Introduction
Liddle syndrome is an autosomal dominant form of
monogenic hypertension with early penetrance. It was first
characterized by Liddle (1) in
1963, and its molecular basis lies in mutations in SCNN1A,
SCNN1B and SCNN1G, which respectively encode the α, β
and γ subunits of the epithelial sodium channel (ENaC). The ENaC
serves an important role in the maintenance of blood pressure
through the reabsorption of sodium and water. Each subunit of the
ENaC has a highly conserved proline-rich sequence in the cytosolic
C-terminus, known as the PY motif (2). Individuals with Liddle syndrome have
gain-of-function mutations, which cause damage to the PY motif,
resulting in excessive sodium absorption and volume expansion
(3). Therefore, Liddle syndrome
responds well to ENaC blockers, but not to spironolactone, which is
a mineralocorticoid receptor antagonist (4).
Typical clinical characteristics of Liddle syndrome
include early-onset hypertension, refractory hypokalemia, low
plasma aldosterone and renin concentrations, and metabolic
alkalosis (5). Previously, Fan
et al (6) summarized the
clinical characteristics of 54 children with Liddle syndrome,
reporting that the median age of typical hypertension onset was
12.75 years and the median age at genetic diagnosis was 14 years.
Although most patients with Liddle syndrome have distinctive
clinical features, certain individuals do not and are thus at risk
of misdiagnosis. Genetic analysis is the gold-standard method for
diagnosing Liddle syndrome at the molecular level. However, the
genotype-phenotype association in Liddle syndrome is still
currently unknown because of the limited number of reported cases
(7).
The present study described a family with Liddle
syndrome, characterized by early-onset hypertension and
hypokalemia. Whole-exome sequencing and Sanger sequencing were used
to identify pathogenic mutations in the family. A systematic review
of follow-up data from patients with Liddle syndrome with
SCNN1B mutations was also conducted to assess the phenotypic
heterogeneity and prognosis of Liddle syndrome after tailored
therapy.
Materials and methods
Compliance with ethical standards
The present study was approved by the Ethics
Committee of Fuwai Hospital, Peking Union Medical College (Beijing,
China; approval no. 2020-1321) and was performed in accordance with
the Declaration of Helsinki. All study participants provided
written informed consent before study enrolment.
Subjects and clinical evaluation
The proband (Fig.
1; III-2) was a 28-year-old male patient who was admitted with
severe hypertension to the Department of Hypertension, Fuwai
Hospital (Beijing, China) in 2021. The hypertension of the patient
had been incidentally diagnosed during a medical examination at the
age of 16 years. During hospitalization, detailed screening tests,
including imaging (echocardiography and computed tomography of the
aortic and renal arteries), physical examination, biochemical
assessments (including plasma serum, aldosterone, renin and urinary
microalbumin concentration measurements) and hormone measurements
(including growth hormone, cortisol and adrenocorticotropic hormone
concentration measurements), were performed to identify the
etiology of hypertension. Moreover, the fundus lesions of
hypertension were evaluated by mydriasis followed by fundoscopy.
The patient had a family history of early-onset hypertension.
Affected family members included the father (II-4) of the patient,
two paternal aunts (II-1 and II-5) and a deceased paternal uncle
(II-7). Therefore, seven biological relatives of the patient (five
females and two males) with a median age of 42 years (interquartile
range 35, 60 years) were enrolled in the present study. They
underwent full clinical evaluation (including blood pressure
measurement, medical history and symptom assessment), biochemical
testing (including plasma serum, aldosterone and renin
concentration measurements) and genetic analysis.
DNA sequencing
DNA was extracted from leukocytes in the peripheral
blood of all enrolled family members using the QIAamp®
DNA Blood Mini kit (cat. no. 51104; Qiagen GmbH) according to the
manufacturer's protocol. The DNA concentration and quality was
measured using a Qubit® 3.0 Fluorometer (Invitrogen;
Thermo Fisher Scientific, Inc.). Additionally, gel electrophoresis
was carried out with a 1% agarose gel to visually inspect the DNA
for intact, high molecular weight bands, to ensure that it was not
degraded or fragmented. Whole-exome sequencing was performed on DNA
from the blood of the proband using the SureSelectXT Human All
ExonV6 kit (cat. no. 5190-8864; Agilent Technologies, Inc.) for
exome capture and the Novaseq 6000 platform (Illumina, Inc.) was
used for genomic DNA sequencing (paired end; 150 bp). The
concentration of the library was measured using a Qubit®
3.0 Fluorometer (Invitrogen; Thermo Fisher Scientific, Inc.) and
then the size distribution of the library was analyzed using a NGS
3K reagent kit (cat. no. CLS960013; PerkinElmer, Inc.) and the
loading concentration of the final library was 17.56 nM. Except for
the proband, Sanger sequencing was performed on the DNA of the
family members to evaluate the candidate variant and to perform
co-segregation analysis. The forward primer used was
5′-CCCACCCAAGAATCACCTCC-3′ and the reverse primer used was
5′-TCAGGACAGGTAGGGACGAG-3′. To ensure that the mutation was
accurately identified, the exon was sequenced forward and backward,
and all products were analyzed using Chromas 2.22 software
(Technelysium Pty Ltd.).
