 dilution; Abcam), IκBα (cat. no. 9242S; 1:1,000 dilution;
Cell Signaling Technology, Inc.), phosphorylated (p)-NF-κB (cat.
no. ab239882; 1:1,000 dilution; Abcam), p-IκBα (cat. no. 2859S;
1:1,000 dilution; Cell Signaling Technology, Inc.), KLF2 (cat. no.
ab236507; 1:2,000 dilution; Abcam) and GAPDH (cat. no. ab181602;
1:10,000 dilution; Abcam) at 4°C overnight. Subsequently, membranes
were incubated with horseradish peroxidase-conjugated secondary
antibodies (cat. no. 7074, 1:5,000; cat. no. 7076, 1:3,000; both
Cell Signaling Technology, Inc.) at room temperature for 2 h.
Following the addition of ECL solution (Shanghai Yeasen
Biotechnology Co., Ltd.), the protein bands were visualized with a
gel imager (C150; Azure Biosystems, Inc.). The gray value of the
protein bands was analyzed using ImageJ (v1.51; National Institutes
of Health) and the relative protein expression levels were
calculated.
Dual-luciferase reporter assay
HT-29 cells were inoculated in a 24-well plate with
1.0×105 cells/well. Once the cell confluence reached
80%, luciferase activity was detected according to the instructions
of a dual-luciferase reporter gene assay kit (cat. no. ab228530;
Abcam). In brief, the pGL3-KLF2 or pGL3 vector (Promega
Corporation) was co-transfected into HT-29 cells together with a
luciferase reporter plasmid containing TRIM22 promoter, or mutated
TRIM22 promoter using Lipofectamine® 3000 (Invitrogen;
Thermo Fisher Scientific, Inc.) in 24-well plates for 24 h, the
culture supernatant was discarded, the cells were collected and
gently rinsed three times with PBS. Subsequently, 120 µl cell lysis
buffer was added to each well of 24-well plates and the plates were
agitated on a horizontal oscillator for 45 min at 110 rpm. Fully
lysed cell mixture (10 µl) was added to a 1.5 ml microplate well
and 50 µl firefly luciferase reagent was added and mixed. Within 10
min, Stop/Glo sealin luciferase reagent (50 µl) was then added,
quickly mixed and luciferase activity was measured. Firefly
Luc/Renilla Luc values were recorded and the TRIM22 promoter
transfer activity was analyzed and normalized to Renilla
luciferase activity. Each group was set up with three multiple
wells, and each experiment was repeated three times.
Enzyme-linked immunosorbent assays
(ELISAs)
The levels of TNF-α (cat. no. PT518), IL-6 (cat. no.
P1330) and IFN-γ (cat. no. P1511) in HT-29 cells were quantified
using ELISA kits (Beyotime Institute of Biotechnology) according to
the manufacturer's instructions.
Chromatin immunoprecipitation (ChIP)
assay
Total genomic DNA was isolated and sonicated using
the EZChip™ Kit (cat. no. 17-371, MilliporeSigma) according to the
manufacturer's protocol. Cell (4×106 cells/well) were
lysed in 400 µl SDS Lysis Buffer (Beyotime Institute of
Biotechnology) and then 800 µl cells lysate was sonicated on ice
(4.5 sec impact, 9 sec per interval, 14 times in total) and the
fragmented DNA was visualized on an agarose gel. Anti-THAP11 (1 µg)
(cat. no. sc-517366, Santa Cruz Biotechnology, Inc.) or IgG
secondary antibodies (Anti-mouse IgG (AS003), ABclonal Technology
Co., Ltd.) were added to interact with the target protein-DNA
complex. For chromatin isolation, the sample was centrifuged again
at 15,000 × g for 10 min at 4°C to remove insoluble material and
10X ChIP dilution buffer (cat. no. abs50034-22T, Absin
Biotechnology Co., Ltd.) was added to the collected supernatant.
The sample was pre-cleared with protein G-agarose beads at 4°C for
1 h with mixing. The samples were centrifuged at 700 × g for 1 min
at 4°C. After boiling, the immunological chromatin samples were
amplified by PCR as aforementioned.
