Introduction
The endoplasmic reticulum (ER) plays an important
role in the folding and maturation of newly produced secretory and
membrane proteins (1). Although
the ER controls the quality of newly generated proteins,
pathological conditions such as renal disease, diabetes and
atherosclerosis result in the accumulation of misfolded and
unfolded proteins in the ER (2).
This leads to a condition called ER stress, and the protective
response to ER stress is called the unfolded protein response
(UPR). The UPR has three branches: Protein kinase R (PKR)-like ER
kinase (PERK), inositol-requiring enzyme 1 and activating
transcription factor (ATF) 6 (3).
In response to ER stress, the UPR upregulates ER chaperones to
restore normal cell function (4).
However, when ER stress is excessive and/or prolonged, the UPR
initiates apoptotic processes (5–7). One
of the key factors responsible for apoptotic induction downstream
of ER stress is the CCAAT/enhancer binding protein (C/EBP)
homologous protein (CHOP). CHOP is upregulated by ATF4, a
transcription factor that responds to ER stress (8).
Glutathione-specific γ-glutamylcyclotransferase 1
(CHAC1) was identified as a UPR-induced gene (9). CHAC1 is a pro-apoptotic factor
downstream of ER stress (9,10).
In addition to its pro-apoptotic function, CHAC1 degrades
intracellular glutathione, contributing to redox homeostasis in the
cell (10,11). Because CHAC1-mediated degradation
of glutathione enhances necroptosis and ferroptosis, which are
previously identified mechanisms of cell death (12), it is considered that CHAC1 has
important roles in cell death. CHAC1 transcription is regulated by
ATF4, ATF3 and C/EBPβ, which are transcription factors responsive
to ER stress (9,11,13).
Thus, CHAC1 is regulated by several transcription factors. However,
the signaling pathways that regulate CHAC1 expression remain
unclear.
Renal ischemia-reperfusion injury (IRI) induces a
remarkable reduction in glutathione levels in the kidney (14). In addition, IRI induces ER stress
in the kidney (15). These results
indicated that CHAC1 is expressed in renal tubular cells. In the
present study, CHAC1 regulation was investigated in tubular cells.
It was observed that 3-(5′-hydroxymethyl-2′-furyl)-1-benzylindazole
(YC-1), an activator of soluble guanylyl cyclase (sGC) (16), increases CHAC1 expression in
cultured human kidney tubular cells (HK-2). Therefore, in the
present study, the signaling pathways that contribute to CHAC1
induction by YC-1 in HK-2 cells were investigated. The results
revealed that the increase in CHAC1 expression by YC-1 occurred via
the cGMP-dependent protein kinase (PKG), ER stress and AKT-mTOR
pathways. It was also observed that CHAC1 upregulation decreased
intracellular glutathione concentration in HK-2 cells. These
findings indicated that CHAC1 is regulated by various signaling
pathways and is involved in cellular redox homeostasis.
Materials and methods
Antibodies, reagents and kits
Anti-CHAC1 antibody (1:1,000; cat. no. AV42623) was
purchased from Sigma-Aldrich; Merck KGaA. Anti-ATF4 was obtained
from Santa Cruz Biotechnology, Inc (1:1,000; cat. no. sc200).
Anti-CHOP (1:1,000; cat. no. 2895) and anti-β-actin (1:5,000: cat.
no. 4970) antibodies were purchased from Cell Signaling Technology,
Inc. The YC-1 (cat. no. 82560) and KT5823 (cat. no. 10010965) were
obtained from Cayman Chemical Company. Tunicamycin (cat. no.
202-08241) was obtained from Fujifilm Wako Chemicals. LY294002
(cat. no. 440202), KU0063794 (cat. no. SML0382) and SB203580 (cat.
no. S8307) were purchased from Sigma-Aldrich; Merck KGaA. BCA
protein assay kit (cat. no. T9300A) was purchased from Takara Bio,
Inc. The DetectX glutathione fluorescence detection kit (cat. no.
K006-F1) was purchased from Arbor Assays. The PathScan
intracellular signaling array kit (cat. no. 7323) was purchased
from Cell Signaling Technology, Inc.
Cell culture and chemical
treatment
HK-2 cell, which is a human kidney tubular cell was
purchased from the American Type Culture Collection (cat. no.
CRL-2190). HK-2 cells were cultured at 37°C in a humidified
atmosphere with 5% CO2 until 80% confluency in DMEM
(Nissui Corporation) containing 10% fetal bovine serum (Serana
Europe GmbH), 100 units/ml penicillin and 100 µg/ml streptomycin.
For the chemical treatment of HK-2 cells, the cells were cultured
in serum-free DMEM for 24 h and then treated with YC-1 or
tunicamycin. When the chemical compounds, including 50 µM LY294002,
10 µM KU0063794 and 10 µM SB203580, were used, the compounds were
added just before YC-1 treatment and were incubated for 4 h at
37°C. The concentrations of LY294002, KU0063794 and SB20358 were
referred to previous studies (17–19).
Almost all cells after 24-h incubation in serum-free medium
attached on the bottom of the culture plate, indicating that the
incubation did not induce apoptosis.
Western blotting
Cells were lysed in lysis buffer [20 mM Tris-HCl (pH
8.0), 0.15 M NaCl, 10 mM EDTA, 1% Triton X-100, 1 mM
phenylmethylsulfonyl fluoride, protease inhibitor mix (GE
Healthcare)]. The protein concentration in the lysates was measured
using a BCA protein assay kit. The proteins (25 µg/lane) were
separated on 7.5 or 15% SDS polyacrylamide gels and transferred
onto polyvinylidene fluoride membranes. The membranes were blocked
with TS solution [150 mM NaCl, 20 mM Tris-HCl (pH 8.0)] containing
1% non-fat dry milk for 30 min at room temperature, and then
incubated with a primary antibody overnight at 4°C and probed with
an HRP-conjugated secondary antibody (KPL, Inc.) for 1 h at room
temperature. Immunoreactive bands were detected using ImmunoStar LD
(FUJIFILM Wako Pure Chemical Corporation) and visualized using a
WSE-6200H LuminoGraph II (ATTO Corporation). The proteins were
quantified by conducting densitometric analysis using ImageJ
software 1.41o (National Institutes of Health).
Reverse transcription-quantitative PCR
(RT-qPCR)
Total RNA was isolated from the HK-2 cells using
ISOGEN (Nippon Gene Co., Ltd.). Total RNA was subjected to qPCR
using the PrimeScript RT reagent Kit with Oligo dT Primer (cat. no.
RR047A; Takara Bio, Inc) and THUNDERBIRD SYBR qPCR Mix (cat. no.
QPS-201; Toyobo). The reverse transcription was performed according
to the manufacturer's instructions; cDNA was synthesized by
incubation at 37°C for 15 min. The following primers were used for
the analyses of CHAC1 mRNA levels: CHAC1 forward,
5′-GTGGTGACGCTCCTTGAAGA-3′ and reverse, 5′-TTCAGGGCCTTGCTTACCTG-3′;
and 36B4 forward, 5′-TCGACAATGGCAGCATCAC-3′ and reverse,
5′-TGATGCAACAGTTGGGTAGC-3′. 36B4 was used for the internal control.
