Introduction
Cardiac hypertrophy is a compensatory reaction to
myocardial stress overload, and is characterized by increased
protein synthesis, myocardial cell volume enlargement and
mesenchymal component alterations (1–3).
Cardiac hypertrophy is an independent risk factor for
cardiovascular disease morbidity and mortality (4). Numerous parameters have been
reported to contribute to this disease, including neurohormonal
factors, mechanical factors, endocrine factors, sympathetic nervous
system activity and nitric oxide production (2,5).
At present, treatment of cardiac hypertrophy mainly involves the
application of angiotensin-converting enzyme inhibitors,
angiotensin II (Ang II) receptor blockers, calcium channel
blockers, β-receptor blockers and diuretics (6–8).
However, the therapeutic effects mediated by these strategies
remain unsatisfactory. Therefore, there is a demand for novel
therapeutic options for cardiac hypertrophy.
Activating transcription factor (ATF) 3 is a member
of the ATF/cAMP-responsive element-binding protein family of
transcription factors (9). ATF3
is a stress response protein that is expressed at low levels in
quiescent cells but is increased under various stress stimuli, such
as injury and toxin exposure, and has been reported to facilitate
the pathological processes of various diseases, including hepatic
ischemia-reperfusion, liver fibrosis and acute lung injury
(10–12). For example, Chen et al
(13) reported that acute hypoxia
promoted the activation of ATF3, which could serve important roles
in the cellular response to stress. It has also been reported that
ATF3 can bidirectionally regulate the transcription of target genes
(14,15). Furthermore, ATF3 may regulate the
inflammatory response, apoptosis and autophagy by modulating the
binding sites of transcription factors, such as transcriptional
activating protein-1, NF-κB and p53 (16,17). Previous studies have reported that
ATF3 serves an important role in the occurrence and development of
various forms of cardiovascular diseases, such as myocardial
ischemia-reperfusion injury, myocardial hypertrophy and heart
failure (18–20). As such, ATF3 deficiency has been
reported to promote pressure overload-induced cardiac hypertrophy,
dysfunction and fibrosis (21).
However, the mechanism underlying the regulatory effects of ATF3
during myocardial hypertrophy remain elusive.
In the present study, the role and mechanism of
action of ATF3 in cardiac hypertrophy were assessed. Ang II
treatment was used to establish an in vitro model of cardiac
hypertrophy before the effects of ATF3 on Ang II-induced
cardiomyocytes, in addition to the association between ATF3 and
cysteine-rich angiogenic protein 61 (Cyr61), were assessed.
Materials and methods
Cell culture and treatment
Primary human cardiac fibroblasts (HCFs; cat. no.
6320) were purchased from ScienCell Research Laboratories, Inc. The
cells were cultured in DMEM (Gibco; Thermo Fisher Scientific, Inc.)
supplemented with 10% FBS (Gibco; Thermo Fisher Scientific, Inc.),
100 µg/ml streptomycin and 100 U/ml penicillin in humidified
conditions with 5% CO2 at 37°C. Cellular hypertrophy of
HCFs was induced using 1 µmol/l Ang II (Sigma-Aldrich; Merck KGaA)
at 37°C for 24 h. The concentration of Ang II was outside the range
that reflects the in vivo situation and has been used in a
similar manner in previous publications (22–24).
Cell transfection
The ATF3-specific pcDNA3.1 overexpression vector
(Oe-ATF3) and empty plasmid (as the corresponding control Oe-NC),
the specific small interfering RNAs (siRNAs) targeting ATF3
(si-ATF3-1, 5′-CGUGCAGUAUCUCAAGAUAUU-3′; si-ATF3-2,
5′-GGUUGUGCUUUCUAGCAAAUA-3′) and Cyr61 (si-Cyr61-1,
5′-GAUUAGUUGGACAGUUUAAAG-3′; si-Cyr61-2,
5′-AGAUUAGUUGGACAGUUUAAA-3′), and the non-targeting corresponding
control (si-NC, 5′-UUCUCCGAACGUGUCACGU-3′) were all purchased from
Shanghai GenePharma Co., Ltd. These vectors (4 µg) and siRNAs (100
nM) were transfected into HCFs (1×105 cells/well) seeded
into 12-well plates using Lipofectamine® 2000 reagent
(Invitrogen; Thermo Fisher Scientific, Inc.) at 37°C for 5 h
according to the manufacturer's protocol. Next, the cells were
incubated in DMEM supplemented with 10% FBS at 37°C for 48 h. At 48
h post-transfection, cells were collected for subsequent
experiments.
Cell Counting Kit-8 (CCK-8) assay
HCFs, with or without transfection, were seeded into
96-well plates at a density of 1×103 cells/well and
cultured in DMEM with 10% FBS, followed by treatment with Ang II.
After 24 h, 10 µl CCK-8 solution (Beijing Solarbio Science &
Technology Co., Ltd.) was added into each well before incubation
for a further 2 h. The absorbance value was detected at a
wavelength of 450 nm using a microplate reader.
Reverse transcription-quantitative PCR
(RT-qPCR)
After treatment, total RNA was extracted from HCFs
using TRIzol® (Invitrogen; Thermo Fisher Scientific,
Inc.) according to the manufacturer's protocol. A
NanoDrop® 3000 spectrophotometer (Thermo Fisher
Scientific, Inc.) was used to confirm the quality and quantity of
total RNA. RT of first-strand cDNA was performed using the
PrimeScript™ RT Master Mix (Perfect Real Time) kit
(Takara Bio, Inc.) according to the manufacturer's protocol.
Amplification of the cDNA was performed by qPCR using the TB Green
Premix Ex Taq II kit (Takara Bio, Inc.). The PCR program was 95°C
for 3 min, followed by 35 cycles of denaturation at 95°C for 30
sec, annealing at 60°C for 30 sec and extension at 72°C for 1 min.
A final extension step at 72°C for 7 min was performed for each PCR
assay. The primer sequences used were as follows: ATF3 forward (F),
5′-AGCACCTTGCCCCAAAATCA-3′ and reverse (R),
5′-AGGGCGTCAGGTTAGCAAAA-3′; Cyr61 F, 5′-AGCGTTTCCCTTCTACAGGC-3′ and
R, 5′-TTCTCCAATCGTGGCTGCAT-3′; and GAPDH F,
5′-GGGAAACTGTGGCGTGAT-3′ and R, 5′-GAGTGGGTGTCGCTGTTGA-3′. The
relative mRNA expression levels were normalized to those of GAPDH
using the 2−ΔΔCq method (25).
