Introduction
The clinical utility of exome sequencing (ES) has
fundamentally deepened our knowledge of the molecular basis of
neurodevelopmental and other rare pediatric disorders. Current data
point to a growing number of genes and leading mechanisms
implicated in rare Mendelian disorders, requiring exome or genome
sequencing and additional diagnostics approaches on transcriptional
level such as RNA sequencing and epigenome analysis (1,2).
The BLM gene (15q26.1), also termed
RECQL3, encodes an intracellular nuclear protein belonging
to the RecQ family of 3′ to 5′ DNA helicases. It is a crucial
component of complex processes of the cell cycle regulation and DNA
repair which maintain the genome stability (3). At present, the ClinVar database
(https://www.ncbi.nlm.nih.gov/clinvar)
summarizes >350 pathogenic and/or likely pathogenic variants
altering the primary sequence of the BLM gene and resulting
in the typical phenotype of Bloom syndrome (retrieved March 27,
2023). The affected individuals manifest a microcephaly, severe
growth restriction, slender physique, general hypotrophy and
abnormal facial appearance including long narrow face, small lower
jaw and prominent nose and ears (4). They often suffer from immune
deficiency and insulin resistance. They have a dramatically
increased susceptibility for early-onset malignancies and lifetime
risk to develop any type of them due to the impaired functioning of
DNA repair machinery (4). The
absence of the functional BLM RecQ-like helicase results in a
chromosomal instability, excessive number of chromosomal breaks
with consequent homologous recombination leading to sister
chromatid exchanges (SCE) (5).
Case report
Case presentation
The proband was a two-year-old girl (born 2020) with
an initial clinical diagnosis of intrauterine growth restriction,
postnatal growth deficiency, general hypotrophy and abnormal facial
appearance. She is an only offspring of non-consanguineous couple
(mother born in 1993, father born in 1990) of Caucasian origin.
They are clinically healthy, both with family history lacking
consanguinity or any abnormal phenotypic features with a relevance
to that of the proband. The prenatal biochemical and ultrasound
screening tests in the first trimester were evaluated as normal.
Since the 24th week of pregnancy she had been monitored due to the
intrauterine growth restriction and amniotic fluid deficiency. She
was delivered prematurely after 36 weeks and 3 days of pregnancy
(1,570 g/39 cm). She had been under the medical supervision for 5
weeks, including 24-h phototherapy due to the icterus neonatorum
and three days in the incubator. She had been breast-fed for four
months. She has been doing Vojta therapy (6) for one year (between February 2020 and
February 2021). She was hospitalized due to a failure to thrive
(not gaining weight) in July 2020. She underwent the clinical
genetics evaluation at the age of 6 (July 2020) and 22 months
(November 2021). The last medical observation at 30 months (June
2022) summarized the short proportionate stature (75.4 cm; Z-score
−3.6) with a general paucity of subcutaneous fat (body weight 6,800
g; Z-score 0), microcephaly and dolichocephaly (head circumference
41 cm), normal intellect, speech and motor development, persistent
failure to thrive and difficulty in feeding. She manifested
distinct facial abnormalities (triangle face, bilateral epicanthus,
mild hypertelorism, narrow long nose, anteverted nostrils and long
philtrum), square-shaped palms and mild 5th finger clinodactyly. A
mild facial symmetric erythema had been observed, as well as two
Café-au-lait spots on the left thigh. The immunologic examination
has shown mildly decreased levels of B lymphocytes and
immunoglobulins (IgA, IgG and IgM). She is under the preventive
medical surveillance at the Clinic of Children's Oncology
(University Hospital Brno, Czech Republic). The recent preventive
examination using magnetic resonance excluded organomegaly, the
levels of tumor markers (alpha-fetoprotein, beta-human chorionic
gonadotropin, neuron-specific enolase) were assessed as normal. She
regularly undergoes medical examinations and counselling at
specialized clinics (including gastroenterology, immunology,
pediatrics and endocrinology). All procedures performed involving
human participants were in accordance with the ethical standards of
the institutional and/or national research committee and with the
1964 Helsinki declaration and its later amendments or comparable
ethical standards. Approval was obtained from the Research Ethics
Committee of Masaryk University (approval no. EKV-2019-056) and
Ethics Committee of University Hospital Brno (approval no.
10-120619/EK). Written informed consent was obtained from the
parents of the patient (proband) before the procedure of genetic
analyses.
Materials and methods
Cytogenetic and molecular cytogenetic
analysis
The peripheral blood samples of proband and her
parents were obtained for cytogenetic analysis of karyotype and for
DNA extraction [using the MagNA Pure 96 System (Roche Diagnostics,
Ltd.) according to the manufacturer's recommendations] for
molecular cytogenetic/genetic analyses. The cytogenetic analysis of
proband's karyotype was performed using the routine G-banding
procedure by Giemsa-Romanowski staining (7). The whole-genome screening of
submicroscopic copy-number variations (CNVs) and copy-number
neutral losses of heterozygosity (cnnLOH) was performed using the
oligonucleotide-based microarray platform SurePrint G3 ISCA v2
CGH+SNP 4X180K Microarray following the manufacturer's
recommendations (Agilent Technologies, Inc.). Microarray data were
extracted and processed to the CGH+SNP profile visualization using
the Agilent Cytogenomics 4.0.3. software (Agilent Technologies,
Inc.). CNVs were called using the ADM-2 algorithm with the settings
of at least three neighboring probes in a genomic region, a minimal
size of 100 kb and minimal absolute log2 ratio 0.25. The regions of
cnnLOH were evaluated at ~10 Mb resolution (LOH score ≥6) across
the entire genome.