The thermocycling conditions consisted of a denaturation at 95°C
for 20 sec, followed by 45 cycles of denaturation at 95°C for 15
sec, annealing at 60°C for 15 sec and extension at 72°C for 20 sec.
CHAC1 expression levels were calculated with the 2−ΔΔCq
method (20).
Analysis of signaling pathways
To investigate the signaling pathway in HK-2 cells
treated with YC-1, a PathScan intracellular signaling array kit
(cat. no. 7323; Cell Signaling Technology, Inc) was used according
to the manufacturer's instructions. HK-2 cells were incubated at
37°C in serum-free DMEM medium for 24 h. Subsequently, cells were
treated with 1 µM YC-1 at 37°C for 2 and 4 h. After washing the
cells with ice cold PBS, HK-2 cells were lysed in lysis buffer
supplemented with 1 mM phenylmethylsulfonyl fluoride and protease
inhibitor mix. The lysates were diluted to 1.0 mg/ml in Array
Diluent Buffer. A total of 70 µl of the lysate were added to each
well of the Array Slide and incubated overnight at 4°C.
Chemiluminescence was detected using a chemiluminescent film (GE
Healthcare). Chemiluminescence intensity was measured using ImageJ
analysis software.
Analysis of the intracellular
concentration of glutathione
A glutathione fluorescence detection kit (K006-F1;
Arbor Assays) was used according to the manufacturer's
instructions. Briefly, HK-2 cells reaching 70% confluency were
washed with PBS and lysed with a 5% 5-sulfo-salicylic acid
solution. After incubating the lysed cells at 4°C for 10 min, they
were centrifuged at 20,600 × g for 10 min at 4°C. The prepared
lysates were diluted 1:5 in an assay buffer and fluorescence
intensities were measured using a Varioskan Flash spectral scanning
multimode reader (Thermo Fisher Scientific, Inc.). Small
interfering (si)CHAC1 (cat. no. sc-90164) and control siRNAs (cat.
no. sc-37007) were obtained from Santa Cruz Biotechnology, Inc. The
sequences of CHAC1 siRNA and control siRNA are not open in the
manufacturer's instraction. After changing culture medium to DMEM
without serum, siRNAs (12.5 nM final concentration) were
simultaneously transfected using Lipofectamine RNAiMAX (Thermo
Fisher Scientific, Inc.) and incubated at 37°C for 24 h.
Statistical analysis
Data are presented as the mean ± standard error of
the mean. GraphPad Prism 6.0d (Graphpad Software: Dotmatics) was
used for statistical analysis. Statistical significance was
assessed using unpaired Student's t-test or one-way ANOVA followed
by Tukey's post hoc test, where values of P<0.05 were considered
to indicate a statistically significant difference.
Results
CHAC1 is induced by YC-1
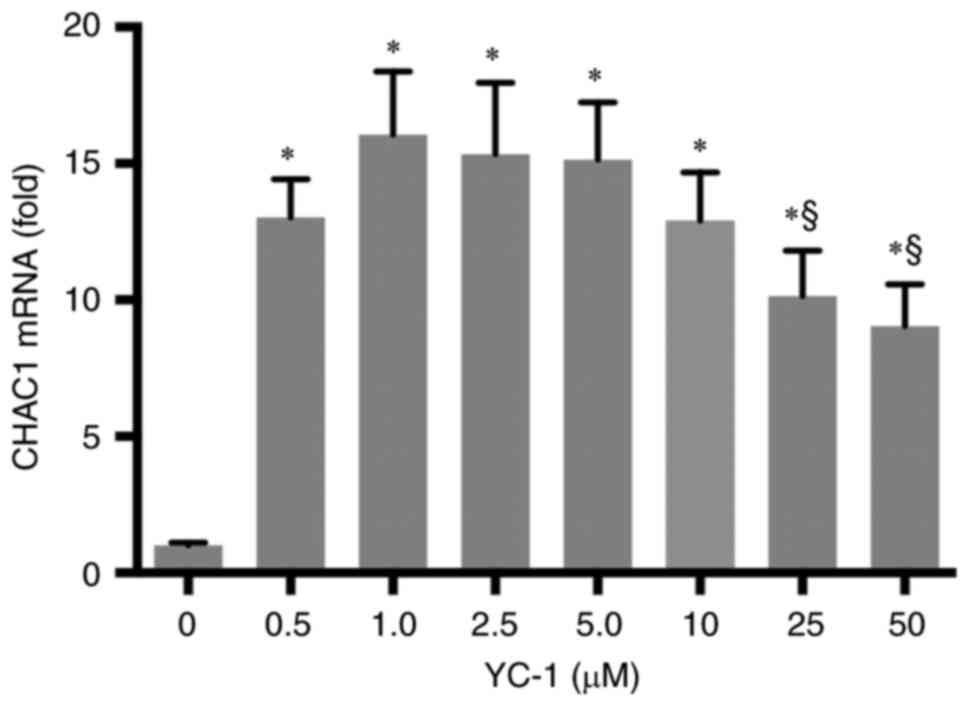
YC-1 treatment upregulated CHAC1 expression in the
human kidney tubular cell line HK-2. First, the upregulation of
CHAC1 expression by YC-1 was confirmed. YC-1 treatment reproducibly
increases CHAC1 expression levels in HK-2. The expression level of
CHAC1 was the highest when treated with 1 µM YC-1. The CHAC1
expression decreased above 25 µM concentration (Fig. 1). Therefore, 1 µM YC-1 was used for
subsequent experiments.