Wound healing assay
The migratory capacity of cells was assessed using
the wound healing assay. Transfected or untransfected cells were
seeded into six-well plates, and cultured to 90% confluence
following treatment with 1 µmol/l Ang II (Sigma-Aldrich; Merck
KGaA). The cell monolayers were then wounded using a 200-µl pipette
tip and washed three times with serum-free medium. After 24 h of
incubation in serum-free medium at 37°C, images were captured using
a light microscope (Leica Microsystems, Inc.) and the migration
rate was calculated using the following formula: (scratch width at
0 h-scratch width at 48 h)/scratch width at 0 h. The wound closure
area of the migrating monolayer of cells was quantified using
ImageJ software (version 1.49; National Institutes of Health).
Immunofluorescence staining
Immunofluorescence staining was used for the
assessment of the expression of α-smooth muscle actin (α-SMA) in
HCFs. Cells were fixed with 4% paraformaldehyde at 37°C for 30 min,
blocked with 5% bovine serum albumin (Beijing Solarbio Science
& Technology Co., Ltd.) at 37°C for 30 min and incubated with
anti-α-SMA antibodies (1:1,000; cat. no. 19245; Cell Signaling
Technology) overnight at 4°C. Subsequently, Alexa Fluor®
488-conjugated secondary antibodies (1:400; cat. no. ab150077;
Abcam) were added for 1 h at room temperature. The nuclei were
stained using 5 µg/ml DAPI solution for 5 min at room temperature.
The cells were subsequently observed and images were captured using
a fluorescence microscope (Olympus Corporation).
Luciferase reporter assay
The 3′UTR fragments of the human Cyr61 promoter were
predicted using the JASPAR database 2022 (https://jaspar.genereg.net). Wild-type (WT) and
corresponding mutant (Mut) fragments of the Cyr61 promoter covering
the predicted DR1 (direct repeat motif with a single nucleotide
spacer) sites were cloned into the firefly luciferase reporter
plasmid pGL3-basic vector (Promega Corporation). Luciferase
activity was then detected using a Dual-Luciferase Reporter Assay
Kit (Promega Corporation) 48 h after transfection of WT/MUT
plasmids and OE-ATF3/OE-NC into HCFs using the Lipofectamine 2000
transfection reagent. Firefly luciferase activity was normalized
against that of the Renilla construct and the relative
luciferase activity in untreated cells was designated as 1.
Chromatin immunoprecipitation (ChIP)
assay
ChIP assays were performed using the EZ
ChIP™ Kit (MilliporeSigma). The cells were first
cross-linked with 1% formaldehyde for 10 min at 37°C and quenched
with 2.5 M glycine for 5 min at room temperature to a final
concentration of 125 µM. The fixed cells were washed twice with
phosphate-buffered saline and were lysed using a lysis buffer [0.1%
sodium dodecyl sulfate (SDS), 0.5% Triton X-100, 20 mM Tris-HCl, pH
8.1] that contained 150 mM NaCl and a protease inhibitor. The lysed
cells were subsequently subjected to sonication in ice water. The
resulting sonicated fragments were within the size range of
200-1,000 bp. Following sonication, the samples were centrifuged at
13,000 × g for 10 min at 4°C, and 100 µl of supernatant was
pre-absorbed by 30 µl protein G magnetic beads (Thermo Fisher
Scientific, Inc.) conjugated to ATF3 antibodies (2 µg; cat. no.
ab254268; 1:30; Abcam) and IgG (as the NC; cat. no. ab172730; 1:50;
Abcam). The immunoprecipitated complex was centrifuged (5,000 × g
for 1 min at 4°C) and washed with low salt, high salt, LiCl and TE
buffers in the kit according to the manufacturer's protocols. The
complex was eluted from the antibody using a solution of 1% SDS,
0.1 mol/l NaHCO3 and 200 mmol/l NaCl. The resultant complex was
incubated in 5 M NaCl and 20 mg/ml proteinase K solution (Cell
Signaling Technology, Inc.) at 65°C for 2 h for the reversal of
crosslinking. After crosslink reversal, precipitated DNA was
analyzed by PCR for the 3′UTR fragments of the Cyr61 promoter. The
input DNA and immunoprecipitated DNA underwent qPCR using
SYBR® Green Real-time PCR Master Mix (Toyobo Life
Science). The primer sequences for PCR were as follows: ATF3
forward, 5′-AGCACCTTGCCCCAAAATCA-3′ and reverse,
5′-AGGGCGTCAGGTTAGCAAAA-3′; Cyr61 forward,
5′-AGCGTTTCCCTTCTACAGGC-3′ and reverse, 5′-TTCTCCAATCGTGGCTGCAT-3′;
and GAPDH forward, 5′-GGGAAACTGTGGCGTGAT-3′ and reverse,
5′-GAGTGGGTGTCGCTGTTGA-3′. The PCR program was 95°C for 3 min,
followed by 35 cycles of denaturation at 95°C for 30 sec, annealing
at 60°C for 30 sec and extension at 72°C for 1 min. A final
extension step was applied at 72°C for 7 min. The data obtained
were normalized to those obtained from the qPCR of the DNA
precipitated by the IgG antibody. The relative mRNA expression
levels were normalized to those of GAPDH using the
2−ΔΔCq method (25).