Relative quantification using
quantitative (q)PCR
Relative quantification using qPCR was then
performed to verify the 15q11.2 microduplication, with two pairs of
DNA primers (Integrated DNA Technologies, Inc.) which were designed
to prime the DNA sequence within and outside the targeted CNV. The
reaction mixtures were prepared with PowerSYBR™ Green PCR MasterMix
(Thermo Fisher Scientific, Inc.) and run on a Light
Cycler® 480 Real-Time PCR System (Roche Diagnostics,
Ltd.) according to the manufacturer's recommendation. The
thermocycling conditions were as follows: 95°C/10 min (initial
denaturation), 40 cycles of 95°C/15 sec, 60°C/1 min (with data
acquisition in every cycle), denaturation at 95°C/15 sec, melting
curve generation from 60°C/1 min to 95°C with continuous data
acquisition, and then cooling. The relative quantification was
performed using the formula 2−ΔΔCq with the threshold
R-value <0.7 for DNA loss and >1.3 for DNA gain in the region
of interest relatively to the reference gene ERH (8). The sequences of the primers are
listed in Table SI (Sheet 1).
Methylation-specific MLPA
The methylation-specific multiplex
ligation-dependent probe amplification (MS-MLPA) analysis was
performed using the SALSA® MLPA® Probemix
ME030 BWS/RSS (MRC Holland). The data were analyzed using the
Coffalyser.Net software according to the manufacturer's
recommendations (MRC Holland).
Exome sequencing
High-quality of genomic DNA samples (~200 ng) were
used for the library preparation with the Human Core Exome kit,
which provides 33 Mb CCDS coverage with 99% ClinVar variants
coverage, with spiked-in Human RefSeq panel (Twist Bioscience) and
custom spiked-in probes for mitochondrial DNA. The DNA libraries
were then sequenced on Illumina NovaSeq 6000 (Illumina, Inc.). All
steps were performed as a commercially available service (Institute
of Applied Biotechnologies A.S.) according to the manufacturer's
recommendations.
Bioinformatic processing of ES
data
Raw sequencing data were processed to obtain
sequence variants, CNVs and cnnLOH, as described previously
(9). Briefly, the quality control
(QC) was performed using the FastQC v0.11.9 (https://www.bioinformatics.babraham.ac.uk/projects/fastqc/).
The low-quality reads and adapter contamination trimming was
performed by the fastp v0.20.1 (10). The remaining reads were aligned to
the reference human genome hg38/GRCh38 primary assembly by a
software package BWA v0.7.17-r1188 (11) with default parameters following by
marking duplicate reads and fix mate information using Picard tools
2.27.5 (http://broadinstitute.github.io/picard/). QC steps and
the coverage were checked using the in-house software Genovesa
(developed by Bioxsys, s.r.o; http://www.bioxsys.eu/#/genovesa). Single-nucleotide
variants (SNVs) and insertion/deletion variants (indels) were
called using the VarScan v2.4.4 (with parameters: Min-coverage, 20;
min-var-freq, 0.1; P-value, 0.5; min-avg-qual, 10) (12). The variant calling process is based
on a Fisher's Exact test, a statistical test procedure that
calculates an exact probability value for the relationship between
two dichotomous variables, as found in a two by two crosstable. The
program calculates the difference between read counts supporting
reference and variant alleles with P-value threshold 0.05. Only
SNVs and indels passing the quality filter (a minimal quality of
coverage ≥20X, base quality ≥10, mapping quality ≥5) and with an
alternative allele frequency ≥10% per sample, P-value (Fisher exact
test) <0.05 were included for further variant filtering.
CNVs were called using two different bioinformatics
pipelines. The first approach was based on the depth calculation
and normalization using the R software v3.6.0 (https://www.r-project.org/) in covered exons (13). Those exons which failed the
mapability criteria (lower than 0.75 defined using 35-mer
mapability score from UCSC genome browser) were excluded from the
analysis. The read depth coverage base line was created using ≥6
samples and then the algorithm compared each sample to each. The
ratio of expected reads to real number of reads was calculated to
estimate a gain or loss in any specific locus defined by
target.
The second approach was a custom pipeline CNVRobot
v3.5 (https://github.com/AnetaMikulasova/CNVRobot). Briefly,
GATK tools v4.2.4.1 (Broad Institute) were used for the processing
of bam files and data denoising. CNVs and losses of heterozygosity
(LOH) were called using a custom R-based segmentation and filtered
by parameters as follows: CNVs; ≥50 bp and two intervals; ≤-0.5
Log2 Ratio (L2R) for losses and >0.3 L2R for gains. LOH; ≥5 Mb
and 10,000 intervals. Unaffected unrelated sex-matching individuals
(31 males and 31 females) served as controls for data
denoising.
Variant prioritization and
classification
The filtering conditions were set to search for only
those sequence variants with ≥20% frequency (% of reads with the
variant) in the proband, the impact ‘moderate’ or ‘high’ based on
the Ensembl Variant Effect Predictor (v105) (14), allele frequencies ≤5% in the
non-Finnish European population (for known variants with
annotations) or with an unknown allele frequency (for novel
variants). Then Locus Reference Genomics (LRG) or Canonical
Transcripts were selected for reporting of clinically relevant
sequence variants. Only variants with a ‘pathogenic’ and ‘likely
pathogenic’ clinical impact based on the current version of the
ClinVar database (15) or
candidate novel variants in OMIM ‘morbid’ genes were considered for
further analysis. The pathogenicity of novel variants in OMIM
‘disease-causing’ genes was evaluated using the VarSome engine
including the current ACMG classification (16). The general information about genes
were obtained from the OMIM database (17). Only clinically relevant causative
variants were then reported to clinicians. CNVs were filtered
according to technical cut-offs: reads ratios ≤0.7 for losses, ≥1.3
for gains; log2 ratios ≤-0.5 for losses and ≥0.35 for gains. Then
CNVs encompassing OMIM ‘morbid’ or candidate ‘morbid’ genes or CNVs
classified as pathogenic or likely pathogenic in the dbVar database
(dbVar Genome Browser; v2.8) were prioritized for further analysis
(18). The presence of cnnLOH was
assessed after the manual curation to evaluate long stretches of
homozygous genotypes from the ES-based sequence variants analysis
and CGH+SNP microarray analysis.