Genetic analysis
The whole-exome sequenced data were first checked
using in-house quality control software to remove low-quality
reads. The sequenced data were then aligned to the reference human
genome (GRCh37/hg19) using Burrows-Wheeler Aligner (version 0.7.12)
(8). SAM tool (version 0.1.19;
http://sourceforge.net/projects/samtools/files/samtools/0.1.19/)
and Sambamba (version 0.8.2; http://github.com/biod/sambamba/releases) were used to
sort bam files and perform duplicate marking, generating the final
bam file (9,10). Single-nucleotide variants and
insertion/deletion variants were identified using the SAM tool
(version 0.1.19) (10) and copy
number variants were detected using CoNIFER (version 0.3) (11). Annotation was performed using
ANNOVAR software (version 322; http://annovar.openbioinformatics.org/en/latest/).
Mutations with a frequency of ≥1% in the 1000 Genomes Project
(1000G_all; http://www.internationalgenome.org/), ESP6500
(ESP6500SIV2_ALL; http://esp.gs.washington.edu/drupal/) and gnomAD
(gnomAD_ALL and gnomAD_EAS; http://gnomad.broadinstitute.org/) databases were
removed. In silico analysis software, including
MutationTaster (MutationTaster2021; http://www.mutationtaster.org/) and Phylogenetic
Analysis with Space/Time Models (phast) Cons (version 1.3;
compgen.bscb.cornell.edu/phast) were used to predict the
pathogenicity of variants. The pathogenicity of the sequence
variants was assessed using the method developed by the American
College of Medical Genetics and Genomics (ACMG) (12).
Systematic review and
genotype-phenotype association analysis
The follow-up data of patients with Liddle syndrome
were analyzed to evaluate the prognosis of patients with Liddle
syndrome after targeted therapy. ‘Liddle syndrome’, ‘Liddle's
syndrome’, ‘pseudoaldosteronism’ and ‘SCNN1B’ were used as
key words to search publications in English in the MEDLINE
(https://www.medline.com/), Cochrane (https://www.cochranelibrary.com/) and Web of
Science (https://clarivate.com/products/scientific-and-academic-research/research-discovery-and-workflow-solutions/webofscience-platform/)
databases from December 1963 to July 2023. Patients that were
genetically diagnosed with Liddle syndrome with available follow-up
data were included in the analysis, while those without genetic
diagnostic information or follow-up data were excluded. As the
prevalence of Liddle syndrome is low, ranging from 0.91–1.52%, the
genotype-phenotype association is still unclear (13,14).
Therefore, a genotype-phenotype association analysis was performed
by reviewing the clinical and biochemical characteristics of
patients carrying the same mutation as the participants in the
present study.
Statistical analysis
Statistical analysis was performed using SPSS
software (version 16.0, SPSS, Inc.). The data are presented as mean
± standard deviation or frequency (%), as applicable. To assess the
differences in blood pressure and serum potassium concentration
before and after treatment with an ENaC inhibitor, paired Student's
two-tailed t-tests were used. To ensure the statistical robustness
of the findings, a minimum of three replicates were conducted for
each experiment. P<0.05 was considered to indicate a
statistically significant difference.
Results
Clinical and biochemical
characteristics
The proband was 28 years old and had a history of
hypertension, which was first diagnosed at the age of 16 years and
had remained refractory to a 12-year combination therapy that
included telmisartan, benidipine and bisoprolol. In December 2021,
the proband was referred to Fuwai Hospital (Beijing, China) for a
detailed evaluation of the hypertension of the patient.
Physical examination at the time of hospital
admission demonstrated that the patient was overweight (body mass
index, 26.2 kg/m2) and had a high blood pressure
(200/140 mmHg; normal range, <140/90 mmHg). Biochemical
assessments demonstrated a serum potassium concentration of 3.18