Western blotting
Total protein was extracted from treated or
untreated HCFs using RIPA buffer (Beyotime Institute of
Biotechnology) and quantified using the bicinchoninic acid method
(Thermo Fisher Scientific, Inc.). Protein samples (40 µg/lane) were
then separated by SDS-PAGE on 10% gels and transferred onto PVDF
membranes. The membranes, which were blocked with 5% skimmed fat
milk overnight at 4°C, were incubated with the following primary
antibodies overnight at 4°C: ATF3 (cat. no. ab254268; 1:1,000;
Abcam), MMP-1 (cat. no. ab134184; 1:1,000; Abcam), MMP-2 (cat. no.
ab92536; 1:1,000; Abcam), connective tissue growth factor (CTGF;
cat. no. ab209780; 1:1,000; Abcam), fibronectin (cat. no. ab268020;
1:1,000; Abcam), collagen I (cat. no. ab138492; 1:1,000; Abcam),
collagen III (cat. no. ab184993; 1:1,000; Abcam), Cyr61 (cat. no.
ab230947; 1:1,000; Abcam), TGF-β (cat. no. ab215715; 1:1,000;
Abcam), phosphorylated (p)-Smad2 (cat. no. ab280888; 1:1,000;
Abcam), p-Smad3 (cat. no. ab52903; 1:2,000; Abcam), Smad2 (cat. no.
ab40855; 1:2,000; Abcam), Smad3 (cat. no. ab40854; 1:1,000; Abcam)
and GAPDH (cat. no. ab9485; 1:2,500; Abcam). Membranes were washed
with TBST (0.1% Tween-20) and incubated with HRP-conjugated
secondary antibodies (cat. no. #7074; 1:3,000; Cell Signaling
Technology, Inc.) for 1 h at room temperature. The immunoreactive
protein bands were visualized using an Amersham ECL Western
Blotting Detection Reagent (Cytiva) and semi-quantified by
densitometry (Quantity One® version 4.5.0; Bio-Rad
Laboratories, Inc.).
Statistical analysis
All experiments were repeated three times
independently. The data were analyzed using SPSS 17.0 software
(SPSS, Inc.) and are presented as the mean ± SD. Unpaired Student's
t-test was used for comparisons between two groups. Differences
among multiple groups were analyzed using one-way ANOVA with the
Bonferroni multiple comparison post hoc test. P<0.05 was
considered to indicate a statistically significant difference.
Results
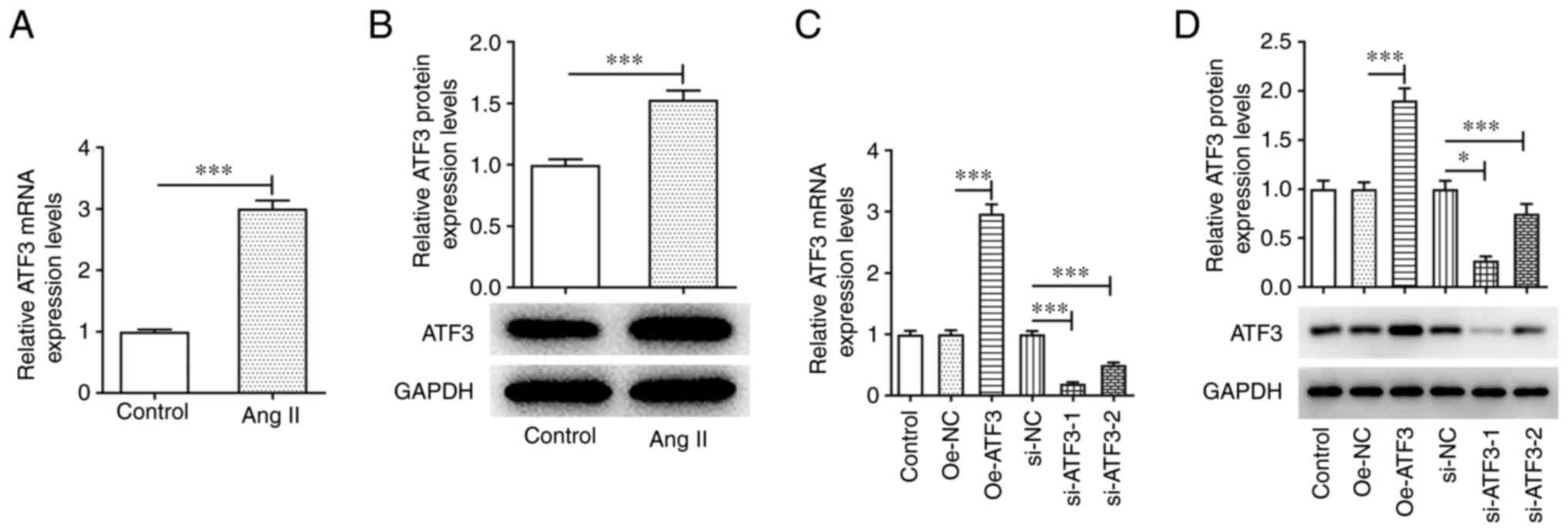
ATF3 is highly expressed in Ang
II-induced HCFs
An in vitro cardiac hypertrophy model was
established by stimulating HCFs with 1 µmol/l Ang II. The results
of RT-qPCR and western blotting demonstrated that the mRNA and
protein expression levels of ATF3 were significantly upregulated in
Ang II-induced HCFs compared with those in the untreated cells
(Fig. 1A and B). To assess the
role of ATF3 in cardiac hypertrophy, ATF3 expression was knocked
down or ATF3 was overexpressed in HCFs. The transfection efficiency
was assessed using RT-qPCR and western blotting. Oe-ATF3
transfection significantly increased the mRNA and protein
expression levels of ATF3, whereas si-ATF3-1/2 transfection
significantly reduced ATF3 mRNA and protein expression, compared
with those in the negative control groups (Fig. 1C and D). Since si-ATF3-1 exhibited
superior transfection efficiency, this siRNA was chosen for
subsequent experiments and is referred to as si-ATF3
thereafter.
Overexpression of ATF3 suppresses Ang
II-induced viability, migration and fibrosis in HCFs
The effect of ATF3 overexpression on Ang II-induced
HCFs was assessed. Ang II treatment significantly enhanced HCF
viability compared with that in the control group; however,
transfection with Oe-ATF3 subsequently reversed this effect
(Fig. 2A). Furthermore, wound
healing assays demonstrated that treatment of HCFs with Ang II
significantly increased the migratory rate compared with that in
the control group in a manner that was reversed by ATF3
overexpression (Fig. 2B).
Additionally, the protein expression levels of MMP-1 and MMP-2 were
significantly increased following Ang II treatment compared with
those in the control group; however, this effect was also reversed
by transfection with Oe-ATF3 (Fig.
2C). Fibrosis was assessed using immunofluorescence staining
and western blotting. Ang II treatment markedly increased the
protein expression levels of α-SMA compared with those in the
control group, whereas the overexpression of ATF3 reversed this
increase in α-SMA expression (Fig.