Sanger sequencing
The presence of pathogenic sequence variants was
then confirmed using Sanger sequencing with two pairs of custom
primers: forward primer BLM_1_F 5′-CTGGGCTGAAACACCAAGAC-3′, reverse
primer BLM_1_R 5′-GCAGCTGTGGAAGATTTGCT-3′ and forward primer
BLM_2_F 5′-GCCCTGCCTGAGTTATGCT-3′, reverse primer BLM_2_R
5′-CCATTTGGGGTTTCTGGATGA-3′ (Integrated DNA Technologies) as
described in detail elsewhere (6).
The sequencing reactions were run on the capillary sequencer ABI
3130 (Thermo Fisher Scientific, Inc.) with further analyses using
the freeware FinchTV (Geospiza, Inc.).
Results
Quality control of technical
parameters of ES
On average, >90 million unique reads were mapped
to the reference genome GRCh38/hg38 primary assembly: ~99% of
targeted bases were covered to at least 30X and the median target
coverage was higher than 100X, reaching an essential quality for a
reliable evaluation and interpretation of ES outputs in the routine
molecular genetic diagnostics. The average proportion of flagged
PCR duplicates was only 16% and the average uniformity assessed
from all samples involved in a research project reached 1.37 which
is a good assumption for CNV analysis. The values of technical
parameters and QC metrics are available in the Table SII.
Cytogenetic analysis and molecular
cytogenetic analysis
The cytogenetic analysis of the proband's karyotype
was performed with a result of normal female karyotype 46,XX. She
immediately underwent the microarray analysis using the
oligonucleotide-based CGH+SNP microarray with a finding of a
recurrent 15q11.2 microduplication (BP1-BP2), classified as likely
benign (19). The parental testing
by qPCR proved its familial origin as it was confirmed in her
father and paternal grandfather. The outputs are available in
Table SI (Sheet 2). The MS-MLPA
analysis was performed with the SALSA® MLPA®
Probemix ME030 BWS/RSS (MRC Holland) due to the severe growth
restriction as a typical phenotypic manifestation of Silver-Russell
(SRS) or Silver-Russell syndrome-like phenotype (SRS-like). The
outputs proved the borderline hypermethylation H19 IC1 locus
(0.73-0.8) on chromosome 11p15. However, this result did not match
the SRS or SRS-like phenotype.
Trio-based ES
As the routine molecular genetic testing
(cytogenetic analysis and CGH+SNP microarray analysis) was
negative, the proband and her unaffected parents were enrolled for
trio-based ES. After the variant filtering and their evaluation
using the VarSome engine, medical and scientific literature and
databases two clinically relevant variants affecting the BLM
gene, c.1642C>T and c.2207_2212delinsTAGATTC (NM_000057.4), were
detected. Their compound heterozygosity was assessed for the
substitution c.1642C>T of maternal origin and deletion-insertion
c.2207_2212delinsTAGATTC of paternal origin (Fig. 1). These findings were then verified
by Sanger sequencing. No incidental reportable findings affecting
the ‘medically-actionable’ genes based on the ACMG recommendation
were detected (20).
The variant c.1642C>T in the exon 7 is a
well-known variant (rs200389141) with a non-Finnish European allele
frequency ~0.03-0.04% but rising to a carrier frequency of ~0.1% in
the Eastern Slavic population, which is the highest observed
frequency (4). Due to the
substitution c.1642C>T, the premature termination codon for a
nonsense variant p.(Gln548Ter) is predicted, however, the aberrant
transcripts are likely to be degraded by a nonsense-mediated mRNA
decay (NMD) pathway.
By contrast, the deletion-insertion
c.2207_2212delinsTAGATTC variant (rs113993962) in the exon 10 of
the BLM gene predominates as a founder allele in Ashkenazi
Jewish population and their descendants (BLMAsh),
reaching an estimated carrier frequency ~1% (21). It alters the DNA sequence of the
exon 10, which is translated to a part of the helicase ATP-binding
domain. The production of the truncated protein due to the
deletion-insertion c.2207_2212TAGATTC, p.(Tyr736LeufsTer5) is
prevented by the NMD pathway based on in silico prediction
tools.
The outputs of in silico prediction tools,
ACMG classification criteria and database records and literature
data confirming the pathogenicity of the BLM variants are
summarized in Table SIII (Sheets
1 and 2). The targeted analyses for the identification of
c.1642C>T and c.2207_2212delinsTAGATTC variants using Sanger
sequencing and the 15q11.2 microduplication (BP1-BP2) using qPCR
were performed in maternal and paternal relatives (Fig. 2). The pathogenicity of the 15q11.2
microduplication has been discussed elsewhere and the current
approach is to classify it as benign and not to report it (19). However, in our case report the
15q11.2 microduplication incidentally serves as a marker of a
probable meiotic crossing-over between it the BLM gene
(15q26.1) during the spermatogenesis of proband's father (Fig. 3).