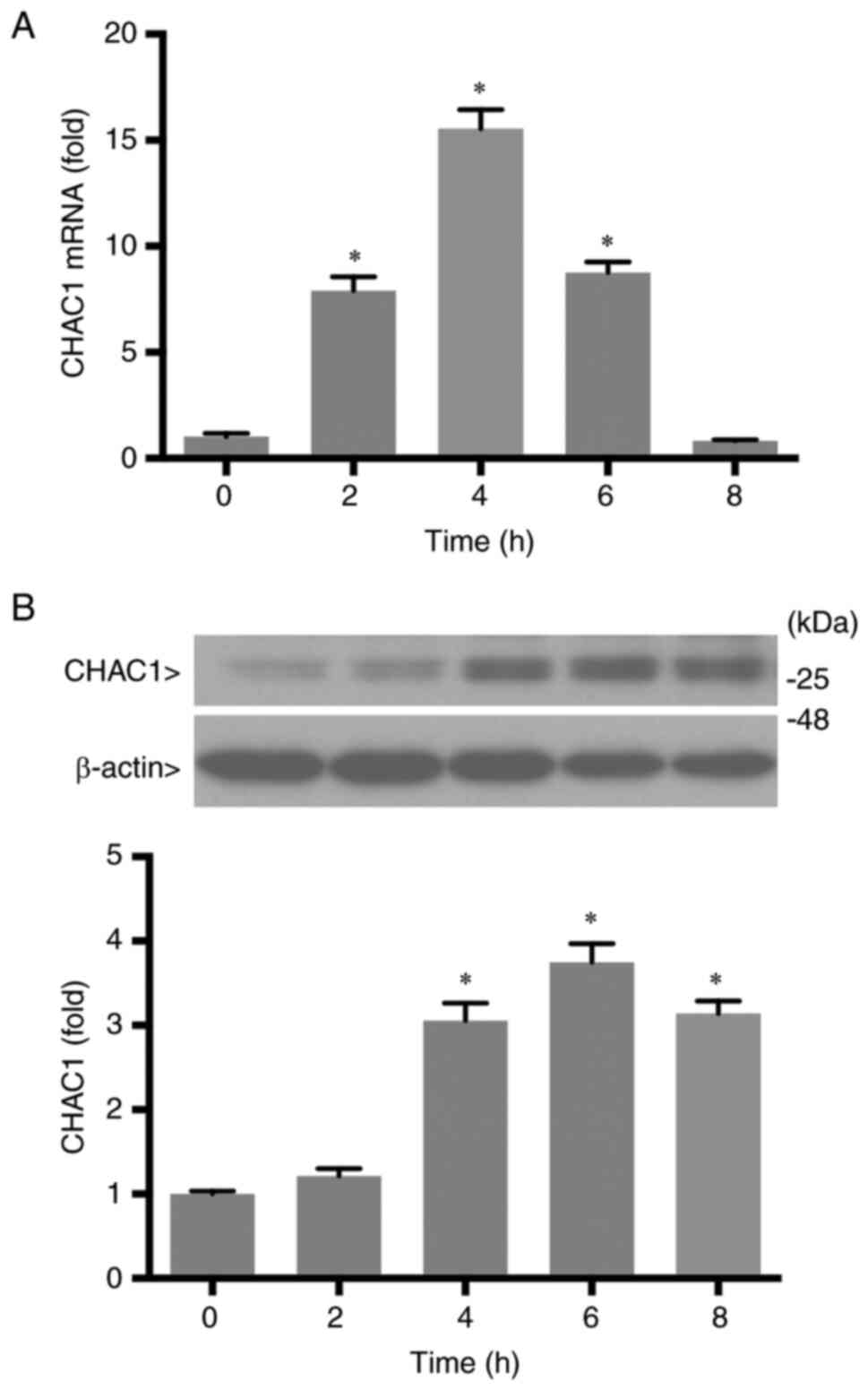
Next, the time-dependent upregulation of CHAC1 upon
treatment with 1 µM YC-1 was investigated. CHAC1 mRNA levels
reached a maximum at 4 h after YC-1 treatment and then gradually
decreased (Fig. 2A). The protein
levels of CHAC1 became detectable 2 h after those of mRNA were
detected, and the maximum level was observed at 6 h (Fig. 2B). Thus, YC-1 increased CHAC1
expression in a concentration- and time-dependent manner.
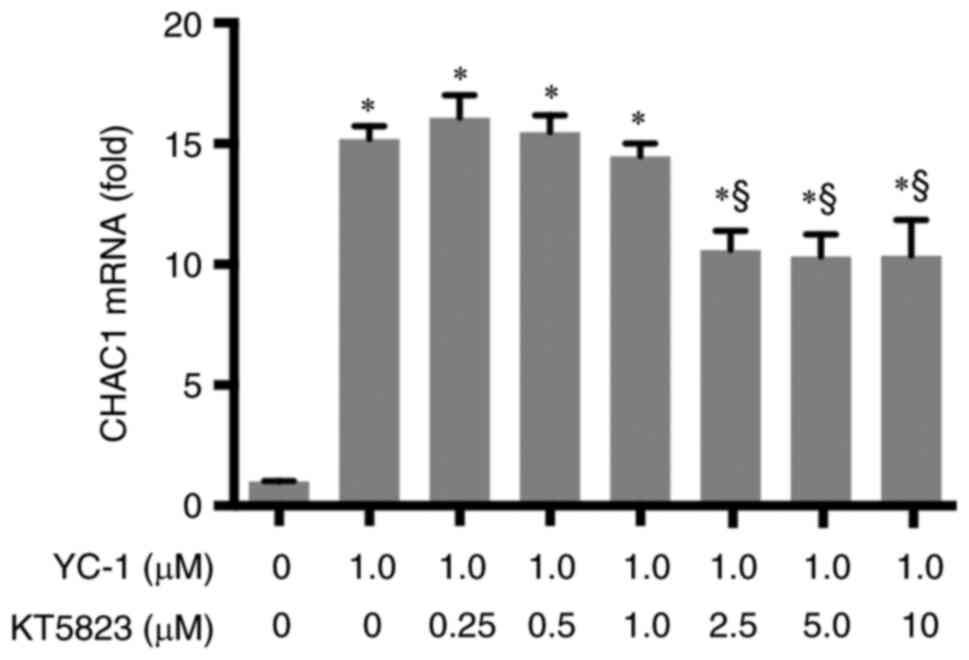
CHAC1 induced by YC-1 is partially
inhibited by treatment with a PKG inhibitor
Because YC-1 is an sGC activator, it has been
suggested that CHAC1 is induced through the sGC-cGMP-PKG signaling
pathway. Therefore, the effect of KT5823, a PKG inhibitor, on CHAC1
induction by YC-1 was studied. HK-2 cells were treated with varying
concentrations of KT5823. As demonstrated in Fig. 3, KT5823 significantly decreased
CHAC1 expression levels at a concentration of 2.5 µM. CHAC1
expression reduced to ~40% of its maximum level. Higher KT5823
concentrations did not result in a further decrease in CHAC1. These
results revealed that YC-1 partially induced CHAC1 expression via
the sGC-cGMP-PKG pathway. In addition, other signaling pathways
contributed to the induction of CHAC1 by YC-1.