Statistical analysis
Data were plotted with GraphPad Prism 8.0 software
(GraphPad Software, Inc.). Data are presented as the mean ±
standard deviation from ≥3 independent experiments. Unpaired
Student's t-test was used for comparisons between two groups, and
one-way ANOVA followed by Tukey's post hoc test was used for
comparisons among multiple groups. The non-parametric
Kruskal-Wallis test was used to analyze the DAI, followed by Dunn's
multiple comparison test. P<0.05 was considered to indicate a
statistically significant difference.
Results
TRIM30 is upregulated in DSS-induced
UC in mice
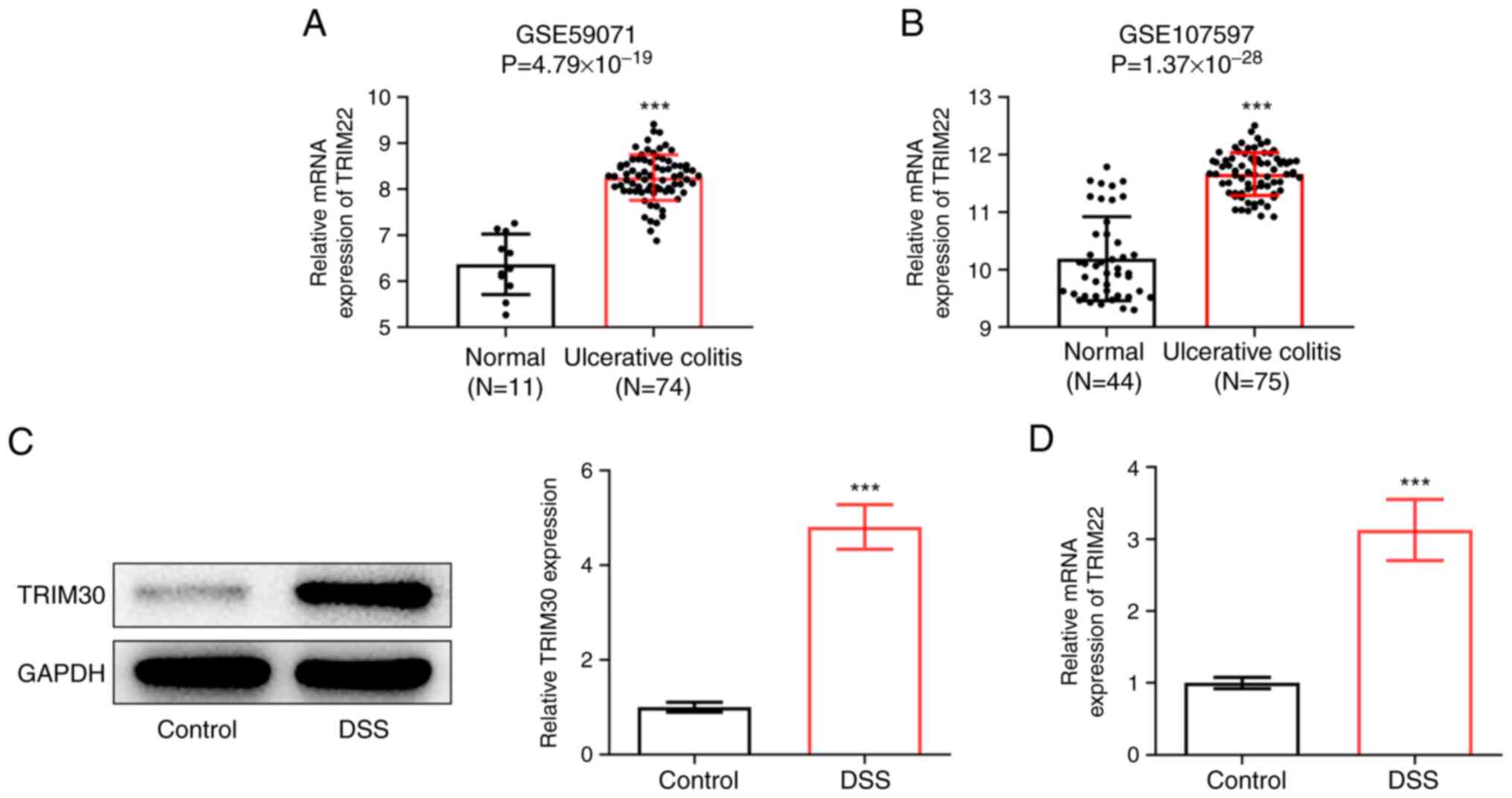
Firstly, GEO2R analysis was performed on the
UC-related datasets GSE59071 (11 control samples, 74 UC samples)
and GSE107597 (44 control samples, 75 UC samples); the results
revealed that TRIM22 was upregulated in the intestinal tissues of
patients with UC (Fig. 1A and B).
