Introduction
Currently, malignant tumors seriously endanger human
life and health. Chemotherapy is considered as the most significant
treatment strategy against cancer (1–4).
Pirarubicin (THP) is a common chemotherapeutic drug used
clinically. However, due to its cardiotoxicity, its clinical
application remains limited (5–8).
It has been reported that cardiotoxicity caused by
pirarubicin (CTP) is closely associated with the occurrence of
oxidative stress in cardiomyocytes (9–11).
Reactive oxygen species (ROS), the key intermediate of oxidative
stress, plays a significant role in CTP (12,13).
NADPH oxidases (NOXs), a major intracellular enzymatic source of
ROS, are transmembrane complexes with electron-transferring ability
that produce ROS (14,15). NOX2 is abundantly expressed in
cardiomyocytes (16,17). A previous study demonstrated that
increased ROS levels promoted mitochondrial dysfunction and it was
therefore considered as a significant factor in
mitochondria-mediated apoptosis (18). In addition, enhanced ROS levels
have also been associated with lipid peroxidation, which in turn
promotes the onset of a unique cell death mode, namely ferroptosis
(19).
Scutellarein (Sc), a flavone monomer with known
anti-inflammatory and antioxidant properties, is widely used in
food and medical products (20–22).
Previous studies demonstrated that Sc could improve oxidative
stress in a diabetes mouse model and superoxide-induced rat
cortical synaptosomes (23,24).
Based on the aforementioned findings, it was hypothesized that food
therapy with Sc ameliorated CTP via inhibition of apoptosis and
ferroptosis through regulation of oxidative stress. However, this
hypothesis has not been confirmed in in vivo or in
vitro studies, while the effect of Sc on NOX2 remains largely
unknown.
The present study aimed to explore the
anti-oxidative stress, anti-ferroptosis and anti-apoptotic
properties of Sc and the effects of the Sc-related key pathways on
regulating oxidative stress, apoptosis and ferroptosis in CTP.
Materials and methods
Materials
The H9c2 cardiomyocyte cell line (cat. no. ZQ0102)
was provided by Shanghai Zhongqiao Xinzhou Biotechnology Co., Ltd.
THP, Sc, dexrazoxane (Dex; a drug particularly approved by the US
Food and Drug Administration for the treatment of CTP), GSK2795039
(GSK), ferrostatin-1 (Fer-1) and erastin were purchased from
MedChemExpress. The brain natriuretic peptide (BNP, cat. no.
H166-1-2), creatine kinase MB (CK-MB, cat. no. H197-1-1) and
cardiac troponin T (cTnT, cat. no. H149-4-2) kits were purchased
from Nanjing Jiancheng Bioengineering Institute. Cell Counting
Kit-8 (CCK-8), ROS (cat. no. S0033M) and TUNEL (cat. no. C10088)
apoptosis assay kits were purchased from Beyotime Institute of
Biotechnology. DMEM and FBS were obtained from Gibco (Thermo Fisher
Scientific, Inc.) and BioAgrio, respectively. The reduced
glutathione (GSH, cat. no. A006-2-1), glutathione peroxidase
(GSH-Px, cat. no. A005-1-2), catalase (CAT, cat. no. A007-1-1),
malondialdehyde (MDA, cat. no. A003-1-2), superoxide dismutase
(SOD, cat. no. A001-3-2), lactate dehydrogenase (LDH, cat. no.
A020-2-2) and total antioxidant capacity (T-AOC, cat. no. A015-2-1)
assay kits were obtained from Nanjing Jiancheng Bioengineering
Institute. An iron assay kit (, cat. no. ab83366) was purchased
from Abcam, while the antibodies against glutathione peroxidase 4
(GPX4), NOX2, NOX4, erythroid 2-related factor 2 (NRF2), Bax,
Bcl-2, GAPDH, caspase 3 and caspase 9 from Proteintech Group,
Inc.