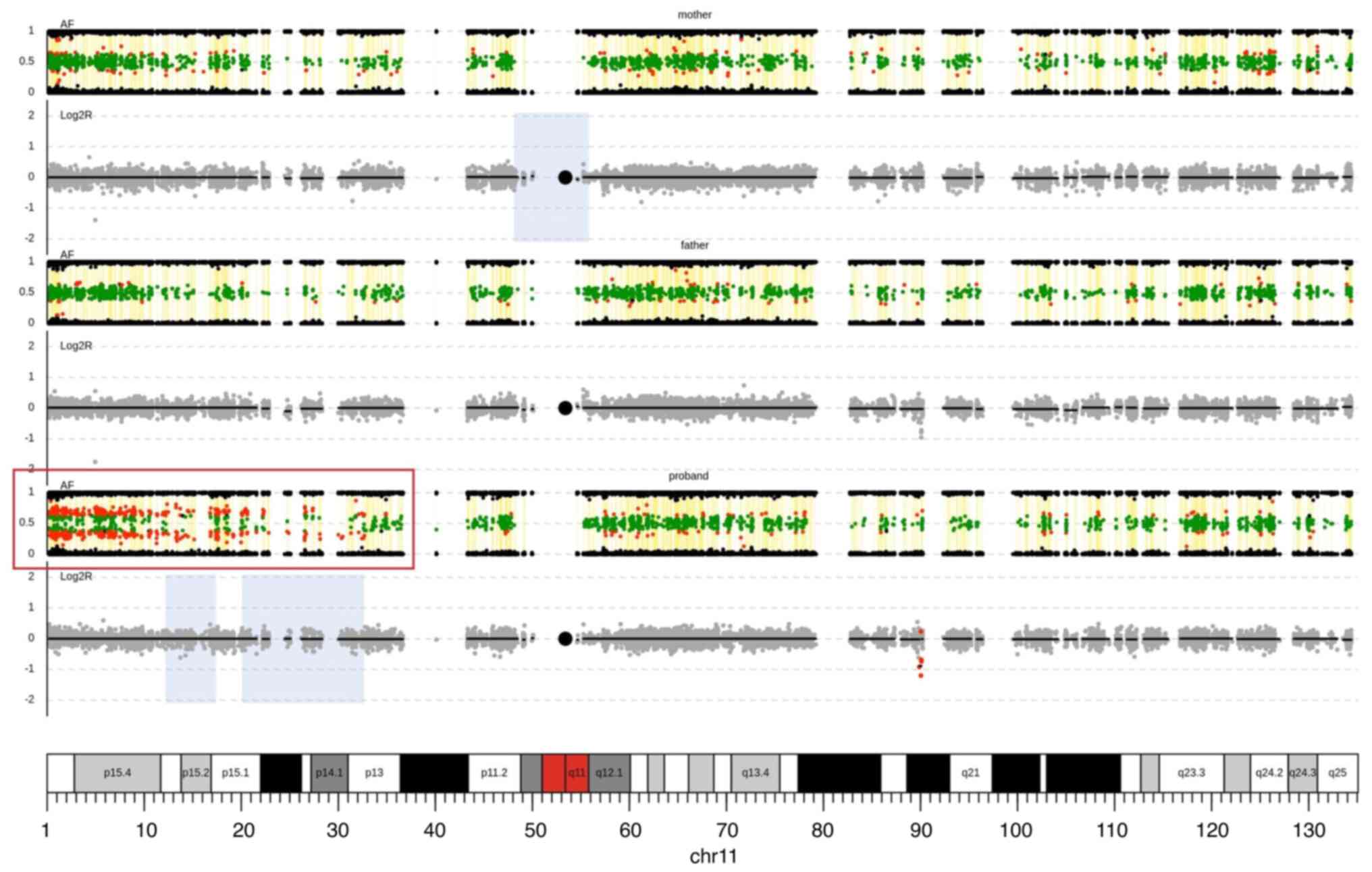
The simultaneous CNV and cnnLOH analysis from ES
data was performed, with a verification of the 15q11.2
microduplication in the proband and her father (Fig. S1), which was initially identified
by the microarray analysis. Moreover, an additional cnnLOH analysis
from ES data uncovered the somatic mosaic cnnLOH of chromosome 11p
which agrees with the output of MS-MLPA analysis; the borderline
hypermethylation H19 IC1 locus (Fig. 4). The mosaic cnnLOH of
chromosome11p is then evident due to the imbalances of the allelic
ratios for heterozygous SNVs on chromosome 11p in proband.
Discussion
The present case report provided a proof of a wide
diagnostic utility of trio-based ES in the molecular genetic
diagnostics of pediatric rare diseases. The simultaneous detection
of sequence variants, CNVs and cnnLOH identified a unique
co-occurrence of compound heterozygous rare pathogenic variants of
the BLM gene and mosaic cnnLOH on chromosome 11p. These two
clinically relevant genetic entities represent important medically
actionable issues for the utility of personalized medicine.
The BLM gene is located on 15q26.1 chromosome
and provides instruction for ATP-dependent RecQ helicase. It is a
component of BRCA1-associated genome surveillance complex, which
plays multiple roles in the DNA damage response to maintain the
genomic stability (4). It unwinds
single-strand (ss)- and double-strand (ds)DNA in a 3′ to 5′
direction, participates in DNA replication and in the repair of
double-strand breaks. The BLM RecQ-like helicase prevents SCE
events, therefore their identification in metaphases is a
cytogenetic marker of Bloom syndrome and other syndromes of the
chromosomal instability (22).