mmol/l (normal, 3.50–5.30 mmol/l), a plasma aldosterone
concentration of 1.0 ng/dl (normal, 3.0–35.3 ng/dl) in the standing
position (Table I), a urinary
microalbumin concentration of 56.7 mg/l (normal, <30.0 mg/l) and
a plasma chloride concentration of 108 mmol/l (normal, 98–106
mmol/l). The plasma renin concentration (5.4 mIU/ml; normal range,
4.4–46.1 mIU/ml), aldosterone/direct renin concentration ratio
[0.19 (ng/dl)/(mIU/ml); normal range, <3.70 (ng/dl)/(mIU/ml)]
and results of a low-dose dexamethasone inhibition test (cortisol,
3.00 µg/dl; normal range, <22.45 µg/dl; and adrenocorticotropic
hormone concentration, <5 pg/ml; normal range <46 pg/ml) were
normal. In addition, the growth hormone concentration (18.40 µg/dl;
normal reference, 5.27–22.45 µg/dl), cortisol concentration
(<0.1 ng/ml; normal reference, 0.0–3.0 ng/ml) and
adrenocorticotropic hormone concentration (10.30 pg/ml; normal
reference, 0.00–46.00 pg/ml) were also within the normal range. The
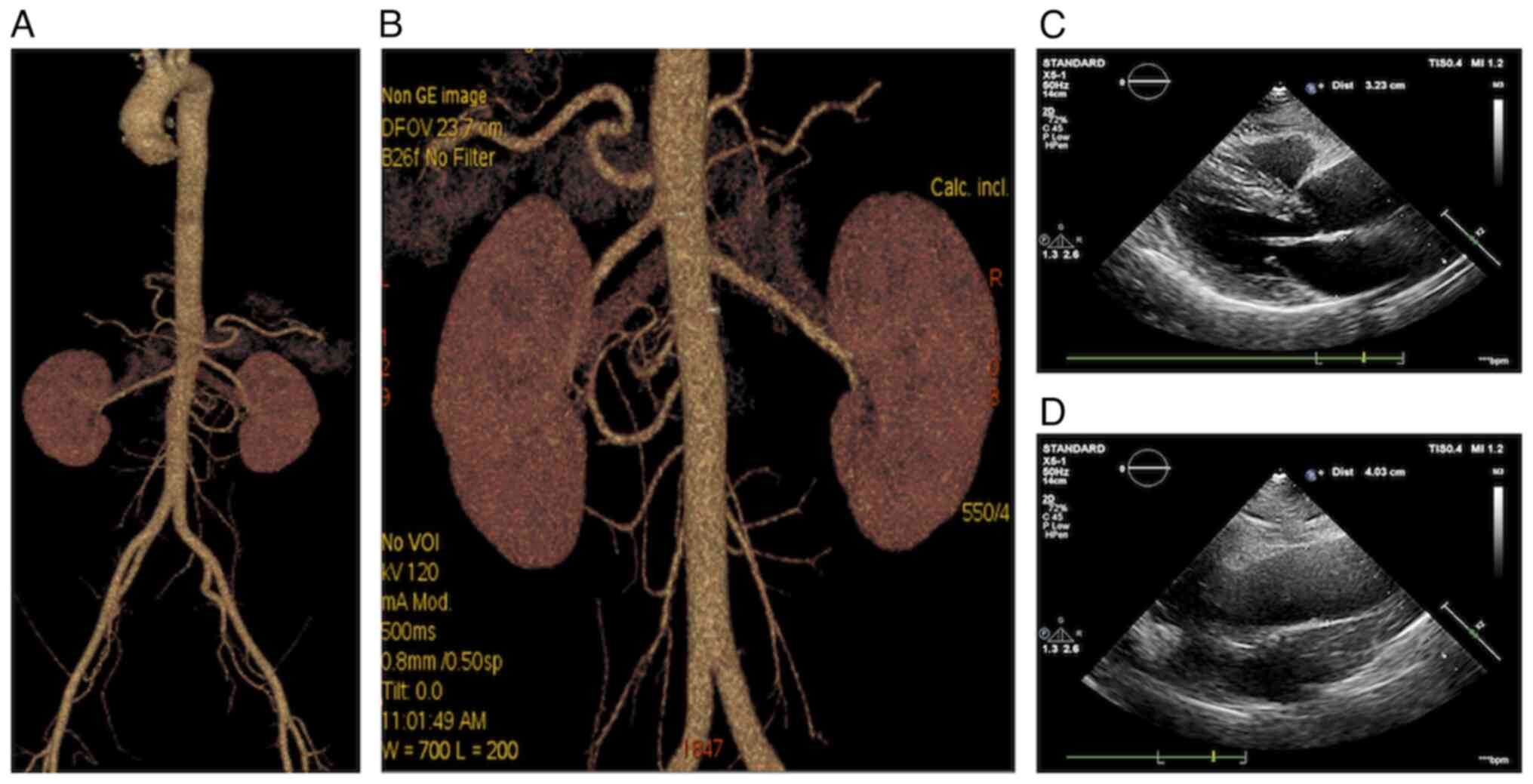
result of aortic and renal artery computed tomography was also
normal (Fig. 1A and B).
Transthoracic echocardiography demonstrated thickening of the left
ventricular posterior wall and interventricular septum and widening
of the ascending aorta (Fig. 1C and
D). Evaluation of the retinal microvasculature suggested
bilateral retinal artery stenosis and grade 1 hypertensive
retinopathy.
 | Table I.Clinical and biochemical data of
enrolled subjects in the present study. |
Table I.
Clinical and biochemical data of
enrolled subjects in the present study.
| A, Affected
patients |
|---|
|
|---|
| Family member | Hypertension
diagnosis | Age at hypertension
diagnosis, years | Blood pressure,
mmHg | Heart rate,
bpm |
Symptoms/cardiovascular
events/comorbidities | Anti-hypertensive
treatment before genetic testing | Serum
K+, mmol/l | Serum plasma
renina, mIU/ml | Serum plasma
aldosteronea,
ng/dl |
|---|
| II-1 | Y | 28 | 151/96 | 70 | Cardiovascular
disease and stroke | Nifedipine and
Bisoprolol | 3.21 | 3.4 | 2.8 |
| II-4 | Y | 18 | 160/100 | 67 | Hyperlipidemia,
hyperuricemia and hyperthyroidism | Nifedipine,
Bisoprolol and Telmisartan | 2.80 | 4.6 | 3.3 |
| II-5 | Y | 32 | 167/103 | 70 | Syncope | Nifedipine,
Losartan potassium hydrochlorothiazide compound tablet and
Carvedilol | 3.02 | 2.1 | 2.4 |
| III-2 | Y | 16 | 200/140 | 75 | - | Nifedipine,
Bisoprolol and Telmisartan | 3.18 | 5.4 | 1.0 |
| III-3 | N | - | 114/85 | 69 | Viral encephalitis
and glioma | - | 3.67 | 4.7 | 1.6 |
|
| B, Unaffected
patients |
|
| Family
member | Hypertension
diagnosis | Age at
hypertension diagnosis, years | Blood pressure,
mmHg | Heart rate,
bpm |
Symptoms/cardiovascular
events/comorbidities |
Anti-hypertensive treatment before
genetic testing | Serum
K+, mmol/l | Serum plasma
renina,
mIU/ml | Serum plasma
aldosteronea,
ng/dl |
|
| II-3 | N | - | 138/89 | 66 | - | - | 4.02 | 40.5 | 16.4 |
| III-1 | N | - | 113/68 | 63 | - | - | 4.50 | 2.7 | 7.0 |
| III-4 | N | - | 124/90 | 72 | - | - | 4.24 | 8.6 | 8.8 |
The father (II-4) of the patient and two paternal
aunts (II-1 and II-5) also had early-onset hypertension and chronic
hypokalemia, for which they intermittently took potassium citrate
supplementation. The paternal grandmother (I-1) of the patient was
diagnosed with hypertension at the age of ~20 years and died of a
stroke at 35 years of age. A paternal uncle (II-7) was reported to
have had hypertension at the age of ~30 years, with little
compliance to blood pressure control and died of cardiac arrest at
the age of 50 years. The mother (II-3) of the proband was
unaffected. Clinical and biochemical profiles of family members are
presented in Table I. The
frequency of early-onset refractory hypertension, hypokalemia and
history of sudden mortality on the paternal side of the family of
the proband was suggestive of an autosomal dominant disease
(Fig. 2).