Animal studies
Animal model and diet
In the present study, a total of 50 male
Sprague-Dawley (SD) rats (weight, 180–200 g) were obtained from the
Experimental Animal Center of Chongqing Medical University. Rats
were maintained under specific pathogen-free conditions at 23±2°C,
55±5% relative humidity, a 12-h light/dark cycle and had free
access to food and water. The rats were randomly divided into the
following five groups (n=10 rats/group): i) The normal diet group
(ND), where rats were fed standard chow and injected with an equal
volume of normal saline via the tail vein, once a week for eight
weeks; ii) the Sc group (Sc), where rats were fed with Sc feed (100
mg/kg) and injected with an equal volume of normal saline via the
tail vein, once a week for eight weeks; iii) the THP group (THP),
where rats were fed with standard chow, while 3 mg/kg THP was
injected into the tail vein once a week for eight weeks (25); iv) the Sc + THP group (Sc + THP),
were rats were fed with Sc feed (100 mg/kg) and 3 mg/kg THP was
injected into the tail vein once a week for eight weeks; and v) the
Dex + THP group (Dex + THP), where rats were fed with standard
chow, while 3 mg/kg THP and 30 mg/kg Dex was injected into the tail
vein and abdominal cavity, respectively, once a week for eight
weeks. The survival of rats was recorded every day, while food
consumption and rat weight were recorded once a week.
Echocardiography
The experiment was completed at week 8. Rats were
first anesthetized by isoflurane inhalation (2% for induction and
2% for maintenance). Subsequently, after removing the chest hair of
rats, the VIVID E95 and L8-18I-D probes (General Electric Company)
were used to perform doppler echocardiography to measure ejection
fraction (EF), fractional shortening (FS), left ventricular
end-diastolic diameter (LVIDd) and left ventricular end-systolic
diameter (LVIDs).
Sample collection, preparation and
biochemical analysis
Following overnight fasting, rats were weighed and
euthanized by cervical dislocation following anesthesia with 1%
pentobarbital (40 mg/kg). Rat hearts were then removed, weighed and
stored at −80°C until further use. A part of the heart tissues was
homogenized and the levels of iron, GSH, GSH-Px, MDA, SOD and T-AOC
were immediately measured, according to the manufacturer's
instructions. Within 2 h, blood samples were collected from the
abdominal aorta and centrifuged at 900 × g for 30 min at room
temperature. The supernatant was then stored at −80°C. The serum
levels of LDH, BNP, CK-MB and cTnT were directly determined using
the corresponding kits.
Cell studies
Cell culture, treatment and
grouping
H9c2 cells were cultured in DMEM supplemented with
10% (v/v) FBS in a humidified incubator with 95% air and 5%
CO2 at 37°C. To establish an in vitro injury
model, H9c2 cells were treated with 5 µmol/l THP for 24 h.
Subsequently, to evaluate the association between oxidative stress,
ferroptosis and apoptosis in CTP, the in vitro experiments
were carried out into two parts. Therefore, cells were grouped as
follows: a) Direction of oxidative stress, including i) the control
(CON) group, where cells were cultured in DMEM; ii) the THP group
(THP), where cells were treated with 5 µmol/l THP for 24 h; iii)
the Sc + THP group (Sc + THP), where cells were pretreated with 100
µmol/l Sc for 1 h followed by treatment with 5 µmol/l THP for 24 h;
iv) the GSK group (GSK), where cells were treated with 25 µmol/l
GSK for 24 h (26); v) the GSK +
THP group (GSK + THP), where cells were co-treated with 5 µmol/l
THP and 25 µmol/l GSK for 24 h; and vi) the Sc + GSK + THP group
(Sc + GSK + THP), where cells were pretreated with 100 µmol/l Sc
for 1 h followed by co-treatment with 5 µmol/l THP and 25 µmol/l
GSK for 24 h. b) Direction of ferroptosis, including i) the CON
group (CON), where cells were cultured in DMEM; ii) the THP group
(THP), where cells were treated with 5 µmol/l THP for 24 h; iii)
the Sc + THP group (Sc + THP), where cells were pretreated with 100
µmol/l Sc for 1 h, followed by treatment with 5 µmol/l THP for 24
h; iv) the Fer-1 group, where cells were treated with 10 µmol/l
Fer-1 for 24 h (27); v) the Fer-1
+ THP group (Fer-1 + THP), where cells were co-treated with 10
µmol/l Fer-1 and 5 µmol/l THP for 24 h; vi) the erastin group
(erastin), where cells were treated with 5 µmol/l erastin for 24 h
(28); and the erastin + Sc group
(erastin + Sc), where cells were pretreated with 100 µmol/l Sc for
1 h, followed by co-treatment with 5 µmol/l erastin for an
additional 24 h.
Cell viability assay
H9c2 cells were seeded in 96-well plates at a
density of 5×103 cells/well for 12 h, prior to use.
Following treatment, a CCK-8 assay kit was used to evaluate cell
viability. Briefly, cells in each well were supplemented with 10 µl
CCK-8 reagent followed by incubation for 2 h. Subsequently, the
absorbance in each well was measured at a wavelength of 450 nm
using a single-wavelength microplate reader.