2D). Marked increases in the protein expression levels of CTGF,
fibronectin, collagen I and collagen III were also observed after
the HCFs were treated with Ang II compared with those in the
control group (Fig. 2E). However,
ATF3 overexpression reversed the effects of Ang II on the protein
expression levels of the aforementioned proteins in HCFs (Fig. 2E).
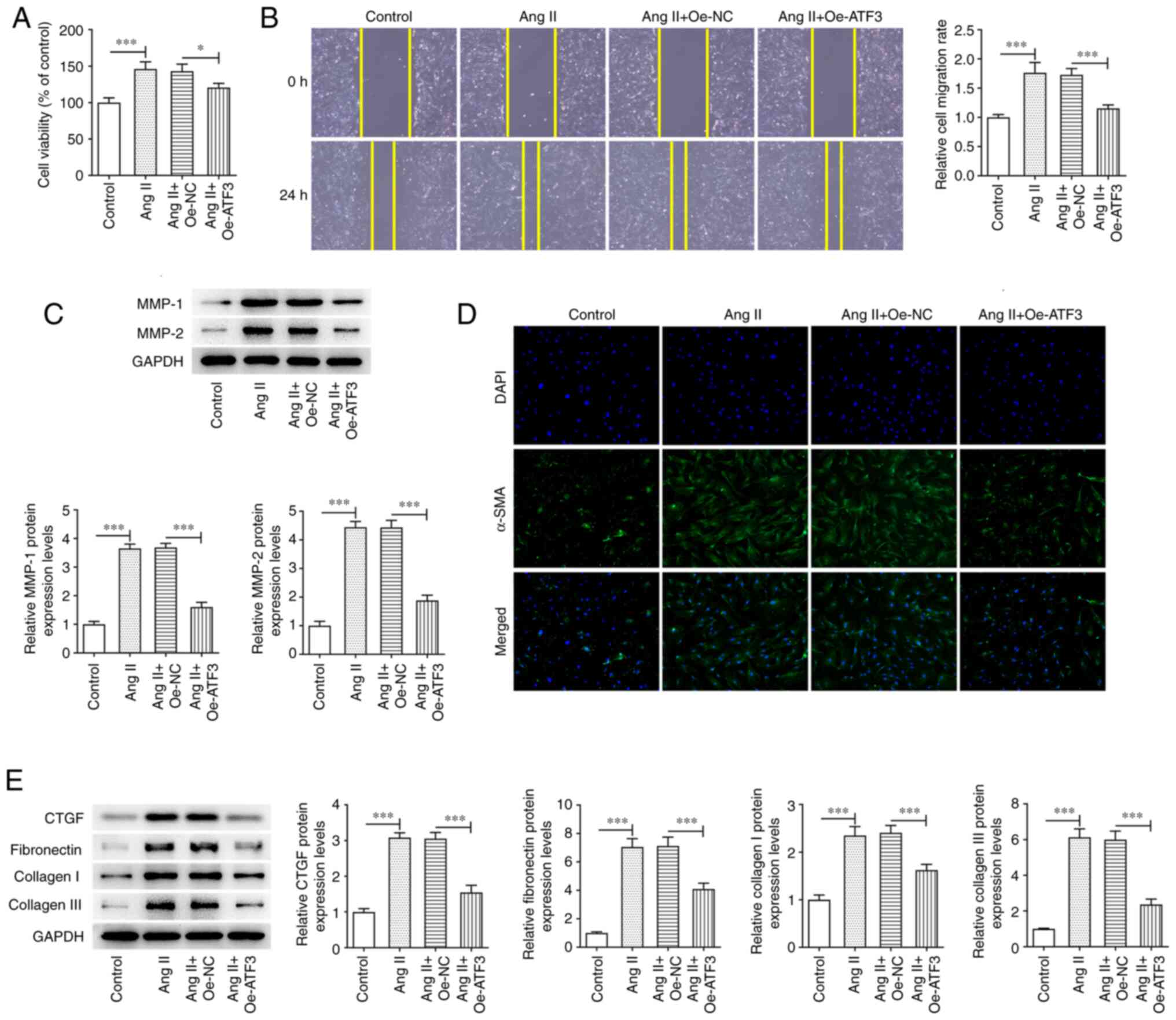 | Figure 2.Overexpression of ATF3 inhibits Ang
II-induced viability, migration and fibrosis in human cardiac
fibroblasts. (A) Cell viability was evaluated using a Cell Counting
Kit-8 assay. (B) Cell migration was assessed using the wound
healing assay (magnification, ×100). (C) Protein expression levels
of MMP-1 and MMP-2 were semi-quantified using western blotting. (D)
Immunofluorescence staining was performed to assess the protein
expression levels of α-SMA (magnification, ×200). (E) Protein
expression levels of CTGF, fibronectin, collagen I and collagen III
were semi-quantified using western blotting. Data are presented as
the mean ± SD. *P<0.05, ***P<0.001. ATF3, activating
transcription factor 3; Ang II, angiotensin II; α-SMA, α-smooth
muscle actin; CTGF, connective tissue growth factor; Oe,
overexpression; NC, negative control. |
Knockdown of ATF3 expression
aggravates Ang II-induced viability, migration and fibrosis of
HCFs
To evaluate the role of ATF3 in Ang II-induced HCFs,
the effects of ATF3 knockdown on Ang II-induced HCFs were assessed.
ATF3 knockdown significantly enhanced Ang II-induced HCF viability
compared with that in cells transfected with the negative control
(Fig. 3A). Cell migration was
also demonstrated to be significantly increased by ATF3 knockdown
compared with that in the si-NC group, and the protein expression
levels of MMP-1 and MMP-2 were significantly potentiated in Ang
II-induced HCFs transfected with si-ATF3 compared with those in the
Ang II + si-NC group (Fig. 3B and
C). Subsequently, α-SMA protein expression was demonstrated to
be increased following transfection with si-ATF3, compared with
that in the Ang II + si-NC cells (Fig. 3D). ATF3 knockdown also
significantly promoted the protein expression levels of CTGF,
fibronectin, collagen I and collagen III in Ang II-induced HCFs
compared with those in the Ang II + si-NC group (Fig. 3E).