Identification of the SCNN1B gene
mutation
The results of whole-exome sequencing in the proband
demonstrated a frameshift variant in SCNN1B (NM_000336:
c.1806dupG, p.Pro603Alafs*5). This mutation led to a heterozygous G
insertion at base 1806 of the β subunit of ENaC. This resulted in
the substitution of alanine in place of proline at codon 603 and a
shortened open reading frame that created a premature stop codon at
position 607. It also abrogated the highly conserved PY motif,
which conformed to the criterion for protein-altering variant
severity (PVS1) (12). This
variant has not been reported in the 1000 Genomes Project or ExAC
(https://exac.broadinstitute.org/)
databases, which matches the criterion for pathogenic moderate 2
(PM2) (12). Sanger sequencing
demonstrated that four affected family members (II-1, II-4, II-5
and III-3) had the same mutation as the proband (Fig. 3), indicating that they matched the
criterion for pathogenic supporting 1 (PP1) (12). Therefore, it was assumed that the
mutation identified in the proband was derived from the paternal
lineage and probably from the grandmother who died at the age of 35
years as a result of early-onset severe hypertension. Moreover, the
c.1806dupG mutation was predicted to be ‘disease causing’ and ‘1’,
determined according to the in silico analysis tools
MutationTaster and phastCons, respectively, and aligning to the
criterion for pathogenic supporting 3 (PP3) (12). Thus, according to the ACMG
guidelines (12), SCNN1B
c.1806dupG was assessed as a pathogenic mutation based on the
evidence of PVS1, PM2, PP1 and PP3.
Precise treatment and follow-up of
patients with Liddle syndrome
All family members with the pathogenic mutation were
prescribed amiloride (5 mg daily, oral administration), except for
III-3, whose blood pressure was within a normal range. Lifestyle
measures, including a low-sodium diet and exercise, were
implemented. Combination antihypertensive therapy (50 mg losartan
potassium/12.5 mg hydrochlorothiazide daily, oral administration)
was prescribed to one family member (II-4), for whom amiloride
alone was ineffective. Moreover, genetic counseling was provided to
the affected individuals and DNA testing by amniocentesis was
recommended. After 3 months of tailored treatment, blood pressure
and electrolyte concentrations in the affected individuals were
reassessed and these were demonstrated to be within normal ranges
(Table II). Subsequently, all of
the patients were followed up for ≥1 year and no abnormal blood
pressure measurements, new cardiovascular events or serious adverse
drug reactions were recorded.
| Table II.Comparison of the phenotypes and
treatment of patients carrying the p.P603Afs*607 mutation in the
studies included in the systematic review. |
Table II.
Comparison of the phenotypes and
treatment of patients carrying the p.P603Afs*607 mutation in the
studies included in the systematic review.
|
|
|
|
|
|
|
|
|
|
|
| Treatment with
epithelial sodium channel inhibitors |
|
|---|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|---|
| First author,
year | Family | Patient | Sex | Onset age of
hypertension, years | Age at diagnosis,
years | Maximum blood
pressure, mmHg | Serum K+,
mmol/l | Plasma aldosterone,
ng/dl | Plasma renin
activity/plasma renin concentration | Notes | Blood pressure,
mmHg | Serum K+,
mmol/l | (Refs.) |
|---|
| Cui et al,
2017 | 1 | A | M | 17 | 45 | 180/120 | ↓ | ↔ | ↓ | Mother of the
patient died of a stroke at the age of 32 years with unknown BP and
serum potassium levels | 120/80 | ↔ | (15) |
|
| 2 | B1 | M | 23 | 31 | 230/150 | ↓ | ↔ | ↓ | Mother of the
patient died suddenly at the of 28 years with unknown BP and serum
potassium levels | 150/85 | ↔ |
|
|
|
| B2 | M | 10 | 16 | 190/120 | ↓ | ↓ | ↓ | - | 125/85 | ↔ |
|
| Fan et al,
2019 | 3 | II-2 | M | 48 | 73 | 220/120 | ↔ | ↔ | ↑ | - | 140/80 | ↔ | (16) |
|
| II-4 | F | 25 | 70 | 200/130 | ↔ | ↓ | ↔ | - | 120/80 | ↔ |
|
|
|
|
| II-7 | M | 36 | 66 | 200/140 | ↓ | ↔ | ↔ | Stroke at the age
of 60 years | 123/75 | ↔ |
|
|
|
| III-3 | M | 19 | 35 | 180/160 | ↔ | ↓ | ↔ | - | 140/100 | ↔ |
|
|
|
| III-8 | F | 16 | 35 | 180/108 | ↔ | ↔ | ↑ | - | 120/90 | ↔ |
|
|
|
| III-15 | F | 16 | 37 | 180/110 | ↔ | ↓ | ↓ | - | 125/96 | ↔ |
|
|
|
| III-16a | M | 15 | 35 | 220/150 | ↓ | ↔ | ↔ | - | 126/88 | ↔ |
|
|
|
| IV-9 | F | 3 | 3 | 130/80 | ↔ | ↔ | ↑ | - | N/A | N/A |
|
|
|
| IV-10 | M | 3 | 10 | 150/130 | ↓ | ↓ | ↓ | - | 100/70 | ↔ |
|
|
|
| II-11 | M | 29 | N/A | N/A | N/A | N/A | N/A | Died of stroke at
the age of 39 years | N/A | N/A |
|
| The present
study | 4 | II-1 | F | 28 | 62 | 151/96 | ↓ | ↓ | ↓ | Stroke at the age
of 58 years | 125/95 | ↔ | - |
|
|
| II-4 | M | 18 | 53 | 160/100 | ↓ | ↔ | ↔ | - | 130/82 | ↔ |
|
|
|
| II-5 | F | 32 | 59 | 167/103 | ↓ | ↓ | ↓ | - | 124/80 | ↔ |
|
|
|
| III-2a | M | 16 | 28 | 200/140 | ↓ | ↓ | ↔ | - | 126/88 | ↔ |
|
|
|
| III-3 | F | - | 40 | 114/85 | ↔ | ↓ | ↔ | - | N/A | N/A |
|
Systematic review and
genotype-phenotype association analysis
A total of 108 patients with Liddle syndrome from 47
families were identified through the literature search. Follow-up
data of these individuals are detailed in Table SI. Median age at hypertension
onset was ~18 years (interquartile range, 14, 23.5 years).