Subsequently, a mouse model of UC was constructed by induction with
2% DSS, and the expression levels of TRIM30 in DSS-treated mice
were detected by western blotting and RT-qPCR. The results showed
that the expression levels of TRIM30 were significantly increased
in DSS-treated mice compared with those in mice given normal
drinking water (Fig. 1C and
D).
Knockdown of TRIM30 alleviates
intestinal injury in DSS-induced UC in mice
To further explore the role of TRIM30 in UC,
sh-TRIM30 was constructed and the knockdown efficiency was verified
by RT-qPCR. Compared with sh-TRIM30-1, the knockdown efficiency of
the sh-TRIM30-2 plasmid was higher (Fig. 2A); therefore, sh-TRIM30-2 was
selected for the subsequent experiments. After 1 week of 2% DSS
treatment, the body weight of the DSS group was significantly lower
than that of the mice given normal drinking water (Fig. 2B), DAI scores were higher than
those of the control group (Fig.
2C) and mice in the DSS group had a shortened colon length with
tissue damage (Fig. 2D and E).
Notably, knockdown of TRIM30 effectively alleviated the
aforementioned inflammatory damage, as evidenced by increased body
weight, decreased DAI score, longer colon length and reduced colon
tissue damage.
Knockdown of TRIM30 inhibits NF-κB
signaling and alleviates inflammation of DSS-induced UC in
mice
To verify whether TRIM30 served a pro-inflammatory
role through the NF-κB signaling pathway, the expression levels of
NF-κB-related proteins were detected by western blotting. Compared
with mice in the normal drinking water group, the protein
expression levels of p-NF-κB and p-IκBα were significantly
increased in DSS-treated mice, which was prevented by knockdown of
TRIM30 (Fig. 3A). In addition,
the levels of inflammation-related factors (TNF-α, IL-6 and IFN-γ)
were examined by ELISA. As shown in Fig. 3B-D, DSS induced an increase in
TNF-α, IL-6 and IFN-γ levels in mice, by contrast, the levels of
inflammation-related factors were significantly decreased following
TRIM30 knockdown, effectively alleviating the inflammatory response
in mice.
TRIM22 is inhibited by KLF2 in
DSS-induced UC in HT-29 cells
Firstly, the expression levels of TRIM22 and KLF2
were detected in DSS-treated HT-29 cells by western blotting and
RT-qPCR. Compared with in the control group, the mRNA and protein
expression levels of TRIM22 were significantly upregulated in
DSS-induced HT-29 cells, whereas the expression levels of KLF2 were
significantly downregulated (Fig. 4A
and B). Subsequently, an overexpression vector and a knockdown
vector of KLF2 were constructed, and transfection efficiency was
detected by RT-qPCR. The results revealed that following
transfection with oe-KLF2 and sh-KLF2-1/2, the expression levels of
KLF2 were significantly increased or decreased, respectively
(Fig. 4C). sh-KLF2-2 had a better
transfection effect than sh-KLF2-1, and was selected for the next
experiments. Subsequently, the expression levels of TRIM22 were
detected and it was revealed that after knockdown of KLF2, the
expression levels of TRIM22 were significantly promoted. By
contrast, following overexpression of KLF2, the expression levels
of TRIM22 were significantly inhibited (Fig. 4D), indicating that TRIM22 was
possibly inhibited by KLF2 transcription. The present study
therefore aimed to further explore the relationship between TRIM22
and KLF2. The results obtained from UCSC and JASPAR databases
implicated that KLF2 could bind to the TRIM22 promoter sequence
(Fig. 4E and F). Luciferase
activity assay results demonstrated that overexpression of KLF2
significantly inhibited the luciferase activity of the
TRIM22-wild-type reporter plasmid, but had no effect on the
luciferase activity of the TRIM22-mutant reporter plasmid (Fig. 4G). Furthermore, the CHIP assay
demonstrated that KLF2 could bind the TRIM22 promoter (Fig. 4H). Therefore, these results
indicated that KLF2 could bind to the TRIM22 promoter to inhibit
the expression of TRIM22.