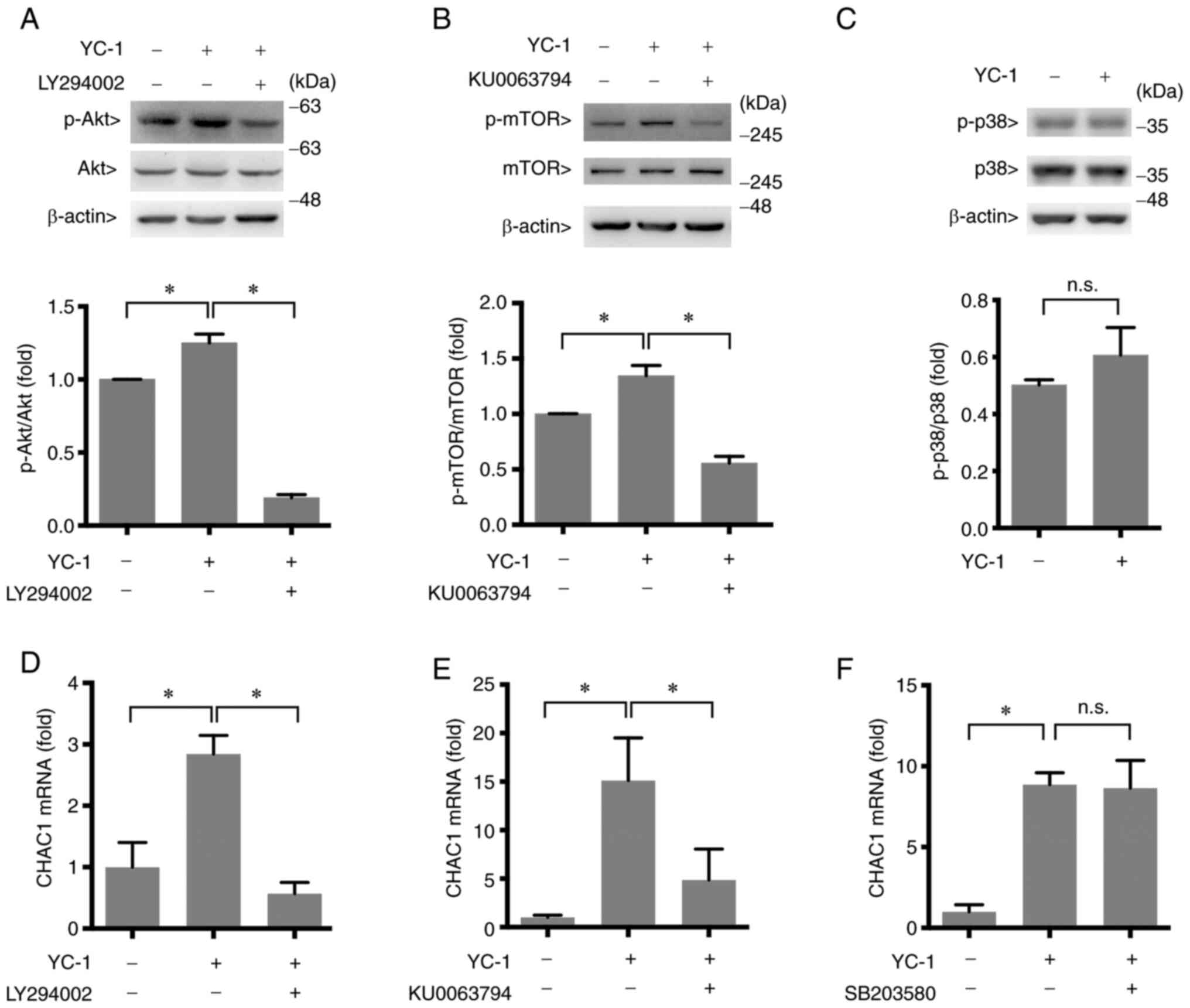
YC-1 upregulates CHAC1 through the
AKT-mTOR pathway
It is possible that YC-1 increases CHAC1 expression
through several signaling pathways. The involvement of other
signaling pathways in the regulation of CHAC1 expression by YC-1
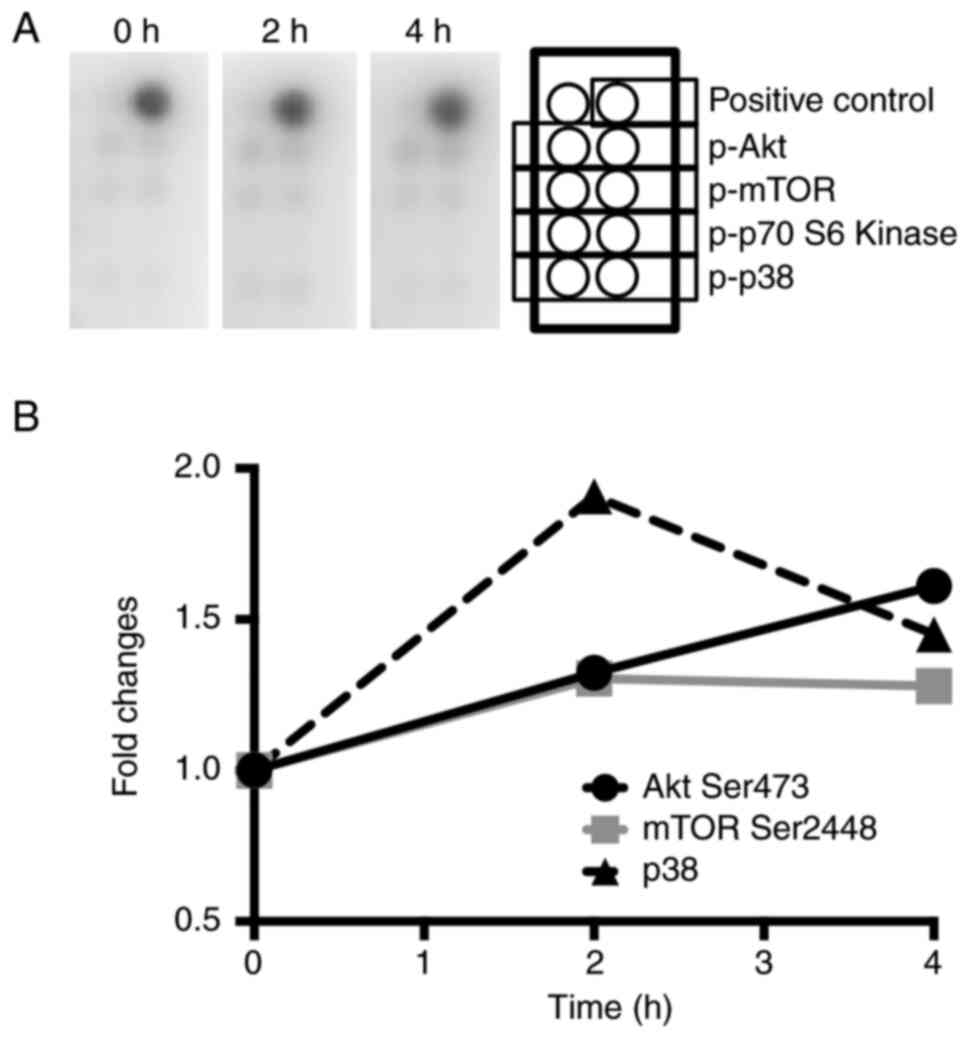
was studied. An antibody array kit that detects signaling factors
was used to observe the signaling pathways activated by YC-1. The
phosphorylation levels of Akt, mTOR and p38 increased following
YC-1 treatment, whereas phosphorylated (p-)p70 S6 kinase was almost
undetectable (Fig. 4A and B).