Incidence of hypertension, hypokalemia, hypoaldosteronism and
hyporeninemia was 97.2, 81.3, 65.1 and 80.2%, respectively. In
patients without targeted therapy, the most common hypertensive
damage was left ventricular hypertrophy (9.6%), followed by stroke
(4.6%). A family history of stroke was reported in ~12% of the
patients. After treatment with an ENaC inhibitor (amiloride or
triamterene), blood pressure significantly decreased from
170.8±23.8/107.6±18.7 to 124.2±9.9/79.4±9.9 mmHg (P<0.001) and
serum potassium concentration significantly increased from 3.14±0.5
to 4.3±0.5 mmol/l (P<0.001). During follow-up, only one
mortality that was unexplained and no cardiovascular events were
reported.
Moreover, 13 patients with Liddle syndrome were
identified from three families, all with c.1806dupG in
SCNN1B (15,16). The relevant clinical data for all
individuals known to have the c.1806dupG mutation, to the best of
our knowledge, are summarized in Table II. Individuals carrying this
mutation had varying degrees of blood pressure elevation, except
for III-3 in the present study (17/18, 94.4%). The median age at
hypertension onset was 18 years (interquartile range, 16, 28 years)
and the mean arterial blood pressure was 180/120 mmHg. Hypokalemia
was identified in 10/17 patients (58.9%), hypoaldosteronism in 9/17
(52.9%) and hyporeninemia in 7/17 (41.1%).
Discussion
The present study demonstrated a frameshift base
variant in SCNN1B (NM_ 000336: c.1806dupG, p.Pro603Alafs*5)
causing Liddle syndrome that was characterized by early-onset
refractory hypertension in a family. This mutation leads to the
insertion of G at base 1806 and results in a truncated β subunit of
the ENaC and deletion of the PY motif, with definite pathogenicity
(16). Genotype-phenotype
association analysis demonstrated wide phenotypic variability,
including certain degrees of hypertension, hypokalemia,
hyporeninemia and hypoaldosteronism. The results indicated that
confirmatory genetic testing and targeted therapy could prevent
early-onset clinical endpoints in patients with Liddle
syndrome.
Liddle syndrome is a form of salt-sensitive
hypertension that is caused by mutations in SCNN1A, SCNN1B
and SCNN1G, which respectively encode the α, β and γ ENaC
subunits. The ENaC serves a role in maintaining blood pressure
homeostasis by controlling sodium and potassium handling in the
kidneys (17). The functions of
the ENaC include reabsorption of sodium ions from the lumen into
epithelial cells and pumping these ions into the interstitial fluid
with the assistance of the sodium/potassium ATPase on the
basolateral membrane, as well as potassium secretion (18). Each subunit of the ENaC contains a
large extracellular domain, two transmembrane regions and
relatively short cytoplasmic domains (N-terminus and C-terminus)
(19). A highly conserved
proline-rich sequence, known as the PY motif, has been identified
in the C-terminus of the β and γ subunits, and is recognized by the
domains of the ubiquitin ligase Nedd4-2. The binding of the ENaC to
Nedd4-2 catalyzes the internalization of the ENaC and its
degradation in proteasomes or lysosomes (18,20).
Gain-of-function mutations in the subunits of ENaC result in
deletion or alteration of the PY motif and subsequent blocking of
ubiquitylation and internalization of ENaC (21). Dense accumulation of the ENaC at
the cell surface increases sodium reabsorption and potassium
secretion, thereby leading to the pathophysiological process of
hypertension.
The prevalence of Liddle syndrome in patients with
hypertension in the general population remains unknown. Previously,
Tapolyai et al (22)
studied 149 hypertensive US veterans and reported a similar
biochemical phenotype to Liddle syndrome in 6% of patients. Wang
et al (13) reported a
Liddle syndrome prevalence of 1.52% in 330 hypertensive patients by
sequencing exon 13 of SCNN1B and SCNN1G. Moreover, a
study including 506 patients with early-onset hypertension reported
a Liddle syndrome prevalence of 0.91% (14). Thus, Liddle syndrome may not be
rare in specific populations with early-onset hypertension.