The BLM gene structure is divided into
several domains providing the key effector functions, which
disruptions lead to an increased cellular sensitivity for DNA
damage. Therefore, the BLM gene is highly expressed in
rapidly proliferating cells and is cell-cycle regulated, reaching
the highest level in the late S and G2 phases (4).
The pathogenic variants including missense and
truncating sequence variants and intragenic deletions of the
BLM gene have been confirmed as the molecular genetic cause
of Bloom syndrome, an autosomal recessive disorder affecting
multiple body tissues and organ systems. The Bloom Syndrome
registry (23) and other medical
literature provide details about ~300 reported cases of individuals
with Bloom syndrome since 1954, when the first case of Bloom
syndrome was documented. To the best of the authors' knowledge, the
BLM gene is the only gene responsible for the clinical
manifestation of Bloom syndrome. However, there are three other
genes RMI1, RMI2 and TOP3A encoding proteins, and
which form a complex with the BLM RecQ-like helicase. Their
pathogenic variants can cause a milder phenotype than is observed
in individuals with Bloom syndrome (‘Bloom syndrome-like’
phenotype) (24). Therefore, it is
suggested that individuals in the Bloom syndrome registry lacking
molecular diagnosis may have causative variants in other genes as
RMI1, RMI2, or TOP3A genes with the overlapping
phenotypic manifestation. Recently, novel deep intronic variant
leading to a pseudo-exon activation has been detected using
RNA-based long-range PCR in an individual with Bloom syndrome and
only one causative variant in the BLM gene which was
detected in the previous analysis (25). Therefore, novel approaches
including genome sequencing or transcriptome analysis may complete
the molecular diagnosis of Bloom syndrome in those individuals with
the phenotypic manifestation of Bloom syndrome in which only one
causative variant in the BLM gene was detected using the
sequencing analysis of its coding region (23,25).
Moreover, a study using single-cell transcriptomic profiling
uncover an altered transcriptional profile and suggested novel
links between BLM helicase dysfunction and aberrant transcription
of condensin complexes genes (26).
To the best of the authors' knowledge, >350
different causative variants of the BLM gene have been
identified as causative, including some founder variants with a
higher frequency in certain populations or ethnic groups (15,23).
The recurrent variant c.1642C>T is enriched in
the Eastern Europe population of the Slavic origin, in which
0.2-0.6% individuals are its carriers. Only a few patients with
Bloom syndrome carrying homozygous c.1642C>T variant are
described in a scientific literature, probably due to the
incomplete phenotypic manifestation lacking the presence of a
typical UV exposure-induced facial erythema (27,28).
It raises the hypothesis of the underdiagnosis of Bloom syndrome at
the clinical and molecular level in this population. The variant
affects the protein region which interacts with the scaffolding
protein involved in DNA repair (SPIDR). SPIDR interconnects BLM and
RAD51 proteins and targets them to sites of DNA damage (29). Certain studies show the association
between the c.1642C>T variant and an increased risk for breast
cancer (0.5–1% breast cancer in Slavic population) (30,31).
The recurrent variant c.2207_2212delinsTAGATTC has
been observed as a founder allele in Ashkenazi Jews and their
descendants with a frequency of 1%, therefore the term
BLMAsh is widely used. Due to the migration and
founder effect, it has been established independently in different
regions worldwide. The estimated prevalence of Bloom syndrome in
the Ashkenazi Jewish population reaches ~1:48 000, but it occurs
extremely rarely in the general population (32). The carriers of the
BLMAsh allele may come up against an increased
risk of developing any type of malignancy; however, no significant
association has been observed so far.
Our proband is a compound heterozygote for these two
founder alleles, maternally inherited c.1642C>T and paternally
inherited c.2207_2212delinsTAGATTC. The frequencies of both
pathogenic variants of BLM gene are rare in the general Caucasian
population, however, they occur in higher frequencies in certain
populations (c.1642C>T in Slavic population and
c.2207_2212delinsTAGATTC in the population of Ashkenazi Jews and
their descendants) (4,21). The compound heterozygosity for
these variants is extremely rare which is documented by a rare
frequency of Bloom syndrome in the general Caucasian population.
The worldwide incidence of Bloom syndrome due to biallelic
causative variants in the BLM gene is unknown, ~300 cases
have been reported so far in databases and in the medical
literature. Although its prevalence in the population of Ashkenazi
Jews is estimated to be ~1:48,000, only ~1/3 of individuals with
Bloom syndrome due to the causative variants in the BLM gene
are of Ashkenazi Jewish descent.
Another documented case of an infant carrying these
two causative BLM gene variants in the compound
heterozygosity has been published recently (33). He was diagnosed with Bloom syndrome
at the age of 9 years, but he developed an infantile fibrosarcoma
at 6 months. His case demonstrates a dramatically increased risk
for childhood malignancies in individuals with Bloom syndrome and
points out the importance of multidisciplinary medical long-term
follow up.