 | Figure 4.TRIM22 is inhibited by KLF2
transcription in a HT-29 cell model of DSS-induced ulcerative
colitis. (A) Western blot analysis and (B) RT-qPCR were used to
detect the expression levels of TRIM22 and KLF2 in DSS-treated
HT-29 cells. mRNA expression levels of (C) KLF2 or (D) TRIM22 in
DSS-treated HT-29 cells transfected with sh-KLF2 or Oe-KLF2, as
determined by RT-qPCR. (E) UCSC and (F) JASPAR databases were used
to predict the binding site of KLF2 to the TRIM22 promoter. (G)
Relative luciferase activity of TRIM22 promoter was detected by
luciferase reporter gene assay. (H) Chromatin immunoprecipitation
was further used to detect the binding ability of KLF2 to TRIM22.
*P<0.05, **P<0.01 and ***P<0.001 vs. Control;
##P<0.01 and ###P<0.001 vs. Oe-NC. DSS,
dextran sulphate sodium; KLF2, Kruppel-like factor 2; Mut, mutant;
NC, negative control; Oe, overexpression; RT-qPCR, reverse
transcription-quantitative PCR; sh, short hairpin; TRIM, tripartite
motif-containing; WT, wild-type. |
Knockdown of KLF2 reverses the effects
of sh-TRIM22 on viability, NF-κB signaling and inflammation in
DSS-treated HT-29 cells
The present study evaluated the effect of the
relationship between KLF2 and TRIM22 on UC. RT-qPCR assay
demonstrated that the expression levels of TRIM22 were
significantly decreased in HT-29 cells post-transfection with
sh-TRIM22 (Fig. 5A).
Subsequently, TUNEL staining and CCK-8 were used to assess the
apoptosis and viability of cells. The results revealed that TRIM22
knockdown could significantly inhibit DSS-induced apoptosis and
viability; however, this effect was reversed by further
interference with KLF2 (Fig.
5B-D). As previously reported, sh-TRIM22 markedly inhibited the
expression levels of NF-κB pathway-related proteins and
inflammation-related factors (Fig. 3A
and B). Notably, the present study interfered with KLF2 in
vitro and demonstrated that sh-KLF2 reversed the inhibitory
effect of sh-TRIM22 on the levels of NF-κB signaling pathway
proteins (Fig. 6A) and
inflammatory factors (Fig. 6B-D)
in DSS-treated HT-29 cells.