Therefore, Akt, mTOR and p38 may participate in the induction of
CHAC1 expression by YC-1. However, the result was observed in the
analysis using just one sample. Therefore, to confirm significance
of the upregulated phosphorylation of Akt, mTOR and p38, western
blot analysis was performed. The results exhibited significant
increase of phosphorylation of Akt and mTOR. However,
phosphorylation of p38 was not significantly increased. These
results indicated that CHAC1 was induced via the Akt-mTOR signaling
pathway (Fig. 5A-C).
To elucidate the involvement of the Akt-mTOR pathway
in CHAC1 induction, the effects of inhibitors on each factor were
assessed. LY294002, an inhibitor of PI-3 kinase upstream of Akt,
and KU0063794, an inhibitor of mTOR, reduced the induction of CHAC1
mRNA transcription levels by YC-1 (Fig. 5D and E), indicating that the
Akt-mTOR pathway participates in the upregulation of CHAC1
expression by YC-1. By contrast, SB203580, an inhibitor of p38, did
not decrease CHAC1 mRNA transcription levels induced by YC-1
(Fig. 5F). These results indicated
that the signaling pathway of p38 is not involved in CHAC1
induction by YC-1.
ER stress is induced by YC-1
CHAC1 is a downstream product of ER stress and ATF4
is a transcription factor that upregulates CHAC1 (11,13).
Therefore, the effects of YC-1 on PERK pathway, which is one of the
branches of ER stress signaling pathway and enhances ATF4, were
investigated. YC-1 induced ATF4 expression; ATF4 levels
significantly increased at 2 h after YC-1 treatment, reached a
maximum at 4 h and then gradually decreased (Fig. 6A). In addition, YC-1 induced CHOP
as well. The increase in CHOP was delayed by ATF4; it significantly
increased 4 h after YC-1 treatment (Fig. 6B). These results indicated that
YC-1 stimulates the PERK pathway, which is one of three branches of
the ER stress. A 4 h treatment with YC-1 induced CHAC1
expression.
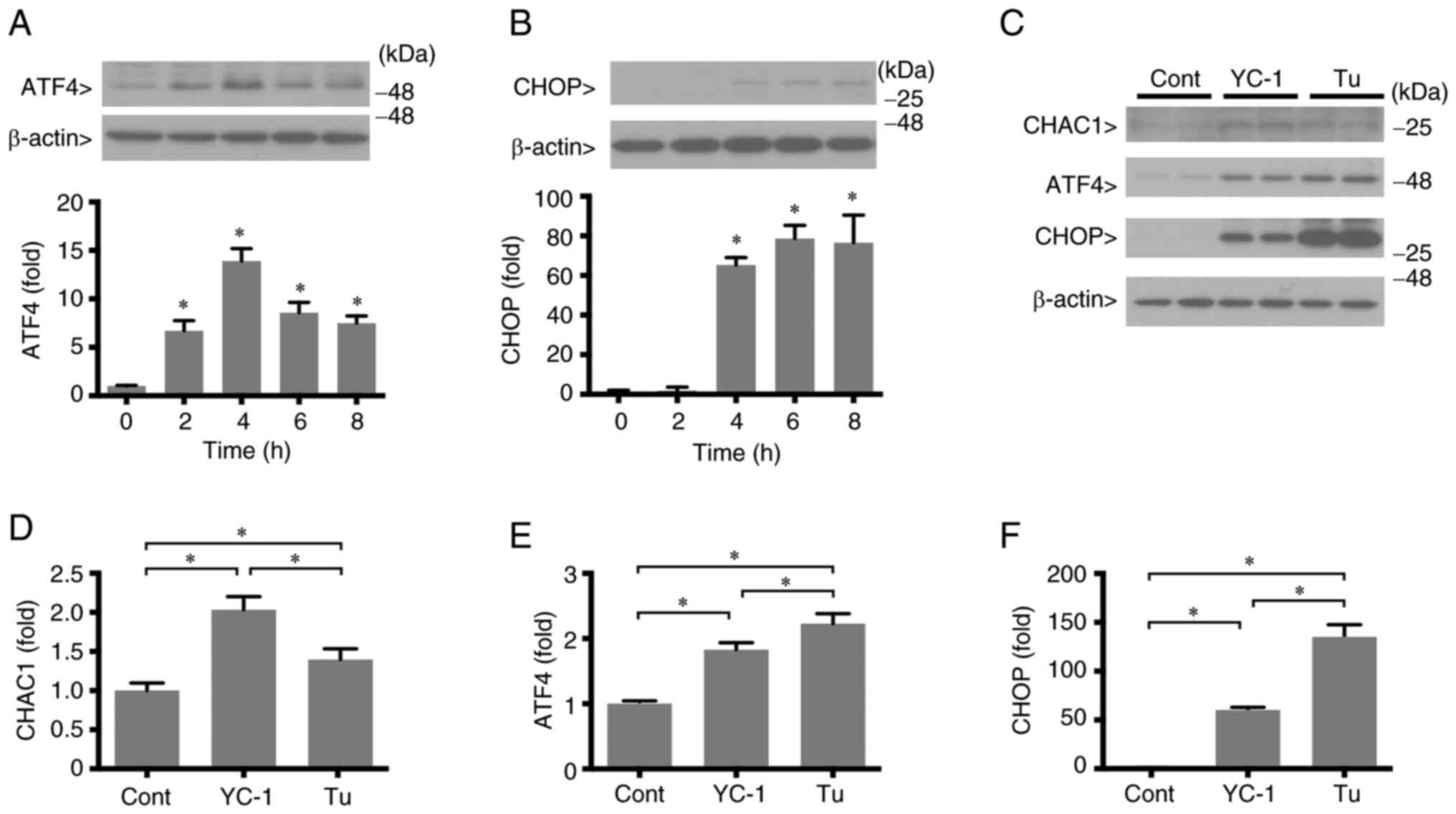 | Figure 6.Endoplasmic reticulum R stress
induction with YC-1 treatment. HK-2 cells reached 70% confluency
and were incubated in serum-free DMEM for 24 h. Subsequently, the
cells were treated with 1 µM YC-1 or 10 µM Tu. (A and B) ATF4 and
CHOP protein levels after treatment with YC-1. (C) ATF4, CHOP and
CHAC1 protein levels after treatment with YC-1 or Tu for 4 h. (D-F)
Data of protein expression levels of ATF4, CHOP and CHAC1, as
observed by western blotting in panel C. All data are presented as
the mean values ± standard error of the mean (n=3). *P<0.05.
YC-1, 3-(5′-hydroxymethyl-2′-furyl)-1-benzylindazole; Tu,
tunicamycin; ATF, activating transcription factor; CHOP, (C/EBP)
homologous protein; CHAC1, glutathione-specific
γ-glutamylcyclotransferase 1; Cont, control. |
As a result of studying detailed mechanisms of CHAC1
regulation by the PERK signaling pathway, the effect of YC-1 was
compared with that of tunicamycin, which is an inducer of ER stress
(21–23). Tunicamycin induced ATF4 and CHOP
expression, and this increase was significantly higher than that
induced by YC-1 (Fig. 6C-F). The
expression levels of CHAC1 with YC-1 are higher than those in the
presence of tunicamycin. These results suggested that CHAC1 is
upregulated via the ER stress pathway.