In the present study, a heterozygous G insertion at
base 1806 of the β subunit of ENaC was identified. This leads to
the substitution of alanine for proline at position 603 and
premature emergence of a stop codon at position 607, resulting in
the loss of 34 amino acids, including the PY motif (16). Further assessment (such as with
biochemical assessments, hormone measurements and image
examination) of the proband was performed due to difficulties in
blood pressure control, although there were indications of Liddle
syndrome based on several early signs, such as refractory
hypertension, hypokalemia and a family history of hypertension. To
the best of our knowledge, 14 frameshift mutations in β-ENaC have
been identified in patients with Liddle syndrome (4,23,24).
The mutation in SCNN1B identified in the present study is
consistent with that in previous reports by Cui et al
(15) and Fan et al
(16). Furthermore, although there
have been reports of a variety of frameshift mutations with certain
base insertion or deletion locations in SCNN1B, including
c.1781dupC (p.Ala595Argfs*13) (25), c.1789dupC (p.Arg597Profs*11)
(26,27) and c.1800_1801insG (p.Thr601Aspfs*7)
(28,29), all introduce a new stop codon at
position 607 that is accompanied by deletion of the proline-rich PY
motif. Missense and non-sense mutations have also been reported to
alter the PY motif (15,30).
Liddle syndrome demonstrates wide phenotypic
variability, including varying degrees of hypertension,
hypokalemia, hyporeninemia and hypoaldosteronism (4,30).
However, owing to its low prevalence, the genotype-phenotype
relationship in Liddle syndrome is still unknown. In 2014, Gong
et al (31) compared the
characteristics of patients with p.Arg566* and p.Arg597Pfs*11
mutations in SCNN1B and reported marked phenotypic
variability between families. The p.Arg563Gln mutation in the
C-terminus of the β subunit of ENaC has also been associated with
low-renin and low-aldosterone hypertension; however, only 2.8% of
individuals with the p.Arg563Gln variant present with the full
Liddle syndrome phenotype (32,33).
A previous study identified the p.Asn530Ser mutation in
SCNN1G in the extracellular loop of the γ subunit, not only
in a patient with the Liddle syndrome phenotype, but also in
healthy controls, although available clinical and laboratory
results were limited (29). A
functional study of the p.Asn530Ser mutation in Xenopus
oocytes reported that the open probability was two-fold higher in
mutant ENaC compared with in wild-type ENaC, but with no marked
change in the cell surface expression of ENaC (29). Moreover, the clinical features of
patients may be similar even with certain mutation sites, as
reported by Cui et al (15).
In the present study, a genotype-phenotype
association analysis was performed to gain new insights into the
phenotypic heterogeneity and clinical management of Liddle
syndrome. A literature search identified 18 patients carrying the
p.Pro603Alafs*5 mutation in four families (15,16).
All of these patients were identified in China, suggesting that
this mutation may be a hotspot variant in the Chinese population.
Phenotypic heterogeneity was evident in patients with the same
mutation, as demonstrated by the proportion of patients with
hypertension (94.4%), hypokalemia (58.9%), hypoaldosteronism
(52.9%) and hyporeninemia (41.1%). As demonstrated in the present
study, although patient III-3 carried the pathogenic mutation, the
blood pressure of the patient was within a normal range, which may
be due to a combination of modifier genes and environmental
factors, such as salt intake. Furthermore, stroke occurred in 3
patients and was fatal in one, and in the two families reported by
Cui et al (15), the
mothers of the two probands died before the age of 35 years and
were strongly suspected to carry the same mutation. The patients
with this mutation responded to targeted ENaC inhibitor therapy,
with a return to normal blood pressure and potassium
concentration.
Refractory early-onset hypertension, hypokalemia and
the effective management of the symptoms of Liddle syndrome with
amiloride are strong indicators of Liddle syndrome; however,
molecular genetic testing is essential to diagnose the condition,
especially in patients with isolated hypertension or hypokalemia
(34). Genetic testing also has
implications for the identification of first-degree relatives
carrying the same pathogenic mutation, such as for guiding targeted
drug use and eugenism. The present review demonstrated that 12% of
patients with Liddle syndrome have a family history of stroke and
are often associated with hypertensive target organ damage, such as
left ventricular hypertrophy or stroke, similar to the findings of
Qu et al (35). With the
exception of one patient who died of an unknown cause, all patients
with Liddle syndrome demonstrated significant improvements in blood
pressure and serum potassium concentration after precise ENaC
inhibitor therapy, and no cardiovascular disease-associated events
were reported.
The present study has several limitations. First, a
functional analysis to assess any increases in the
amiloride-sensitive Na+ current of Xenopus
oocytes expressing p.Pro603Alafs*5 was not performed. Considering
that the p.Pro603Alafs*5 variant results in the deletion of the
final 34 amino acids in the β subunit of ENaC, including the PY
motif, it is hypothesized that this mutation is pathogenic and
leads to an increased channel activity and excessive sodium
retention (2,7,16,36).
Second, the effects of amiloride in patients with Liddle syndrome
were based on middle-term treatment only and so a limited duration
of treatment was analyzed; long-term efficacy requires further
follow-up studies. Third, the present study was a small-scale
study. Therefore, large multicenter clinical studies should be
conducted to screen genes for Liddle syndrome in patients with
hypertension and to determine the prevalence of their involvement.
Regular follow-up of patients should also be conducted to determine
the long-term outcomes of patients with Liddle syndrome. In
addition, the lack of available images of the eye was a limitation
of the present study, which was due to the image storage
capabilities of the fundus examination equipment not being fully
implemented.