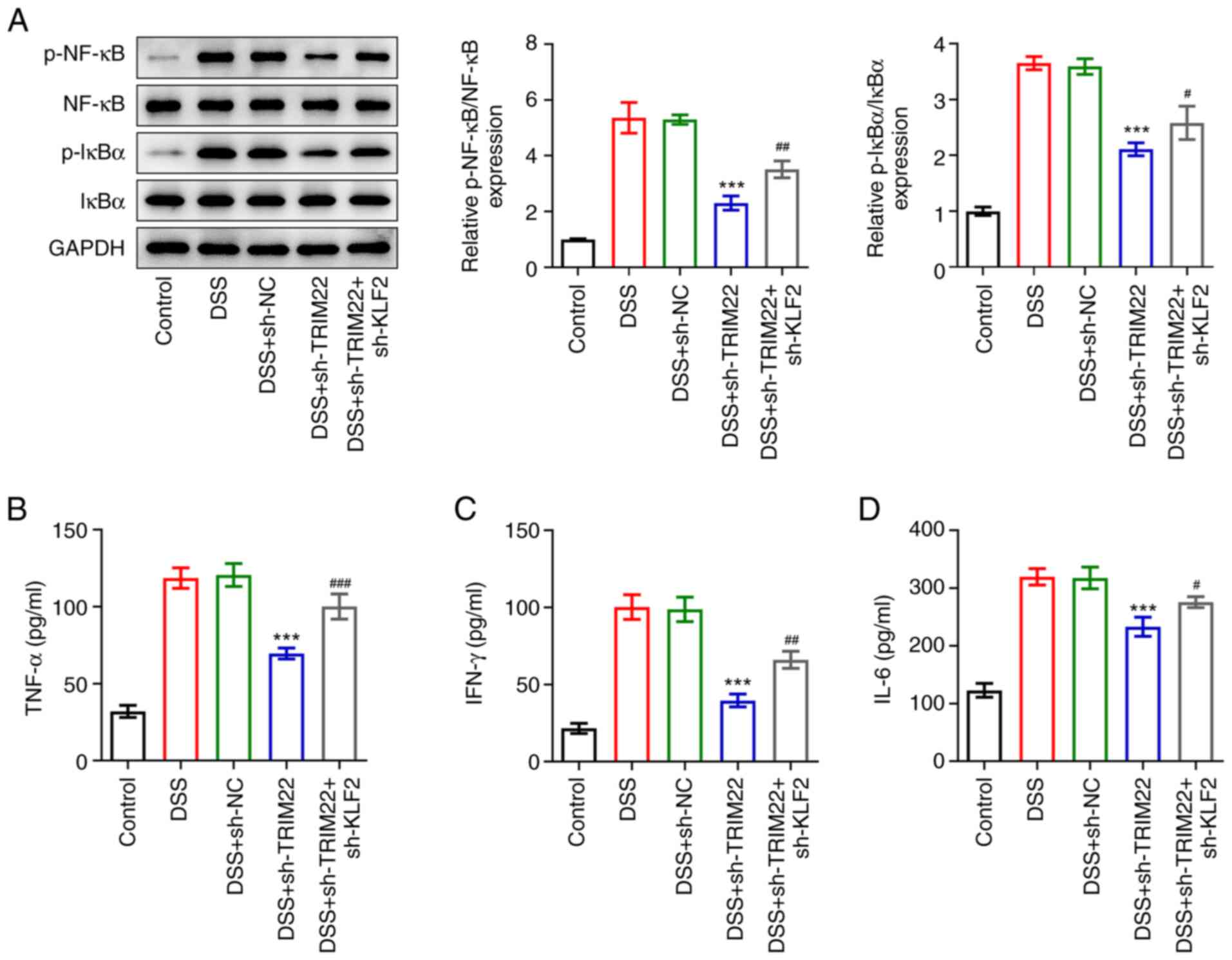 | Figure 6.Knockdown of KLF2 reverses the
inhibitory effects of sh-TRIM22 on NF-κB signaling and inflammation
in DSS-treated HT-29 cells. (A) Expression levels of NF-κB
pathway-related proteins (p-NF-κB NF-κB, IκBα and p-IκBα) were
detected by western blotting. ELISA was used to detect the levels
of (B) TNF-α, (C) IL-6, and (D) IFN-γ. ***P<0.001 vs. DSS +
sh-NC; #P<0.05, ##P<0.01 and
###P<0.001 vs. DSS + sh-TRIM22. DSS, dextran sulphate
sodium; KLF2, Kruppel-like factor 2; NC, negative control; p,
phosphorylated; sh, short hairpin; TRIM, tripartite
motif-containing. |
Discussion
UC is a chronic non-specific inflammatory bowel
disease, which mainly occurs in the intestinal mucosa and submucosa
(1). UC is characterized by
long-term disease, recurrence and the risk of cancer, which
seriously affects the quality of life of patients (3,26).
In recent years, evidence has suggested that variations in TRIM22
expression may be related to inflammatory bowel disease (27). Overexpression of TRIM22 has been
shown to inhibit the growth of monocytes, whereas downregulation of
TRIM22 could protect neurons against oxygen-glucose
deprivation/re-oxygenation-induced apoptosis and inflammation
(11). The present study revealed
that TRIM30 was upregulated in an in vivo model of UC and
knockdown of TRIM30 alleviated DSS-induced intestinal injury in
mice with UC.