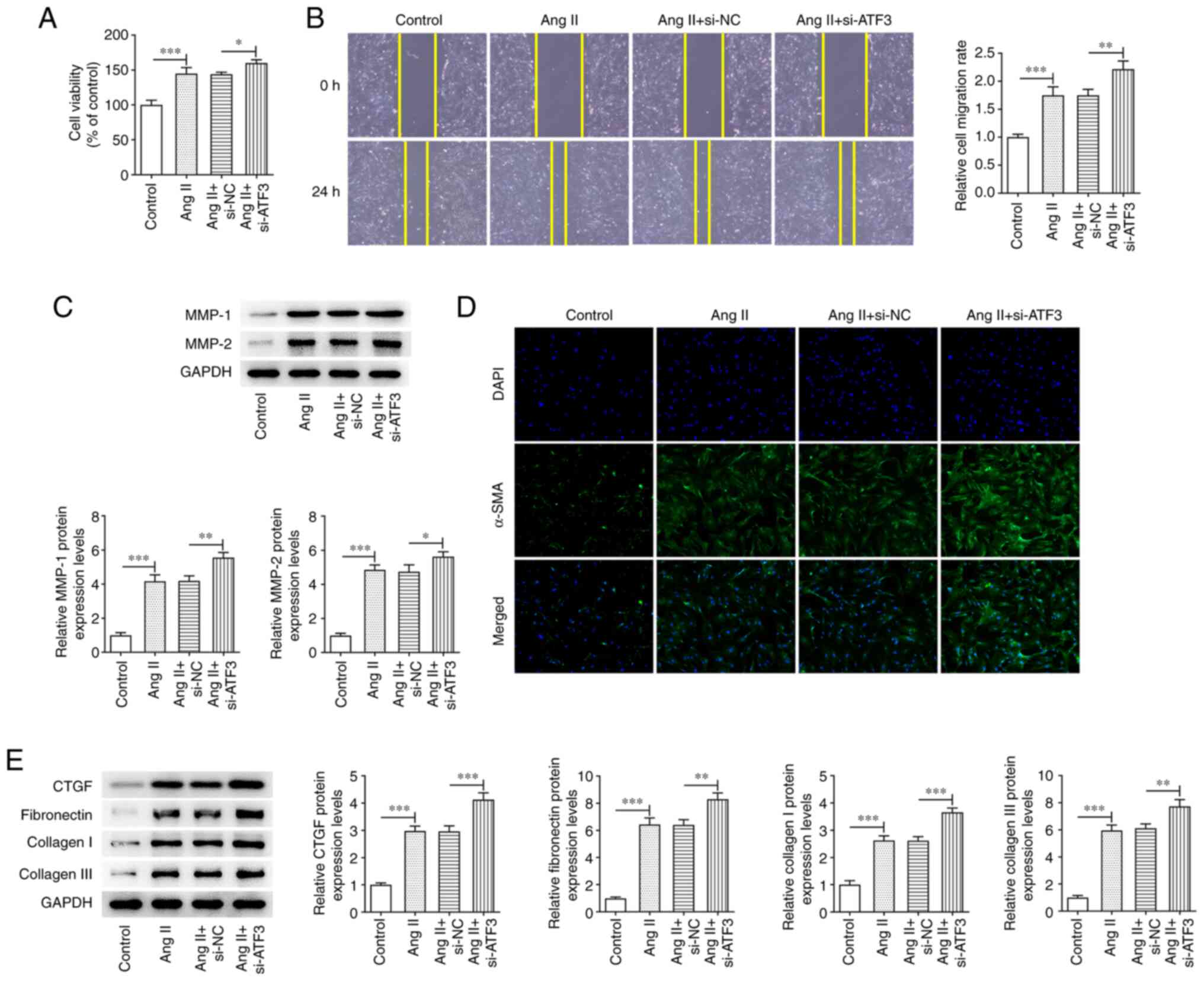 | Figure 3.Knockdown of ATF3 promotes Ang
II-induced viability, migration and fibrosis of cardiac
fibroblasts. (A) Cell viability was evaluated using a Cell Counting
Kit-8 assay. (B) Cell migration was evaluated using the wound
healing assay (magnification, ×100). (C) Protein expression levels
of MMP-1 and MMP-2 were semi-quantified using western blotting. (D)
Immunofluorescence staining was performed to assess the protein
expression levels of α-SMA (magnification, ×200). (E) Protein
expression levels of CTGF, fibronectin, collagen I and collagen III
were semi-quantified using western blotting. Data are presented as
the mean ± SD. *P<0.05, **P<0.01, ***P<0.001. ATF3,
activating transcription factor 3; Ang II, angiotensin II; α-SMA,
α-smooth muscle actin; CTGF, connective tissue growth factor; si,
small interfering RNA; NC, negative control. |
ATF3 promotes the transcriptional
activation of Cyr61
The potential mechanisms by which ATF3 regulated Ang
II-induced HCFs were evaluated. Compared with those in their
corresponding negative controls, ATF3 overexpression significantly
increased the mRNA and protein expression levels of Cyr61 in HCFs,
whereas they were significantly decreased by ATF3 silencing
(Fig. 4A and B). A binding site
was predicted using the JASPAR database (Fig. 4C). The luciferase reporter assay
demonstrated that the luciferase activity of the WT Cyr61 promoter
was significantly increased by ATF3 overexpression compared with in
the Oe-NC group, whereas no notable changes in the luciferase
activity of the Mut Cyr61 promoter were observed (Fig. 4D). Furthermore, the ChIP assay
verified that compared with the Oe-NC group, a significant increase
was observed in the enrichment of Cyr61 promoter in ATF3 antibody
in the Oe-ATF3 group, implying that ATF3 could bind to the
predicted Cyr61 binding site (Fig.
4E).
ATF3 affects Ang II-induced HCFs and
TGF-β signaling by regulating Cyr61
To further explore the role of Cyr61 in Ang
II-induced HCFs following ATF3 manipulation, Cyr61 expression was
knocked down by transfection with si-Cyr61-1/2. The transfection
efficiency was assessed using RT-qPCR and western blotting. Since
si-Cyr61-1 transfection resulted in superior transfection
efficiency (Fig. 5A and B),
si-Cyr61-1 was chosen for use in subsequent experiments and is
referred to as si-Cyr61 thereafter. CCK-8 assay results
demonstrated that Cyr61 knockdown increased the Oe-ATF3-reduced
cell viability (Fig. 5C).