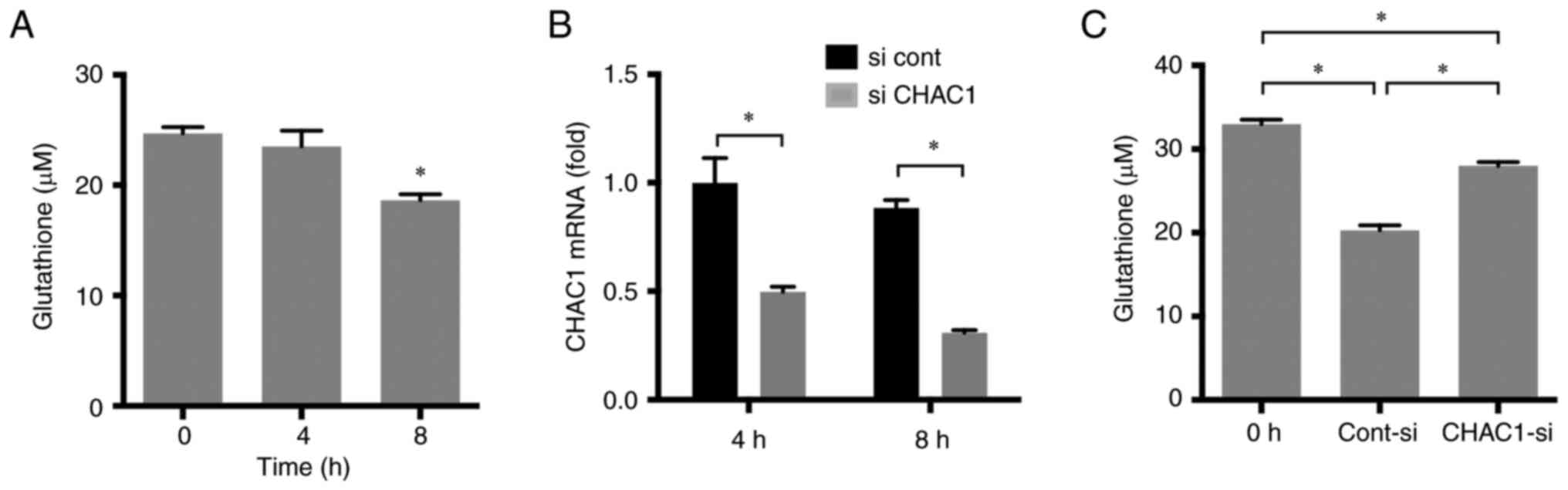
CHAC1 reduces intracellular
glutathione concentration
CHAC1 is a glutathione catabolic enzyme that reduces
intracellular glutathione concentration. Therefore, the
intracellular glutathione concentration in HK-2 cells treated with
YC-1 was measured. A total of 1 µM YC-1 reduced intracellular
glutathione concentration 8 h after treatment (Fig. 7A). siCHAC1 was introduced into HK-2
cell using Lipofectamine RNAiMAX. The siRNA significantly reduced
CHAC1 expression (Fig. 7B). In
HK-2 cells transfected with siCHAC1, the degradation of glutathione
by YC-1 was reduced (Fig. 7C).
These results demonstrated that CHAC1 acts as a glutathione
catabolic enzyme in tubular cells.
Discussion
Although the regulation of CHAC1 expression by
several transcription factors has been previously investigated
(9,10,13),
the signaling pathways that regulate CHAC1 expression remain
unclear. YC-1 increased CHAC1 in HK-2 cells. In the present study,
the signaling pathways involved in CHAC1 induction by YC-1 in
cultured human tubular HK-2 cells were investigated.
YC-1 is an sGC activator (16); therefore, it is possible that YC-1
increases CHAC1 expression via the sGC-cGMP-PKG pathway. The
results of the present study revealed the involvement of the
sGC-cGMP-PKG pathway in the induction of CHAC1 by YC-1. cGMP is
produced downstream of the nitric oxide (NO) signaling pathway. IRI
induces inducible NO synthase (iNOS), which produces NO in the
kidney (24–26), indicating that CHAC1 is induced
through the NO signaling pathway. However, the effect of the PKG
inhibitor was only partial. Therefore, the involvement of other
signaling pathways in CHAC1 induction by YC-1 was predicted.
Signaling pathway analysis using an antibody array demonstrated
that YC-1 activated p38, AKT and mTOR. Among the signaling pathways
induced by YC-1, the AKT-mTOR pathway participates in the
upregulation of CHAC1. Although the phosphorylation of p38 appeared
to be increased on antibody array, western blot analysis did not
reveal significant increase of phosphorylation of p38 and the
inhibitor SB203580 did not inhibit the upregulation of CHAC1. The
signal intensities of p-p38 on antibody array were weak. Therefore,
it is suggested that the effect of YC-1 on p38 phosphorylation is
slight. In the present study, the phosphorylation of p70 S6 kinase
was hardly detectable, although it was downstream of the AKT-mTOR
pathway. These results indicated that CHAC1 was induced through the
AKT-mTOR signaling pathway via downstream signals other than p70 S6
kinase. In addition, YC-1 induced the PERK pathway which is one of
the branches of the three ER stress pathways, indicating that the
PERK pathway contributes to the upregulation of CHAC1 expression.
Because it is known that CHAC1 is regulated by ATF4 (11,13),
PERK pathway-induced ATF4 expression may upregulate CHAC1.
Tunicamycin, which is a strong inducer of ER stress, was used to
clear detailed regulation mechanisms of CHAC1 and compare the
effects of tunicamycin on PERK pathway with that of YC-1. A
comparison of YC-1 with tunicamycin revealed that tunicamycin
induces ATF4 and CHOP more highly than YC-1, indicating that the
induction of the PERK pathway by YC-1 is not as strong as that of
tunicamycin. However, YC-1 induced CHAC1 expression to a greater
extent than tunicamycin. These findings support the hypothesis that
CHAC1 is involved in several signaling pathways in addition to the
PERK pathway. CHOP, a gene downstream of ER stress, is known
to induce apoptosis and is regulated by ATF4 (27). Tunicamycin, a strong inducer of the
PERK pathway, caused higher induction of CHOP than CHAC1.
Meanwhile, YC-1, a weak inducer of the PERK pathway, induced CHAC1
more than CHOP. These results indicated that CHOP is mainly
regulated by ER stress, whereas CHAC1 is regulated by numerous
signaling pathways. Additionally, it may be involved in the
inhibitory effects of CHOP on CHAC1 transcription (28).