The NF-κB pathway has long been considered a typical
pro-inflammatory signaling pathway (28). It has been reported that TRIM22
may be used as a potential activator of the NF-κB signaling pathway
(29). Notably, TRIM22 has been
shown to activate the NF-κB signaling pathway by increasing
degradation of the NF-κB inhibitor IκBα (29). Kang et al (11) revealed that knocking down TRIM22
alleviated inflammation and apoptosis induced by cerebral
ischemia-reperfusion by inhibiting the NF-κB/NLRP3 axis. Therefore,
the present study examined the expression levels of NF-κB
pathway-associated proteins and inflammatory factors in a model of
UC. The results indicated that knockdown of TRIM30 significantly
inhibited NF-κB signaling and alleviated DSS-induced intestinal
inflammation in mice with UC, which was consistent with the
findings of Kang et al (11), suggesting that TRIM30 may induce
inflammation through activation of the NF-κB pathway.
The present study also assessed the mechanism
underlying the effects of TRIM22 on UC and it was revealed that
KLF2, as the upstream transcription factor of TRIM22, could
effectively bind to the TRIM22 promoter sequence according to
bioinformatics analysis. This finding was confirmed by
dual-luciferase reporter assay and ChIP. Since TRIM22 induced
inflammation by activating NF-κB, given the association of KLF2
with TRIM22 promoter binding, it is hypothesized that KLF2 also
regulates inflammation, which is consistent with previous studies
on the regulatory role of KLF2 in various inflammatory diseases
(17,30). According to the results of a
previous study, KLF2 could serve a regulatory role in acute lung
injury as a transcription inhibitor of HSPH1 (18). In addition, simvastatin has been
reported to upregulate the expression of KLF2 in vascular
endothelial cells susceptible to atherosclerosis and relieve
vascular inflammation (31).
These results indicated that KLF2 and TRIM22 may have opposite
regulatory roles in UC; therefore, the cells were co-transfected
with sh-TRIM22 and sh-KLF2. Notably, KLF2 knockdown inhibited cell
viability, promoted apoptosis, and enhanced NF-κB signaling and
inflammation in DSS-treated HT-29 cells transfected with sh-TRIM22.
These results indicated that TRIM22 may have a role in promoting
disease progression by activating the NF-κB signaling pathway in UC
but could be inhibited by its upstream transcription factor
KLF2.
In conclusion, DSS significantly increased the
expression levels of TRIM22 in HT-29 cells and TRIM30 in mice,
whereas KLF2 knockdown reversed the inhibitory effect of TRIM22 on
viability and the enhancing effects of TRIM22 on inflammation in
DSS-treated HT-29 cells. These results suggested that TRIM22 and
KLF2 may be potential targets for the treatment of UC, and also
provided information that may improve the understanding of the
pathogenesis of UC.
Acknowledgements
Not applicable.
Funding
The present study was supported by the Project and the Suzhou
Science and Technology Development Plan Project (grant no.
2018.55).
Availability of data and materials
The datasets used and/or analyzed during the current
study are available from the corresponding author on reasonable
request.
Authors' contributions
BY and ZL conceptualized and designed the current
study. BY acquired, analyzed and interpreted the data. ZL drafted
the manuscript and revised it critically for important intellectual
content. Both authors agreed to be held accountable for the current
study in ensuring questions related to the integrity of any part of
the work are appropriately investigated and resolved. BY and ZL
confirm the authenticity of all the raw data. Both authors read and
approved the final manuscript.
Ethics approval and consent to
participate
Animal experiments in the present study were
approved by the Ethics Committee of Suzhou Hospital affiliated to
Nanjing Medical University.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Adams SM and Bornemann PH: Ulcerative
colitis. Am Fam Physician. 87:699–705. 2013.PubMed/NCBI
|
|
2
|
Keshteli AH, Madsen KL and Dieleman LA:
Diet in the pathogenesis and management of ulcerative colitis; a
review of randomized controlled dietary interventions. Nutrients.