Furthermore, ATF3 overexpression significantly suppressed cell
migration, and markedly declined MMP-1 and MMP-2 expression in Ang
II-treated HCFs, which was reversed by Cyr61 silencing (Fig. 5D and E). α-SMA protein expression
levels were reversed by Cyr61 silencing compared with the Oe-ATF3
group when assessed using immunofluorescence staining, and the
protein expression levels of CTGF, fibronectin, collagen I and
collagen III were elevated in Cyr61-silenced cells compared with
those in the Ang II+Oe-ATF3 group (Fig. 5F and G). Furthermore, the protein
expression levels of TGF-β, p-Smad2/Smad2 and p-Smad3/Smad3 were
significantly increased by stimulation with Ang II, and were in
turn significantly reduced by ATF3 overexpression (Fig. 5H). However, Cyr61 knockdown
significantly reversed the effects of ATF3 overexpression on the
protein expression levels of these three aforementioned
proteins.
 | Figure 5.ATF3 regulates Ang II-induced
development of cardiac fibroblasts and the TGF-β signaling pathway
through binding to Cyr61. (A) mRNA and (B) protein expression
levels of Cyr61 were detected using reverse
transcription-quantitative PCR and western blotting respectively.
(C) Cell viability was evaluated using a Cell Counting Kit-8 assay.
(D) Cell migration was evaluated using a wound healing assay
(magnification, ×100). (E) Protein expression levels of MMP-1 and
MMP-2 were semi-quantified using western blotting. (F)
Immunofluorescence staining was performed to assess the protein
expression levels of α-SMA (magnification, ×200). Protein
expression levels of (G) CTGF, fibronectin, collagen I and collagen
III, and (H) TGF-β, p-Smad2, p-Smad3, Smad2 and Smad3 were
semi-quantified using western blotting. Data are presented as the
mean ± SD. *P<0.05, **P<0.01, ***P<0.001. ATF3, activating
transcription factor 3; Ang II, angiotensin II; α-SMA, α-smooth
muscle actin; Cyr61, cysteine-rich angiogenic protein 61; p,
phosphorylated; CTGF, connective tissue growth factor; Oe,
overexpression; NC, negative control; si, small interfering
RNA. |
Discussion
During cardiac hypertrophy, the regulation of
cardiac fibroblasts by Ang II and other factors results in
excessive proliferation and the production of excessive quantities
of fibrin, which leads to cardiac fibrosis and can further
aggravate cardiac hypertrophy (26–28). Therefore, inhibition of the
proliferation and fibrosis of cardiac fibroblasts may serve as a
viable strategy for treating cardiac hypertrophy. The present study
aimed to evaluate the therapeutic potential of ATF3 for alleviating
cardiac viability and fibrosis in addition to assessing the
potential molecular mechanism underlying its function using an
in vitro model.
ATF3 is a stress response protein, the expression of
which rapidly increases following stress stimulation by endogenous
and exogenous factors, in order to regulate the expression of
target genes (29). Li et
al (30) reported that
cardiac fibroblasts were the main cell type that express ATF3 in
response to stimulation. ATF3 expression has been reported to be
upregulated in Ang II- and aortic constriction-induced mouse
myocardium, whereas ATF3 knockdown could worsen Ang II-induced
cardiac fibrosis and hypertrophy (31). These findings suggested that ATF3
may serve an important regulatory role in myocardial fibrosis. In
the present study, HCFs were stimulated with Ang II and it was
demonstrated that Ang II treatment significantly increased the mRNA
and protein expression levels of ATF3. ATF3 overexpression also
exerted a significant inhibitory effect on Ang II-induced increases
in HCF viability, migration and fibrosis. ATF3 expression was
subsequently silenced and it was demonstrated that it mediated the
opposite effects on Ang II-induced HCF viability, migration and
fibrosis compared with ATF3 overexpression, which suggested a
possible protective role for ATF3 against Ang II-induced stress in
HCFs. These results were consistent with those of a previous study,
which reported that reduced ATF3 expression may promote stress
overload-induced cardiac hypertrophy, dysfunction and fibrosis
(21).
It has previously been reported that ATF3 can
transcriptionally upregulate Cyr61 expression in hepatocellular
carcinoma (32). Cyr61, also
known as cellular communication network factor (CCN)1, is a member
of the CCN family of proteins that serves as an angiogenic factor
(33). Previous studies have
reported that Cyr61 expression is elevated during chronic heart
failure and is associated with Ang (34–36). In the present study, a binding
site of ATF3 on the Cyr61 promoter was predicted using the JASPAR
database, which was verified experimentally using a combination of
luciferase reporter and ChIP assays. You et al (37) reported that Cyr61/CCN1 expression
was regulated by the cooperation of c-Jun/AP-1 and hypoxia
inducible factor-1α under hypoxic conditions in retinal vascular
endothelial cells. Furthermore, Cyr61 has been reported to suppress
myocardial fibrosis and improve cardiac function (38). The present study silenced Cyr61
expression in Ang II-induced HCFs and demonstrated that it
significantly reversed the effects of ATF3 overexpression on cell
viability, migration and fibrosis, which suggested that ATF3
regulated HCFs induced by Ang II via transcriptional activation of
Cyr61.
Numerous studies have reported that Ang II can
induce the proliferation of cardiac fibroblasts through multiple
signaling pathways, including the TGF-β/MAPK and TGF-β/Smad
signaling pathways (39,40). It has previously been reported
that the CCN protein family can moderate the production of growth
factors and cytokine signaling (41). Borkham-Kamphorst et al
(42) reported that CCN1 exerted
anti-fibrotic effects through the induction of reactive oxygen
species, which in turn attenuated TGF-β signaling by scavenging the
TGF-β ligand. In the present study, significant increases in the
protein expression levels of TGF-β, p-Smad2/Smad2 and p-Smad3/Smad3
were detected in Ang II-induced HCFs. ATF3 overexpression
significantly reversed this increase in the expression levels of
these proteins. However, Cyr61 knockdown significantly negated the
effects induced by ATF3 overexpression on the increased levels of
TGF-β, p-Smad2 and p-Smad3. These results suggested that the
TGF-β/Smad pathway may be involved in the modulation of
ATF3/Cyr61-mediated viability and fibrosis of HCFs. Notably, there
were several limitations in the present study. Only in vitro
experiments were performed and further in vivo experiments
are required to confirm the results in future studies. Furthermore,
the potential mechanisms and key pathways require elucidation in
future studies.