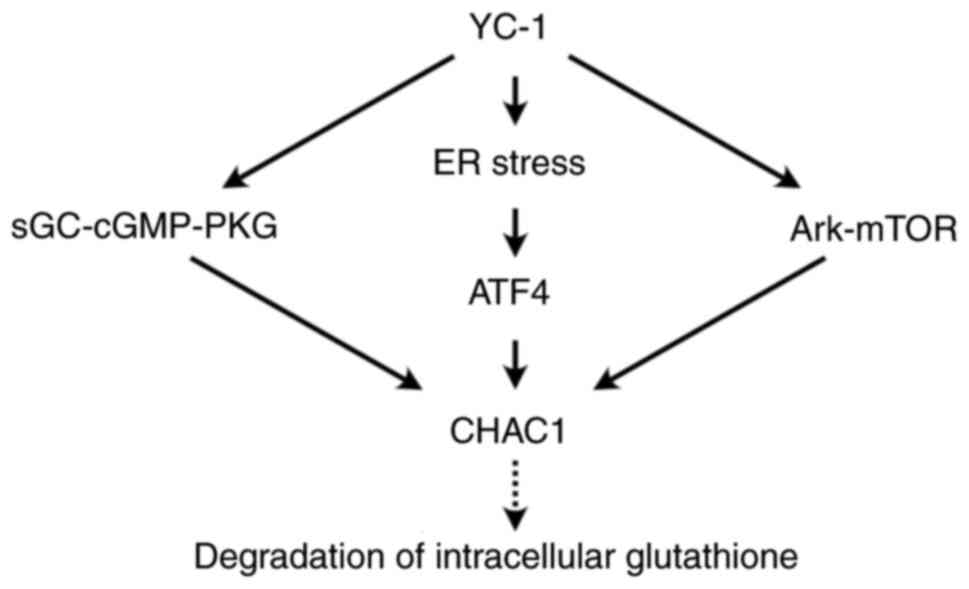
Induction of CHAC1 by YC-1 in HK-2 cells reduced
intracellular glutathione levels, indicating that CHAC1 regulates
redox homeostasis. YC-1 has several effects on HK-2 cells and
induces CHAC1 expression through the sGC-cGMP-PKG, AKT-mTOR and ER
stress pathways (Fig. 8).
Acute kidney injury (AKI) is a common clinical
problem characterized by an abrupt decrease in the glomerular
filtration rate and is associated with high morbidity and mortality
(29). IRI is the most common
cause of AKI (30,31). CHAC1 was found to be upregulated in
renal tubular cells of an IRI mouse model. Reactive oxygen species,
which are generated by an abrupt supply of oxygen after reperfusion
(32–34), tend to damage the ER and cause ER
stress (16). NO levels are also
increased via iNOS induction in IRI (24–26).
Therefore, the induction of CHAC1 in tubular cells after IRI was
probably due to ER stress and NO signaling.
Ferroptosis is a previously described form of
programmed cell death triggered by oxidative damage (35). IRI induces ferroptosis in the
kidney, and cell death is one of the causes of renal dysfunction
(36). The findings of the present
study demonstrated that CHAC1 degraded glutathione in HK-2 cells.
In addition, it was previously revealed that knockdown of CHAC1
rescued the cysteine-starvation-induced reduction of glutathione
levels and ferroptosis in human breast cancer cell lines (12). Therefore, it is possible that
upregulated CHAC1 degrades glutathione and induces ferroptosis in
renal IRI. In AKI, ferroptosis plays an important role in the
pathogenesis and is directly related to the post-ischemic renal
necrosis of renal tubules (36,37).
Therefore, ferroptosis is a potential therapeutic target for IRI.
The findings of the present study about regulation mechanisms of
CHAC1 are informative for development of therapeutic strategies
through regulation of ferroptosis in IRI.
In addition to the renal IRI, ferroptosis is a key
factor that leads to IRI and organ failure, myocardial and cerebral
IRI (37). Therefore, the findings
of the present study that investigated the regulation pathways of
CHAC1 probably contributed to elucidation of the induction
mechanisms of ferroptosis and the future discovery of therapeutic
targets of numerous ischemic injuries.
Acknowledgements
Not applicable.
Funding
The present study was partially supported by KAKENHI (grant no.
23K07711).
Availability of data and materials
All data generated or analyzed in the present study
are included in this published article.
Authors' contributions
YK and YF contributed to the conception and design
of the study, acquired and analyzed the data, and drafted the
manuscript. YK and YF confirm the authenticity of all the raw data.
ST and ES contributed to the study design and revision of the
manuscript. All authors read and approved the final version of the
manuscript.
Ethics approval and consent to
participate
Not applicable.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Anelli T and Sitia R: Protein quality
control in the early secretory pathway. EMBO J. 27:315–27. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Tabas I and Ron D: Integrating the
mechanisms of apoptosis induced by endoplasmic reticulum stress.
Nat Cell Biol. 13:184–90. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Walter P and Ron D: The unfolded protein
response: From stress pathway to homeostatic regulation. Science.
334:1081–1086. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Schröder M and Kaufman RJ: The mammalian
unfolded protein response. Annu Rev Biochem. 74:739–789. 2005.
View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Oyadomari S, Araki E and Mori M:
Endoplasmic reticulum stress-mediated apoptosis in pancreatic
beta-cells. Apoptosis. 7:335–345. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Okada K, Minamino T, Tsukamoto Y, Liao Y,
Tsukamoto O, Takashima S, Hirata A, Fujita M, Nagamachi Y, Nakatani
T, et al: Prolonged endoplasmic reticulum stress in hypertrophic
and failing heart after aortic constriction: Possible contribution
of endoplasmic reticulum stress to cardiac myocyte apoptosis.