In addition, our proband is a carrier of a mosaic
cnnLOH of chromosome 11p corresponding to the borderline imprinting
center 1 (IC1) hypermethylation (0.73-0.8). The IC1 (H19
gene) hypermethylation of chromosome 11p15 increases the risk for
Wilms tumor due to the biallelic expression of the IGF2 gene
(34). The IC1 hypermethylation
has been observed in 5–10% of Beckwith-Wiedemann patients and among
the molecular subgroups of BWS represents an increased risk to
develop a malignancy of a kidney (Wilms tumor) or liver
(hepatoblastoma) (35). Although
the cnnLOH analysis from ES data indicates the cnnLOH of chromosome
11p15p13, mosaic IC2 (KCNQ1OT1) hypomethylation using the
MS-MLPA analysis was not detected most likely due to the inability
of MS-MLPA to identify low-level mosaic imprinting defects
(36). Most individuals affected
by BWS or SRS are affected by mosaic imbalances of IC1 and IC2 on
chromosome 11p15 (37). The
borderline mosaic cnnLOH 11p may be a result of a defective
homologous recombination due to the aberrant double-strand break
repair caused by the dysfunction of the BLM RecQ-like helicase. The
increased frequency of SCE events and mosaic cnnLOH are typical
markers of Bloom syndrome (22,38).
The initial clinical diagnosis of our proband was
SRS due to the severe prenatal and postnatal growth restriction.
The microarray CGH+SNP array and BWS/RSS MS-MLPA excluded that
diagnosis. The subsequent analysis using trio-based ES elucidated
the diagnosis of Bloom syndrome. As well as previously documented
in some cases of Bloom syndrome, some of the cases do not manifest
typical features, such as sun-induced, butterfly-shaped skin
lesions, which would have led to a clinical misdiagnosis (27,39).
The mosaicism for IC1 (H19 gene) hypermethylation and
differences of has been observed in a subset of patients with BWS
in a risk for embryonal tumors in early childhood (40). The distribution of chromosome 11p15
mosaicism for methylation changes can significantly vary between
tissues, so additional tissue-specific testing may be valuable in
personalized medical intervention (41).
A diagnosis of Bloom syndrome carries a greatly
increased risk to develop early-onset malignancies and then an
increased life-time risk to develop multiple malignancies due to
the genome instability. Therefore, the co-existence of
cancer-predisposing Bloom syndrome and risk factors resulting from
the IC1 11p15 hypermethylation due to the mosaic cnnLOH of
chromosome 11p could classify our proband as a highly-risk
individual requiring the multidisciplinary medical and therapeutic
observation and prospective medical intervention (42).
A rapid molecular genetic diagnostics using
trio-based ES for the simultaneous detection of sequence variants,
CNVs and cnnLOH improves the quality of medical care due to the
early medical surveillance, interventions and optimal setting of a
specialized healthcare of pediatric patients with rare diseases
with an adverse prognosis.
Supplementary Material
Supporting Data
Supporting Data
Supporting Data
Supporting Data
Acknowledgements
Not applicable.
Funding
The present study was financially supported by Ministry of
Health of the Czech Republic, grant no. NU20-07-00145.
Availability of data and materials
The datasets generated for the present case report,
including raw and processed ES outputs of proband and her parents,
microarray analysis and qPCR are not publicly available due to the
protection of individuals' privacy, but are available from the
corresponding author on reasonable request. The full visualization
of presented BLM gene variants (IGV software v2.8.13), Sanger
sequencing data, including the chromatograms of the proband, her
parents and their relatives for the BLM pathogenic variants and the
MS-MLPA outputs for the borderline H19 hypermethylation in the
proband are stored in the Figshare online digital repository under
DOI: doi.org/10.6084/m9.figshare.19727653.
Authors' contributions
MW, VV, PB, AM, DM, HDF, JS, AT, RG and PK
contributed to the study conception and design. MW, VV and DM
analyzed and interpreted the ES data regarding the proband
phenotype using the relevant literature, databases and in
silico tools. MW was a major contributor in the writing of the
manuscript. PB processed raw ES data using advanced bioinformatics
tools through the in-house developed pipeline for sequence variants
and CNV calling and the variant annotation. AM performed CNV and
cnnLOH calling from processed ES data. PB and AM wrote and revised
the part of the manuscript regarding to the bioinformatic
processing of ES data. VV and JS performed and interpreted the
microarray analysis. DM and HDF performed Sanger sequencing, qPCR
and MLPA verification analyses. AT and RG provided the specialized
genetic counselling for the proband and her family and interpreted
the laboratory findings in the clinical context. PK contributed
towards the interpretation of data and performed the general
scientific supervision and general critical revision of the
manuscript. MW and PB confirm the authenticity of all the raw data.
All authors discussed the results, reviewed and approved the final
manuscript.
Ethics approval and consent to
participate
All procedures performed involving human
participants were in accordance with the ethical standards of the
institutional and/or national research committee and with the 1964
Helsinki declaration and its later amendments or comparable ethical
standards. Approval was obtained from the Research Ethics Committee
of Masaryk University (approval no. EKV-2019-056) and Ethics
Committee of University Hospital Brno (approval no. 10-120619/EK).
Written informed consent was obtained from the parents of the
patient (proband) and all the adult relatives before the procedure
of genetic analyses.
Patient consent for publication
The patient's legal guardian and all the adult
relatives signed the informed consent form, including the consent
for publication and for the use of the data.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Posey JE: Genome sequencing and
implications for rare disorders. Orphanet J Rare Dis. 14:1532019.
View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Cummings BB, Marshall JL, Tukiainen T, Lek
M, Donkervoot S, Foley AR, Bolduc V, Waddell LB, Sandaradura SA,
O'Grady GL, et al: Improving genetic diagnosis in Mendelian disease
with transcriptome sequencing. Sci Transl Med. 9:eaal52092017.
View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Patel DS, Misenko SM, Her J and Bunting
SF: BLM helicase regulates DNA repair by counteracting RAD51
loading at DNA double-strand break sites. J Cell Biol.