In conclusion, the present study demonstrated that
ATF3 overexpression could suppress the viability, migration and
fibrosis of HCFs through the transcriptional activation of Cyr61.
These findings may provide novel insights into anti-viability and
anti-fibrosis strategies for the treatment of cardiac
hypertrophy.
Acknowledgements
Not applicable.
Funding
The present study was supported by the Scientific Research and
Cultivation Project of Meizhou People's Hospital, China (grant no.
PY-C20210026).
Availability of data and materials
The datasets used and/or analyzed during the current
study are available from the corresponding author on reasonable
request.
Authors' contributions
YZ, HW, HL and CH designed the study and performed
the experiments. YZ, HW, HL and YL wrote the manuscript and
analyzed the data. YZ supervised the experiments and revised the
manuscript. YZ and HW confirm the authenticity of all the raw data.
All authors read and approved the final manuscript.
Ethics approval and consent to
participate
Ethics approval for the use of human cardiac
fibroblasts was waived by Meizhou People's Hospital.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Nakamura M and Sadoshima J: Mechanisms of
physiological and pathological cardiac hypertrophy. Nat Rev
Cardiol. 15:387–407. 2018. View Article : Google Scholar
|
|
2
|
Shimizu I and Minamino T: Physiological
and pathological cardiac hypertrophy. J Mol Cell Cardiol.
97:245–262. 2016. View Article : Google Scholar
|
|
3
|
Zhu L, Li C, Liu Q, Xu W and Zhou X:
Molecular biomarkers in cardiac hypertrophy. J Cell Mol Med.
23:1671–1677. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Gallo S, Vitacolonna A, Bonzano A,
Comoglio P and Crepaldi T: ERK: A key player in the pathophysiology
of cardiac hypertrophy. Int J Mol Sci. 20:21642019. View Article : Google Scholar
|
|
5
|
Wang L, Wang J, Li G and Xiao J:
Non-coding RNAs in physiological cardiac hypertrophy. Adv Exp Med
Biol. 1229:149–161. 2020. View Article : Google Scholar
|
|
6
|
Tham YK, Bernardo BC, Ooi JY, Weeks KL and
McMullen JR: Pathophysiology of cardiac hypertrophy and heart
failure: Signaling pathways and novel therapeutic targets. Arch
Toxicol. 89:1401–1438. 2015. View Article : Google Scholar
|
|
7
|
Bisping E, Wakula P, Poteser M and Heinzel
FR: Targeting cardiac hypertrophy: Toward a causal heart failure
therapy. J Cardiovasc Pharmacol. 64:293–305. 2014. View Article : Google Scholar
|
|
8
|
Hou J and Kang YJ: Regression of
pathological cardiac hypertrophy: Signaling pathways and
therapeutic targets. Pharmacol Ther. 135:337–354. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Persengiev SP and Green MR: The role of
ATF/CREB family members in cell growth, survival and apoptosis.
Apoptosis. 8:225–228. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Zabala V, Boylan JM, Thevenot P, Frank A,
Senthoor D, Iyengar V, Kim H, Cohen A, Gruppuso PA and Sanders JA:
Transcriptional changes during hepatic ischemia-reperfusion in the
rat. PLoS One. 14:e02270382019. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Shi Z, Zhang K, Chen T, Zhang Y, Du X,
Zhao Y, Shao S, Zheng L, Han T and Hong W: Transcriptional factor
ATF3 promotes liver fibrosis via activating hepatic stellate cells.
Cell Death Dis. 11:10662020. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Qian L, Zhao Y, Guo L, Li S and Wu X:
Activating transcription factor 3 (ATF3) protects against
lipopolysaccharide-induced acute lung injury via inhibiting the
expression of TL1A. J Cell Physiol. 232:3727–3734. 2017. View Article : Google Scholar
|
|
13
|
Chen SC, Liu YC, Shyu KG and Wang DL:
Acute hypoxia to endothelial cells induces activating transcription
factor 3 (ATF3) expression that is mediated via nitric oxide.
Atherosclerosis. 201:281–288. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Bueno M, Brands J, Voltz L, Fiedler K,
Mays B, St Croix C, Sembrat J, Mallampalli RK, Rojas M and Mora AL:
ATF3 represses PINK1 gene transcription in lung epithelial cells to
control mitochondrial homeostasis. Aging Cell. 17:e127202018.
View Article : Google Scholar
|
|
15
|
Zhao W, Sun M, Li S, Chen Z and Geng D:
Transcription factor ATF3 mediates the radioresistance of breast
cancer. J Cell Mol Med. 22:4664–4675. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Ku HC and Cheng CF: Master regulator
activating transcription factor 3 (ATF3) in metabolic homeostasis
and cancer. Front Endocrinol (Lausanne). 11:5562020. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Kumar M, Majumder D, Mal S, Chakraborty S,
Gupta P, Jana K, Gupta UD, Ghosh Z, Kundu M and Basu J: Activating
transcription factor 3 modulates the macrophage immune response to
Mycobacterium tuberculosis infection via reciprocal regulation of
inflammatory genes and lipid body formation. Cell Microbiol.
22:e131422020. View Article : Google Scholar
|
|
18
|
Zhou H, Li N, Yuan Y, Jin YG, Guo H, Deng
W and Tang QZ: Activating transcription factor 3 in cardiovascular
diseases: A potential therapeutic target. Basic Res Cardiol.
113:372018. View Article : Google Scholar
|
|
19
|
Qin W, Yang H, Liu G, Bai R, Bian Y, Yang
Z and Xiao C: Activating transcription factor 3 is a potential
target and a new biomarker for the prognosis of atherosclerosis.
Hum Cell. 34:49–59. 2021. View Article : Google Scholar
|
|
20
|
Li YL, Hao WJ, Chen BY, Chen J and Li GQ:
Cardiac fibroblast-specific activating transcription factor 3
promotes myocardial repair after myocardial infarction. Chin Med J
(Engl). 131:2302–2309. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Zhou H, Shen DF, Bian ZY, Zong J, Deng W,
Zhang Y, Guo YY, Li H and Tang QZ: Activating transcription factor
3 deficiency promotes cardiac hypertrophy, dysfunction, and
fibrosis induced by pressure overload. PLoS One. 6:e267442011.