11:14982019. View Article : Google Scholar
|
|
3
|
Du L and Ha C: Epidemiology and
pathogenesis of ulcerative colitis. Gastroenterol Clin North Am.
49:643–654. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Kaenkumchorn T and Wahbeh G: Ulcerative
colitis: Making the diagnosis. Gastroenterol Clin North Am.
49:655–669. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
da Silva BC, Lyra AC, Rocha R and Santana
GO: Epidemiology, demographic characteristics and prognostic
predictors of ulcerative colitis. World J Gastroenterol.
20:9458–9467. 2014. View Article : Google Scholar
|
|
6
|
Huang Y, Chen J, Wong T and Liow JL:
Experimental and theoretical investigations of non-Newtonian
electro-osmotic driven flow in rectangular microchannels. Soft
Matter. 12:6206–6213. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Nowarski R, Jackson R, Gagliani N, de
Zoete MR, Palm NW, Bailis W, Low JS, Harman CC, Graham M, Elinav E
and Flavell RA: Epithelial IL-18 equilibrium controls barrier
function in colitis. Cell. 163:1444–1456. 2015. View Article : Google Scholar
|
|
8
|
Pugliese D, Felice C, Papa A, Gasbarrini
A, Rapaccini GL, Guidi L and Armuzzi A: Anti TNF-α therapy for
ulcerative colitis: Current status and prospects for the future.
Expert Rev Clin Immunol. 13:223–233. 2017. View Article : Google Scholar
|
|
9
|
Li L, Qi Y, Ma X, Xiong G, Wang L and Bao
C: TRIM22 knockdown suppresses chronic myeloid leukemia via
inhibiting PI3K/Akt/mTOR signaling pathway. Cell Biol Int.
42:1192–1199. 2018. View Article : Google Scholar
|
|
10
|
Lou J, Wang Y, Zheng X and Qiu W: TRIM22
regulates macrophage autophagy and enhances Mycobacterium
tuberculosis clearance by targeting the nuclear factor-multiplicity
κB/beclin 1 pathway. J Cell Biochem. 119:8971–8980. 2018.
View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Kang C, Lu Z, Zhu G, Chen Y and Wu Y:
Knockdown of TRIM22 relieves oxygen-glucose
deprivation/reoxygenation-induced apoptosis and inflammation
through inhibition of NF-κB/NLRP3 axis. Cell Mol Neurobiol.
41:341–351. 2021. View Article : Google Scholar
|
|
12
|
Li Q, Lee CH, Peters LA, Mastropaolo LA,
Thoeni C, Elkadri A, Schwerd T, Zhu J, Zhang B, Zhao Y, et al:
Variants in TRIM22 that affect NOD2 signaling are associated with
very-early-onset inflammatory bowel disease. Gastroenterology.
150:1196–1207. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Liu R, Zhao W, Wang H and Wang J: Long
noncoding RNA LINC01207 promotes colon cancer cell proliferation
and invasion by regulating miR-3125/TRIM22 axis. Biomed Res Int.
2020:12163252020. View Article : Google Scholar
|
|
14
|
Chen C, Zhao D, Fang S, Chen Q, Cheng B,
Fang X and Shu Q: TRIM22-mediated apoptosis is associated with bak
oligomerization in monocytes. Sci Rep. 7:399612017. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Ji F and Sadreyev RI: RNA-seq: Basic
bioinformatics analysis. Curr Protoc Mol Biol. 124:e682018.
View Article : Google Scholar
|
|
16
|
Canzoneri R, Lacunza E and Abba MC:
Genomics and bioinformatics as pillars of precision medicine in
oncology. Medicina (B Aires). 79:(Spec 6/1). 587–592.