In the present study, results from whole-exome
sequencing demonstrated a frameshift mutation (NM_000336:
c.1806dupG, p.Pro603Alafs*5) in the β subunit of ENaC that was
associated with Liddle syndrome in a family. The mutation in the
extracellular domain leads to the substitution of alanine in place
of proline at codon 603 and a shortened open reading frame which
creates a premature stop codon at position 607 and results in a
truncated β subunit of ENaC with deletion of the PY motif.
Confirmatory genetic testing is essential and targeted therapy is
necessary to prevent severe sequelae early in life (16). However, further studies are needed
to characterize the long-term prognosis and reasons for the
phenotypic heterogeneity in patients with Liddle syndrome.
Supplementary Material
Supporting Data
Acknowledgements
Not applicable.
Funding
The present work was supported by CAMS Innovation Fund for
Medical Sciences (grant nos. 2022-I2M-C&T-A-010 and
2022-I2M-C&T-A-011).
Availability of data and materials
The datasets generated and/or analyzed during the
current study are available in the Sequence Read Archive
repository, https://www.ncbi.nlm.nih.gov/bioproject/PRJNA1020912/
and all other datasets used and/or analyzed during the current
study are available from the corresponding author on reasonable
request.
Authors' contributions
YLu, XL, YLi and XZ designed the study and modified
the manuscript. YLu, LS, DZ, KY, PF and LZ collected clinical
information and performed data analysis. YLu and XL wrote the
manuscript. YLu and XL confirm the authenticity of all the raw
data. All authors read and approval the final version of the
manuscript.
Ethics approval and consent to
participate
The present study was approved by the Ethics
Committee of Fuwai Hospital (approval no. 2020-1321). All of the
participants provided written informed consent before study
enrolment.
Patient consent for publication
Written informed consent was obtained from the
individual(s) and minor(s)' legal guardian/next of kin for the
publication of any potentially identifiable patient information
included in the present article.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Liddle GW: A familial renal disorder
simulating primary aldosteronism but with negligible aldosterone
secretion. Trans Assoc Am Phys. 76:199–213. 1963.
|
|
2
|
Schild L, Lu Y, Gautschi I, Schneeberger
E, Lifton RP and Rossier BC: Identification of a PY motif in the
epithelial Na channel subunits as a target sequence for mutations
causing channel activation found in Liddle syndrome. EMBO J.
15:2381–2387. 1996. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Yang KQ, Xiao Y, Tian T, Gao LG and Zhou
XL: Molecular genetics of Liddle's syndrome. Clin Chim Acta.
436:202–206. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Tetti M, Monticone S, Burrello J,
Matarazzo P, Veglio F, Pasini B, Jeunemaitre X and Mulatero P:
Liddle syndrome: review of the literature and description of a new
case. Int J Mol Sci. 19:8122018. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Khandelwal P and Deinum J: Monogenic forms
of low-renin hypertension: Clinical and molecular insights. Pediatr
Nephrol. 37:1495–1509. 2021. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Fan P, Pan XC, Zhang D, Yang KQ, Zhang Y,
Tian T, Luo F, Ma WJ, Liu YX, Wang LP, et al: Pediatric liddle
syndrome caused by a novel SCNN1G variant in a chinese family and
characterized by early-onset hypertension. Am J Hypertens.
33:670–675. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Jeunemaitre X, Bassilana F, Persu A,
Dumont C, Champigny G, Lazdunski M, Corvol P and Barbry P:
Genotype-phenotype analysis of a newly discovered family with
Liddle's syndrome. J Hypertens. 15:1091–1100. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Li H and Durbin R: Fast and accurate short
read alignment with Burrows-Wheeler transform. Bioinformatics.
25:1754–1760. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Tarasov A, Vilella AJ, Cuppen E, Nijman IJ
and Prins P: Sambamba: Fast processing of NGS alignment formats.
Bioinformatics. 31:2032–2034. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Li H, Handsaker B, Wysoker A, Fennell T,
Ruan J, Homer N, Marth G, Abecasis G and Durbin R; 1000 Genome
Project Data Processing Subgroup, : The SEquence Alignment/Map
format and SAMtools. Bioinformatics. 25:2078–2079. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Krumm N, Sudmant PH, Ko A, O'Roak BJ,
Malig M and Coe BP; NHLBI Exome Sequencing Project, ; Quinlan AR,
Nickerson DA and Eichler EE: Copy number variation detection and
genotyping from exome sequence data. Genome Res. 22:1525–1532.
2012. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Richards S, Aziz N, Bale S, Bick D, Das S,
Gastier-Foster J, Grody WW, Hegde M, Lyon E, Spector E, et al:
Standards and guidelines for the interpretation of sequence
variants: A joint consensus recommendation of the American college
of medical genetics and genomics and the association for molecular
pathology. Genet Med. 17:405–424. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Wang LP, Yang KQ, Jiang XJ, Wu HY, Zhang
HM, Zou YB, Song L, Bian J, Hui RT, Liu YX and Zhou XL: Prevalence
of liddle syndrome among young hypertension patients of
undetermined cause in a Chinese population. J Clin Hypertens
(Greenwich). 17:902–907. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Liu K, Qin F, Sun X, Zhang Y, Wang J, Wu
Y, Ma W, Wang W, Wu X, Qin Y, et al: Analysis of the genes involved
in Mendelian forms of low-renin hypertension in Chinese early-onset
hypertensive patients. J Hypertens. 36:502–509. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Cui Y, Tong A, Jiang J, Wang F and Li C:
Liddle syndrome: Clinical and genetic profiles. J Clin Hypertens.