View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Zhou Y, Xie Y, Li T, Zhang P, Chen T, Fan
Z and Tan X: P21-activated kinase 1 mediates angiotensin II-induced
differentiation of human atrial fibroblasts via the JNK/c-Jun
pathway. Mol Med Rep. 23:2072021. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Gwathmey TM, Pendergrass KD, Reid SD, Rose
JC, Diz DI and Chappell MC:
Angiotensin-(1–7)-angiotensin-converting enzyme 2 attenuates
reactive oxygen species formation to angiotensin II within the cell
nucleus. Hypertension. 55:166–171. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Yang LL, Li DY, Zhang YB, Zhu MY, Chen D
and Xu TD: Salvianolic acid A inhibits angiotensin II-induced
proliferation of human umbilical vein endothelial cells by
attenuating the production of ROS. Acta Pharmacol Sin. 33:41–48.
2012. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Livak KJ and Schmittgen TD: Analysis of
relative gene expression data using real-time quantitative PCR and
the 2(−Delta Delta C (T)) method. Methods. 25:402–408. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Zhai CG, Xu YY, Tie YY, Zhang Y, Chen WQ,
Ji XP, Mao Y, Qiao L, Cheng J, Xu QB and Zhang C: DKK3
overexpression attenuates cardiac hypertrophy and fibrosis in an
angiotensin-perfused animal model by regulating the ADAM17/ACE2 and
GSK-3β/β-catenin pathways. J Mol Cell Cardiol. 114:243–252. 2018.
View Article : Google Scholar
|
|
27
|
Sheng R, Gu ZL, Xie ML, Zhou WX and Guo
CY: EGCG inhibits proliferation of cardiac fibroblasts in rats with
cardiac hypertrophy. Planta Med. 75:113–120. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Ji Y, Qiu M, Shen Y, Gao L, Wang Y, Sun W,
Li X, Lu Y and Kong X: MicroRNA-327 regulates cardiac hypertrophy
and fibrosis induced by pressure overload. Int J Mol Med.
41:1909–1916. 2018.PubMed/NCBI
|
|
29
|
Nyunt T, Britton M, Wanichthanarak K,
Budamagunta M, Voss JC, Wilson DW, Rutledge JC and Aung HH:
Mitochondrial oxidative stress-induced transcript variants of ATF3
mediate lipotoxic brain microvascular injury. Free Radic Biol Med.
143:25–46. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Li Y, Li Z, Zhang C, Li P, Wu Y, Wang C,
Bond Lau W, Ma XL and Du J: Cardiac fibroblast-specific activating
transcription factor 3 protects against heart failure by
suppressing MAP2K3-p38 signaling. Circulation. 135:2041–2057. 2017.
View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Pan J, Xu Z, Guo G, Xu C, Song Z, Li K,
Zhong K and Wang D: Circ_nuclear factor I X (circNfix) attenuates
pressure overload-induced cardiac hypertrophy via regulating
miR-145-5p/ATF3 axis. Bioengineered. 12:5373–5385. 2021. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Chen C, Ge C, Liu Z, Li L, Zhao F, Tian H,
Chen T, Li H, Yao M and Li J: ATF3 inhibits the tumorigenesis and
progression of hepatocellular carcinoma cells via upregulation of
CYR61 expression. J Exp Clin Cancer Res. 37:2632018. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Chaqour B: Regulating the regulators of
angiogenesis by CCN1 and taking it up a Notch. J Cell Commun
Signal. 10:259–261. 2016. View Article : Google Scholar
|
|
34
|
Bonda TA, Kamiński KA, Dziemidowicz M,
Litvinovich S, Kożuch M, Hirnle T, Dmitruk I, Chyczewski L and
Winnicka MM: Atrial expression of the CCN1 and CCN2 proteins in
chronic heart failure. Folia Histochem Cytobiol. 50:99–103. 2012.
View Article : Google Scholar
|
|
35
|
Wang J, Fu D, Senouthai S, Jiang Y, Hu R
and You Y: Identification of the transcriptional networks and the
involvement in Angiotensin II-induced injury after
CRISPR/Cas9-mediated knockdown of Cyr61 in HEK293T cells. Mediators
Inflamm. 2019:86972572019. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Hilfiker A, Hilfiker-Kleiner D, Fuchs M,
Kaminski K, Lichtenberg A, Rothkötter HJ, Schieffer B and Drexler
H: Expression of CYR61, an angiogenic immediate early gene, in
arteriosclerosis and its regulation by angiotensin II. Circulation.
106:254–260. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
You JJ, Yang CM, Chen MS and Yang CH:
Regulation of Cyr61/CCN1 expression by hypoxia through cooperation
of c-Jun/AP-1 and HIF-1α in retinal vascular endothelial cells. Exp
Eye Res. 91:825–836. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Meyer K, Hodwin B, Ramanujam D, Engelhardt
S and Sarikas A: Essential role for premature senescence of
myofibroblasts in myocardial fibrosis. J Am Coll Cardiol.
67:2018–2028. 2016. View Article : Google Scholar
|
|
39
|
Li L, Fan D, Wang C, Wang JY, Cui XB, Wu
D, Zhou Y and Wu LL: Angiotensin II increases periostin expression
via Ras/p38 MAPK/CREB and ERK1/2/TGF-β1 pathways in cardiac
fibroblasts. Cardiovasc Res. 91:80–89. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Wu X, Liu Y, An J, Li J, Lv W, Geng S and
Zhang Y: Piperlongumine inhibits angiotensin II-induced
extracellular matrix expression in cardiac fibroblasts. J Cell
Biochem. 119:10358–10364. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Leask A and Abraham DJ: All in the CCN
family: Essential matricellular signaling modulators emerge from
the bunker. J Cell Sci. 119:4803–4810. 2006. View Article : Google Scholar
|
|
42
|
Borkham-Kamphorst E, Schaffrath C, Van de
Leur E, Haas U, Tihaa L, Meurer SK, Nevzorova YA, Liedtke C and
Weiskirchen R: The anti-fibrotic effects of CCN1/CYR61 in primary
portal myofibroblasts are mediated through induction of reactive
oxygen species resulting in cellular senescence, apoptosis and
attenuated TGF-β signaling. Biochim Biophys Acta. 1843:902–914.
2014. View Article : Google Scholar
|