Circulation. 110:705–712. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Szegezdi E, Duffy A, O'Mahoney ME, Logue
SE, Mylotte LA, O'brien T and Samali A: ER stress contributes to
ischemia-induced cardiomyocyte apoptosis. Biochem Biophys Res
Commun. 349:1406–1411. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Fawcett TW, Martindale JL, Guyton KZ, Hai
T and Holbrook NJ: Complexes containing activating transcription
factor (ATF)/cAMP-responsive-element-binding protein (CREB)
interact with the CCAAT/enhancer-binding protein (C/EBP)-ATF
composite site to regulate Gadd153 expression during the stress
response. Biochem J. 339:135–141. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Mungrue IN, Pagnon J, Kohannim O,
Gargalovic PS and Lusis AJ: CHAC1/MGC4504 is a novel proapoptotic
component of the unfolded protein response, downstream of the
ATF4-ATF3-CHOP cascade. J Immunol. 182:466–476. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Kumar A, Tikoo S, Maity S, Sengupta S,
Sengupta S, Kaur A and Bachhawat AK: Mammalian proapoptotic factor
ChaC1 and its homologues function as γ-glutamyl cyclotransferases
acting specifically on glutathione. EMBO Rep. 13:1095–1101. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Crawford RR, Prescott ET, Sylvester CF,
Higdon AN, Shan J, Kilberg MS and Mungrue IN: Human CHAC1 protein
degrades glutathione, and mRNA induction is regulated by the
transcription factors ATF4 and ATF3 and a bipartite ATF/CRE
regulatory element. J Biol Chem. 290:15878–15891. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Chen MS, Wang SF, Hsu CY, Yin PH, Yeh TS,
Lee HC and Tseng LM: CHAC1 degradation of glutathione enhances
cystine-starvation-induced necroptosis and ferroptosis in human
triple negative breast cancer cells via the GCN2-eIF2α-ATF4
pathway. Oncotarget. 8:114588–114602. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Oh-Hashi K, Nomura Y, Shimada K, Koga H,
Hirata Y and Kiuchi K: Transcriptional and post-translational
regulation of mouse cation transport regulator homolog 1. Mol Cell
Biochem. 380:97–106. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Younis NS and Ghanim AMH: The protective
role of celastrol in renal ischemia-reperfusion injury by
activating Nrf2/HO-1, PI3K/AKT signaling pathways, modulating NF-κb
signaling pathways, and inhibiting ERK phosphorylation. Cell
Biochem Biophys. 80:191–202. 2022. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Hoppins S and Nunnari J: Mitochondrial
dynamics and apoptosis-the ER connection. Science. 337:1052–1054.
2012. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Martin E, Lee YC and Murad F: YC-1
activation of human soluble guanylyl cyclase has both
heme-dependent and heme-independent components. Proc Natl Acad Sci
USA. 98:12938–12942. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Vlahos CJ, Matter WF, Hui KY and Brown RF:
A specific inhibitor of phosphatidylinositol 3-kinase,
2-(4-morpholinyl)-8-phenyl-4H-1-benzopyran-4-one (LY294002). J Biol
Chem. 269:5241–5248. 1994. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
García-Martínez JM, Moran J, Clarke RG,
Gray A, Cosulich SC, Chresta CM and Alessi DR: Ku-0063794 is a
specific inhibitor of the mammalian target of rapamycin (mTOR).
Biochem J. 421:29–42. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Cuenda A, Rouse J, Doza YN, Meier R, Cohen
P, Gallagher TF, Young PR and Lee JC: SB 203580 is a specific
inhibitor of a MAP kinase homologue which is stimulated by cellular
stresses and interleukin-1. FEBS Lett. 364:229–233. 1995.
View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Livak KJ and Schmittgen TD: Analysis of
relative gene expression data using real-time quantitative PCR and
the 2(−Delta Delta C(T)) method. Methods. 25:402–408. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Surani MA: Glycoprotein synthesis and
inhibition of glycosylation by tunicamycin in preimplantation mouse
embryos: Compaction and trophoblast adhesion. Cell. 18:217–227.
1979. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Yoo J, Mashalidis EH, Kuk ACY, Yamamoto K,
Kaeser B, Ichikawa S and Lee SY: GlcNAc-1-P-transferase-tunicamycin
complex structure reveals basis for inhibition of N-glycosylation.
Nat Struct Mol Biol. 25:217–224. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Hakulinen JK, Hering J, Brändén G, Chen H,
Snijder A, Ek M and Johansson P: MraY-antibiotic complex reveals
details of tunicamycin mode of action. Nat Chem Biol. 13:265–267.
2017. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Liu F, Ni W, Zhang J, Wang G, Li F and Ren
W: Administration of curcumin protects kidney tubules against renal
ischemia-reperfusion injury (RIRI) by modulating nitric oxide (NO)
signaling pathway. Cell Physiol Biochem. 44:401–411. 2017.
View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Walker LM, Walker PD, Imam SZ, Ali SF and
Mayeux PR: Evidence for peroxynitrite formation in renal
ischemia-reperfusion injury: Studies with the inducible nitric
oxide synthase inhibitor L-N(6)-(1-Iminoethyl)lysine. J Pharmacol
Exp Ther. 295:417–422. 2000.PubMed/NCBI
|
|
26
|
Chatterjee PK, Patel NS, Sivarajah A,
Kvale EO, Dugo L, Cuzzocrea S, Brown PA, Stewart KN, Mota-Filipe H,
Britti D, et al: GW274150, a potent and highly selective inhibitor
of iNOS, reduces experimental renal ischemia/reperfusion injury.
Kidney Int. 63:853–865. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Luo J, Xia Y, Luo J, Li J, Zhang C, Zhang
H, Ma T, Yang L and Kong L: GRP78 inhibition enhances ATF4-induced
cell death by the deubiquitination and stabilization of CHOP in
human osteosarcoma. Cancer Lett. 410:112–123. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Nomura Y, Sylvester CF, Nguyen LO, Kandeel
M, Hirata Y, Mungrue IN and Oh-Hashi K: Characterization of the
5′-flanking region of the human and mouse CHAC1 genes. Biochem
Biophys Rep. 24:1008342020.PubMed/NCBI
|
|
29
|
Schiffl H, Lang SM and Fischer R: Daily
hemodialysis and the outcome of acute renal failure. N Engl J Med.
346:305–310. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Edelstein CL, Ling H and Schrier RW: The
nature of renal cell injury. Kidney Int. 51:1341–1351. 1997.
View Article : Google Scholar : PubMed/NCBI
|
|
31
|
DuBose TD Jr, Warnock DG, Mehta RL,
Bonventre JV, Hammerman MR, Molitoris BA, Paller MS, Siegel NJ,
Scherbenske J and Striker GE: Acute renal failure in the 21st
century: Recommendations for management and outcomes assessment. Am
J Kidney Dis. 29:793–799. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Basile DP: The endothelial cell in
ischemic acute kidney injury: Implications for acute and chronic
function. Kidney Int. 72:151–156. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Bonventre JV and Yang L: Cellular
pathophysiology of ischemic acute kidney injury. J Clin Invest.
121:4210–4221. 2011. View
Article : Google Scholar : PubMed/NCBI
|
|
34
|
Kloner RA, Przyklenk K and Whittaker P:
Deleterious effects of oxygen radicals in ischemia/reperfusion.
Resolved and unresolved issues. Circulation. 80:1115–1127. 1989.
View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Cao JY and Dixon SJ: Mechanisms of
ferroptosis. Cell Mol Life Sci. 73:2195–209. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Linkermann A, Skouta R, Himmerkus N, Mulay
SR, Dewitz C, De Zen F, Prokai A, Zuchtriegel G, Krombach F, Welz
PS, et al: Synchronized renal tubular cell death involves
ferroptosis. Proc Natl Acad Sci USA. 111:16836–16841. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Li X, Ma N, Xu J, Zhang Y, Yang P, Su X,
Xing Y, An N, Yang F, Zhang G, et al: Targeting ferroptosis:
pathological mechanism and treatment of ischemia-reperfusion
injury. Oxid Med Cell Longev. 2021:15879222021. View Article : Google Scholar : PubMed/NCBI
|