2019.PubMed/NCBI
|
|
17
|
Wang ZL, Wang YD, Wang K, Li JA and Li L:
KFL2 participates in the development of ulcerative colitis through
inhibiting inflammation via regulating cytokines. Eur Rev Med
Pharmacol Sci. 22:4941–4948. 2018.
|
|
18
|
Liang Y, Luo J, Yang N, Wang S, Ye M and
Pan G: Activation of the IL-1β/KLF2/HSPH1 pathway promotes STAT3
phosphorylation in alveolar macrophages during LPS-induced acute
lung injury. Biosci Rep. 40:BSR201935722020. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Vanhove W, Peeters PM, Staelens D, Anica
S, der Goten JV, Cleynen I, De Schepper S, Van Lommel L, Reynaert
NL, Schuit F, et al: Strong upregulation of AIM2 and IFI16
inflammasomes in the mucosa of patients with active inflammatory
bowel disease. Inflamm Bowel Dis. 21:2673–2682. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Ouahed J, Gordon W, Canavan JB, Zhou HY,
Du S, von Schack D, Phillips K, Wang L, Dunn WA III, Field M, et
al: Mucosal gene expression in pediatric and adult patients with
ulcerative colitis permits modeling of ideal biopsy collection
strategy for transcriptomic analysis. Inflamm Bowel Dis.
24:2565–2578. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Cheng F, Li Q, Wang J, Zeng F, Wang K and
Zhang Y: Identification of differential intestinal mucosa
transcriptomic biomarkers for ulcerative colitis by bioinformatics
analysis. Dis Markers. 2020:88765652020. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Cardon AD, Bailey MR and Bennett BT: The
animal welfare act: From enactment to enforcement. J Am Assoc Lab
Anim Sci. 51:301–305. 2012.
|
|
23
|
Kihara N, de la Fuente SG, Fujino K,
Takahashi T, Pappas TN and Mantyh CR: Vanilloid receptor-1
containing primary sensory neurones mediate dextran sulphate sodium
induced colitis in rats. Gut. 52:713–719. 2003. View Article : Google Scholar
|
|
24
|
Chen Q, Fang X, Yao N, Wu F, Xu B and Chen
Z: Suppression of miR-330-3p alleviates DSS-induced ulcerative
colitis and apoptosis by upregulating the endoplasmic reticulum
stress components XBP1. Hereditas. 157:182020. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Livak KJ and Schmittgen TD: Analysis of
relative gene expression data using real-time quantitative PCR and
the 2(−Delta Delta C(T)) method. Mehods. 25:402–408. 2001.
|
|
26
|
Yamamoto-Furusho JK, Gutiérrez-Grobe Y,
López-Gómez JG, Bosques-Padilla F and Rocha-Ramírez JL; Grupo del
Consenso Mexicano de Colitis Ulcerosa Crónica Idiopática, : The
Mexican consensus on the diagnosis and treatment of ulcerative
colitis. Rev Gastroenterol Mex (Engl Ed). 83:144–167. 2018.(In
English, Spanish). PubMed/NCBI
|
|
27
|
Ashton JJ, Mossotto E, Stafford IS,
Haggarty R, Coelho TAF, Batra A, Afzal NA, Mort M, Bunyan D,
Beattie RM and Ennis S: Genetic sequencing of pediatric patients
identifies mutations in monogenic inflammatory bowel disease genes
that translate to distinct clinical phenotypes. Clin Transl
Gastroenterol. 11:e001292020. View Article : Google Scholar
|
|
28
|
Lawrence T: The nuclear factor NF-kappaB
pathway in inflammation. Cold Spring Harb Perspect Biol.
1:a0016512009. View Article : Google Scholar
|
|
29
|
Ji J, Ding K, Luo T, Zhang X, Chen A,
Zhang D, Li G, Thorsen F, Huang B, Li X and Wang J: TRIM22
activates NF-κB signaling in glioblastoma by accelerating the
degradation of IκBα. Cell Death Differ. 28:367–381. 2021.
View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Jha P and Das H: KLF2 in regulation of
NF-κB-mediated immune cell function and inflammation. Int J Mol
Sci. 18:23832017. View Article : Google Scholar
|
|
31
|
Zhuang T, Liu J, Chen X, Zhang L, Pi J,
Sun H, Li L, Bauer R, Wang H, Yu Z, et al: Endothelial Foxp1
suppresses atherosclerosis via modulation of Nlrp3 inflammasome
activation. Circ Res. 125:590–605. 2019. View Article : Google Scholar
|