216:3521–3534. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Cunniff C, Bassetti JA and Ellis NA:
Bloom's syndrome: Clinical spectrum, molecular pathogenesis, and
cancer predisposition. Mol Syndromol. 8:4–23. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Traverso G, Bettergowda C, Kraus J,
Speicher MR, Kinzler KW, Vogelstein B and Lengauer C:
Hyper-recombination and genetic instability in BLM-deficient
epithelial cells. Cancer Res. 63:8578–8581. 2003.PubMed/NCBI
|
|
6
|
Vojta V: Rehabilitation des spastischen
infantilen syndroms. Eigene Methodik. Orthop Traumat. 12:557–562.
1965.
|
|
7
|
Howe B, Umrigar A and Tsien F: Chromosome
preparation from cultured cells. J Vis Exp.
28:e502032014.PubMed/NCBI
|
|
8
|
Livak KJ and Schmittgen TD: Analysis of
relative gene expression data using real-time quantitative PCR and
the 2(−Delta Delta-C(T)) method. Methods. 25:402–408. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Wayhelova M, Vallova V, Broz P, Mikulasova
A, Loubalova D, Filkova H, Smetana J, Drabova K, Gaillyova R and
Kuglik P: Novel de novo pathogenic variant in the GNAI1 gene as a
cause of severe disorders of intellectual development. J Hum Genet.
67:209–214. 2022. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Chen S, Zhou Y, Chen Y and Gu J: fastp: An
ultra-fast-all-in-one FASTQ preprocessor. Bioinformatics.
34:i884–i890. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Li H and Durbin R: Fast and accurate short
read alignment with Burrows-Wheeler transform. Bioinformatics.
25:1754–1760. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Koboldt DC, Chen K, Wylie T, Larson DE,
McLellan MD, Mardis ER, Weinstock GM, Wilson RK and Ding L:
VarScan: Variant detection in massively parallel sequencing of
individual and pooled samples. Bioinformatics. 25:2283–2285. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
13
|
R Core Team (2020), . R: A language and
environment for statistical computing. R Foundation for Statistical
Computing; Vienna, Austria:
|
|
14
|
McLaren W, Gil L, Hunt SE, Riat HS,
Ritchie GRS, Thormann A, Flicek P and Cunningham F: The ensembl
variant effect predictor. Genome Biol. 17:1222016. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Landrum MJ, Lee JM, Benson B, Brown GR,
Chao C, Chitipiralla S, Gu B, Hart J, Hoffman D, Jang W, et al:
ClinVar: Improving access to variant interpretations and supporting
evidence. Nucleic Acids Res. 46:D1062–D1067. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Kopanos C, Tsiolkas V, Kouris A, Chapple
CE, Aguillera MA, Meyer R and Massouras A: VarSome: The human
genomic variant search engine. Bioinformatics. 35:1978–1980. 2019.
View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Hamosh A, Scott AF, Amberger JS, Bocchini
CA and McKusick VA: Online mendelian inheritance in man (OMIM), a
knowledgebase of human genes and genetic disorders. Nucleic Acids
Res. 33:D514–D517. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Lappalainen I, Lopez J, Skipper L,
Hefferson T, Spalding JD, Garner J, Chen C, Maguire M, Corberr M,
Zhou G, et al: DbVar and DGVa: Public archives for genomic
structural variation. Nucleic Acids Res. 41:D936–D941. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Kendall KM, Bracher-Smith M, Fitzpatrick
H, Lynham A, Rees E, Escott-Price V, Owen MJ, O'Donovan MC, Walters
JTR and Kirov G: Cognitive performance and functional outcomes of
pathogenic copy number variants: Analysis of the UK Biobank. Br J
Psychiatry. 214:297–304. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Miller DT, Lee K, Abul-Husn NS, Amendola
LM, Brothers K, Chung WK, Gollob MH, Gordon AS, Harrison SM,
Hershberger RE, et al: ACMG SF v3.1 list for reporting of secondary
findings in clinical exome and genome sequencing: A policy
statement of the American College of medical genetics and genomics
(ACMG). Genet Med. 24:1407–1414. 2022. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Li L, Eng C, Desnick RJ, German J and
Ellis NA: Carrier frequency of the Bloom syndrome blmAsh mutation
in the Ashkenazi Jewish population. Mol Genet Metab. 64:286–290.
1998. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Montenegro MM, Quaio CR, Palmeira P,
Gasparini Y, Rangel-Santos A, Damasceno J, Novak EM, Gimenez TM,
Yamamoto GL, Ronjo RS, et al: Gene expression profile suggesting
immunological dysregulation in two Brazilian Bloom's syndrome
cases. Mol Genet Genomic Med. 8:e11332020. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
German J, Sanz MM, Ciocci S, Ye TZ and
Ellis NA: Syndrome-causing mutations of the BLM gene in persons in
the Bloom's syndrome registry. Hum Mutat. 28:743–753. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Gönenc II, Elcioglu NH, Grijalva CM, Aras
S, Großmann N, Praulich I, Altmüller J, Kaulfuß S, Li Y, Nürnberg
P, et al: Phenotypic spectrum of BLM- and RMI1-related Bloom
syndrome. Clin Genet. 101:559–564. 2022. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Backers L, Parton B, De Bruyne M,
Tavernier SJ, Van Den Bogaert K, Lambrecht BN, Haerynck F and Claes
KBM: Missing heritability in Bloom syndrome: First report of a deep
intronic variant leading to pseudo-exon activation in the BLM gene.