19:524–529. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Fan P, Lu CX, Yang KQ, Lu PP, Hao SF, Luo
F, Zhang HM, Song L, Wu HY, Cai J, et al: Truncated epithelial
sodium channel β subunit responsible for liddle syndrome in a
Chinese family. Kidney Blood Press Res. 44:942–949. 2019.
View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Pitzer AL, Van Beusecum JP, Kleyman TR and
Kirabo A: ENaC in salt-sensitive hypertension: Kidney and beyond.
Curr Hypertens Rep. 22:692020. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Hanukoglu I and Hanukoglu A: Epithelial
sodium channel (ENaC) family: Phylogeny, structure-function, tissue
distribution, and associated inherited diseases. Gene. 579:95–132.
2016. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Snyder PM: Minireview: Regulation of
epithelial Na+ channel trafficking. Endocrinology. 146:5079–5085.
2005. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Rotin D and Staub O: Role of the ubiquitin
system in regulating ion transport. Pflugers Arch. 461:1–21. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Bhalla V and Hallows KR: Mechanisms of
ENaC regulation and clinical implications. J Am Soc Nephrol.
19:1845–1854. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Tapolyai M, Uysal A, Dossabhoy NR, Zsom L,
Szarvas T, Lengvárszky Z and Fülöp T: High prevalence of liddle
syndrome phenotype among hypertensive US Veterans in Northwest
Louisiana. J Clin Hypertens (Greenwich). 12:856–860. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Ding X, Jia N, Zhao C, Zhong Y, Dai D,
Zhao Y, Xu C, Cai J, Wang Q and He Q: A family with Liddle's
syndrome caused by a new c.1721 deletion mutation in the epithelial
sodium channel β-subunit. Exp Ther Med. 17:2777–2784.
2019.PubMed/NCBI
|
|
24
|
Fan P, Lu CX, Zhang D, Yang KQ, Lu PP,
Zhang Y, Meng X, Hao SF, Luo F, Liu YX, et al: Liddle syndrome
misdiagnosed as primary aldosteronism resulting from a novel
frameshift mutation of SCNN1B. Endocr Connect. 7:1528–1534. 2018.
View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Findling JW, Raff H, Hansson JH and Lifton
RP: Liddle's syndrome: prospective genetic screening and suppressed
aldosterone secretion in an extended kindred. J Clin Endocrinol
Metab. 82:1071–1074. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Nakano Y, Ishida T, Ozono R, Matsuura H,
Yamamoto Y, Kambe M, Chayama K and Oshima T: A frameshift mutation
of beta subunit of epithelial sodium channel in a case of isolated
Liddle syndrome. J Hypertens. 20:2379–2382. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Awadalla M, Patwardhan M, Alsamsam A and
Imran N: Management of liddle syndrome in pregnancy: A case report
and literature review. Case Rep Obstet Gynecol.
2017:62794602017.PubMed/NCBI
|
|
28
|
Ma X, Tian Y, Gao Y and Guo X: A study of
mutation(s) of the epithelial sodium channel gene in a Liddle's
syndrome family. Zhonghua Nei Ke Za Zhi. 40:390–393. 2001.(In
Chinese). PubMed/NCBI
|
|
29
|
Hiltunen TP, Hannila-Handelberg T,
Petäjäniemi N, Kantola I, Tikkanen I, Virtamo J, Gautschi I, Schild
L and Kontula K: Liddle's syndrome associated with a point mutation
in the extracellular domain of the epithelial sodium channel gamma
subunit. J Hypertens. 20:2383–2390. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Bogdanović R, Kuburović V, Stajić N,
Mughal SS, Hilger A, Ninić S, Prijić S and Ludwig M: Liddle
syndrome in a Serbian family and literature review of underlying
mutations. Eur J Pediatr. 171:471–478. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Gong L, Chen J, Shao L, Song W, Hui R and
Wang Y: Phenotype-genotype analysis in two Chinese families with
Liddle syndrome. Mol Biol Rep. 41:1569–1575. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Rayner BL, Owen EP, King JA, Soule SG,
Vreede H, Opie LH, Marais D and Davidson JS: A new mutation, R563Q,
of the beta subunit of the epithelial sodium channel associated
with low-renin, low-aldosterone hypertension. J Hypertens.
21:921–926. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Jones ES, Owen EP, Davidson JS, Van Der
Merwe L and Rayner BL: The R563Q mutation of the epithelial sodium
channel beta-subunit is associated with hypertension. Cardiovasc J
Afr. 22:241–244. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Brower RK, Ghlichloo IA, Shabgahi V,
Elsholz D, Menon RK and Vyas AK: Liddle syndrome due to a novel
c.1713 deletion in the epithelial sodium channel β-subunit in a
normotensive adolescent. AACE Clin Case Rep. 7:65–68. 2021.
View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Qu Y, Lu Y, Zhang D, Liu X, Fan P, Chen J,
Zhang H, Yang K, Tian T, Zhou Y, et al: Identification of a novel
frameshift mutation in the SCNN1B causing Liddle syndrome. Sci Bull
(Beijing). 68:383–387. 2023. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Hansson JH, Nelson-Williams C, Suzuki H,
Schild L, Shimkets R, Lu Y, Canessa C, Iwasaki T, Rossier B and
Lifton RP: Hypertension caused by a truncated epithelial sodium
channel gamma subunit: genetic heterogeneity of Liddle syndrome.
Nat Genet. 11:76–82. 1995. View Article : Google Scholar : PubMed/NCBI
|