Clin Genet. 99:292–297. 2021. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Gönenc II, Wolff A, Schmidt J, Zibat A,
Müller C, Cyganek L, Argyriou L, Räschle M, Yigit G and Wollnik B:
Single-cell transcription profiles in Bloom syndrome patients link
BLM deficiency with altered condensin complex expression
signatures. Hum Mol Genet. 31:2185–2193. 2022. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Suspitsin EN, Sibgatullina FI, Lyazina LV
and Imyanitov EN: First two cases of Bloom syndrome in Russia: Lack
of skin manifestation in a BLM c.1642C>T (p.Q548X) homozygote as
a likely cause of underdiagnosis. Mol Syndromol. 8:103–106. 2017.
View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Trizuljak J, Petruchová T, Blaháková I,
Vrzalová Z, Hořínová V, Doubková M, Michalka J, Mayer J,
Pospíšilová Š and Doubek M: Diagnosis of Bloom syndrome in a
patient with short stature, recurrence of malignant lymphoma, and
consanguineous origin. Mol Syndromol. 11:73–82. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Wan L, Han J, Liu T, Dong S, Xie F, Chen H
and Huang J: Scaffolding protein SPIDR/KIAA0146 connects the Bloom
syndrome helicase with homologous recombination repair. Proc Natl
Acad Sci USA. 110:10646–10651. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Sokolenko AP, Iyevleva AG,
Preobrazhenskaya EV, Mitiushkina NV, Abysheva SN, Suspitsin EN,
Kuligina ES, Gorodnova TV, Pfeifer W, Togo AV, et al: High
prevalence and breast cancer predisposing role of the BLM
c.1642C>T (Q548X) mutation in Russia. Int J Cancer.
130:2867–2873. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Prokofyeva D, Bogdanova N, Dubrowinskaja
N, Bermisheva M, Takhirova Z, Antonenkova N, Turmanov N, Datsyuk I,
Gantsev S, Christiansen H, et al: Nonsense mutation p.Q548X in BLM,
the gene mutated in Bloom's syndrome, is associated with breast
cancer in Slavic population. Breast Cancer Res Treat. 137:533–539.
2013. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Shahrabani-Gargir L, Shomrat R, Yaron Y,
Orr-Urteger A, Groden J and Legum C: High frequency of a common
Bloom syndrome Ashkenazi mutation among Jews of polish origin.
Genet Test. 2:293–296. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Huson SM, Staab T, Pereira M, Ward H,
Paredes R, Evans DG, Baumhoer D, O'Sullivan J, Cheesman E,
Schindler D and Meyer S: Infantile fibrosarcoma with TPM3-NTRK1
fusion in a boy with Bloom syndrome. Fam Cancer. 21:85–90. 2022.
View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Fiala EM, Ortiz MV, Kennedy JA, Glodzik D,
Fleischut MH, Duffy KA, Hathaway ER, Heaton T, Gerstle JT,
Steinherz P, et al: 11p15.5 epimutations in children with Wilms
tumor and hepatoblastoma detected in peripheral blood. Cancer.
126:3114–3121. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Ibrahim A, Kirby G, Hardy C, Dias RP, Tee
L, Lim D, Berg J, MacDonald F, Nightingale P and Maher ER:
Methylation analysis and diagnostics of Beckwith-Wiedemann syndrome
in 1,000 subjects. Clin Epigenetics. 6:112014. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Van Veghel-Plandsoen MM, Wouters CH,
Kromosoeto JNR, den Ridder-Klünnen MC, Halley DJJ and van den
Ouweland AMW: Multiplex ligation-depending probe amplification is
not suitable for detection of low-grade mosaicism. Eur J Hum Genet.
19:1009–1012. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Brioude F, Kalish JM, Mussa A, Foster AC,
Bliek J, Ferrero GB, Boonen SE, Cole T, Baker R, Bertoletti M, et
al: Expert consensus document: Clinical and molecular diagnosis,
screening and management of Beckwith-Wiedemann syndrome: An
international consensus statement. Nat Rev Endocrinol. 14:229–249.
2018. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
LaRocque JR, Stark JM, Oh J, Bojilova E,
Yusa K, Horie K, Takeda J and Jasin M: Interhomolog recombination
and loss of heterozygosity in wild-type and Bloom syndrome helicase
(BLM)-deficient mammalian cells. Proc Natl Acad Sci USA.
108:11971–11976. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Bouman A, van Koningsbruggen S,
Karakullukcu MB, Schreuder WH and Lakeman P: Bloom syndrome does
not always present with sun-sensitive facial erythema. Eur J Med
Genet. 61:94–97. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
MacFarland SP, Duffy KA, Bhatti TR,
Bagatell R, Balamuth NJ, Brodeur GM, Ganguly A, Mattei PA, Surrey
LF, Balis FM and Kalish JM: Diagnosis of beckwith-wiedemann
syndrome in children presenting with wilms tumor. Pediatr Blood
Cancer. 65:e272962018. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Alders M, Maas SM, Kadouch DJM, var der
Lip K, Bliek J, van der Horst CMAM and Mannens MMAM: Methylation
analysis in tongue tissue of BWS patients identifies the
(EPI)genetic cause in 3 patients with normal methylation levels in
blood. Eur J Med Genet. 57:293–297. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Campbell MB, Campbell WC, Rogers J, Rogers
N, Rogers Z, van den Hurk AM, Webb A, Webb T and Zaslaw P: Bloom
syndrome: Research and data priorities for the development of
precision medicine as identified by some affected families. Cold
Spring Harb Mol Case Stud. 4:a0028162018. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Robinson JT, Thorvaldsdóttir H, Winckler
W, Guttman M, Lander ES, Getz G and Mesirov JP: Integrative
Genomics Viewer. Nat Biotechnol. 29:24–26. 2011. View Article : Google Scholar : PubMed/NCBI
|