ROS staining
Cells were seeded into 24-well plates and after
reaching 50–60% confluency, they were treated with the indicated
compounds. Subsequently, cells were stained with DCFH-DA dye (37°C,
20 min), provided by the ROS kit, according to the manufacturer's
instructions (Beyotime Institute of Biotechnology). Cells were
observed under a fluorescence microscope and the positive stained
area was measured using ImageJ v1.53c software (National Institutes
of Health).
Cell apoptosis
For cell apoptosis assessment, cells were seeded
into 24-well plates and after reaching 50–60% confluency, the cells
were treated as previously described. Subsequently, cell apoptosis
was assessed using a TUNEL apoptosis assay kit, according to the
manufacturer's instructions. Briefly, following fixing at room
temperature for 30 min (Immunostaining fixative, Beyotime Institute
of Biotechnology, cat. no. P0098), H9c2 cells were washed with
ice-cold PBS and stained with DAPI (at room temperature for 5 min,
Beyotime Institute of Biotechnology, cat. no. C1005) and TUNEL dyes
(protected from light, at 37°C for 60 min). After washing,
fluorescent images were captured under a fluorescence microscope
and analyzed using ImageJ v1.53c software.
Immunofluorescence staining
Cells were fixed with 4% formaldehyde (at room
temperature for 15 min), washed with PBS and were then
permeabilized with 0.2% Triton X-100 (at room temperature for 20
min). Following blocking with goat serum (cat. no. C0265; Beyotime
Institute of Biotechnology) at room temperature for 30 min, the
cells were first incubated at 4°C, overnight with primary
antibodies against NOX2 (cat. no. 19013-1-AP; 1:200; Proteintech
Group, Inc.) and then with the corresponding secondary antibody
[FITC-labeled goat anti-rabbit IgG (H+L); cat. no. A0562; 1:500;
Beyotime Institute of Biotechnology] at room temperature for 90
min. Finally, the cell nuclei were stained with DAPI (at room
temperature for 5 min) and images were captured under a
fluorescence microscope.
Western blot analysis
The protein expression levels of GAPDH (cat. no.
10494-1-AP; 1:5,000), NOX2 (cat. no. 19013-1-AP; 1:1,000), NOX4
(cat. no. 14347-1-AP; 1:1,000), NRF2 (cat. no. 16396-1-AP;
1:2,000), GPX4 (cat. no. 30388-1-AP; 1:500), Bax (cat. no.
50599-2-Ig; 1:2,000), Bcl-2 (cat. no. 26593-1-AP; 1:500), cleaved
and total caspase 3 (cat. no. 19677-1-AP; 1:500), and cleaved and
total caspase 9 (cat. no. 10380-1-AP; 1:500) were detected by
western blot analysis. Briefly, H9c2 cells or cardiac tissue were
lysed in RIPA lysis buffer (Beyotime Institute of Biotechnology)
with 1% (v/v) phenylmethylsulfonyl fluoride. Following
centrifugation at 13,700 × g for 15 min at 4°C, the supernatants
were collected and then the protein concentration was measured
using a BCA protein determination kit. An equal amount of protein
extracts (30 µg) was separated by 12% SDS-PAGE and proteins were
then transferred onto a PVDF membrane. Following blocking with 5%
(w/v) skimmed milk (at room temperature for 2 h), the membrane was
cut into strips according to the molecular weight of each
target-protein. Subsequently, the membrane was first incubated with
primary antibodies overnight at 4°C and then with the corresponding
horseradish peroxidase-conjugated secondary antibodies (cat. no.
SA00001-2; Proteintech Group, Inc.; 1:5,000). The protein bands
were visualized using an ECL reagent (Biosharp life sciences, cat.
no. BL520B). GAPDH served as an internal control for protein
loading and analysis. ChemiDoc™ XRS+ with Image Lab Software
(BIO-RAD) was used for densitometry.
Statistical analysis
GraphPad Prism 8.0 (Dotmatics) was used for
statistical analysis. All experiments were repeated at least three
times. The data are expressed as the mean ± standard deviation.
First, the normal distribution and homogeneity of variance of the
data were assessed. The differences between groups were compared
using one- or two-way ANOVA, followed by Tukey's multiple
comparison post hoc test. P<0.05 was considered to indicate a
statistically significant difference.
Results
Sc effectively improves the
THP-mediated changes in food intake, body weight, heart indexes and
survival in SD rats
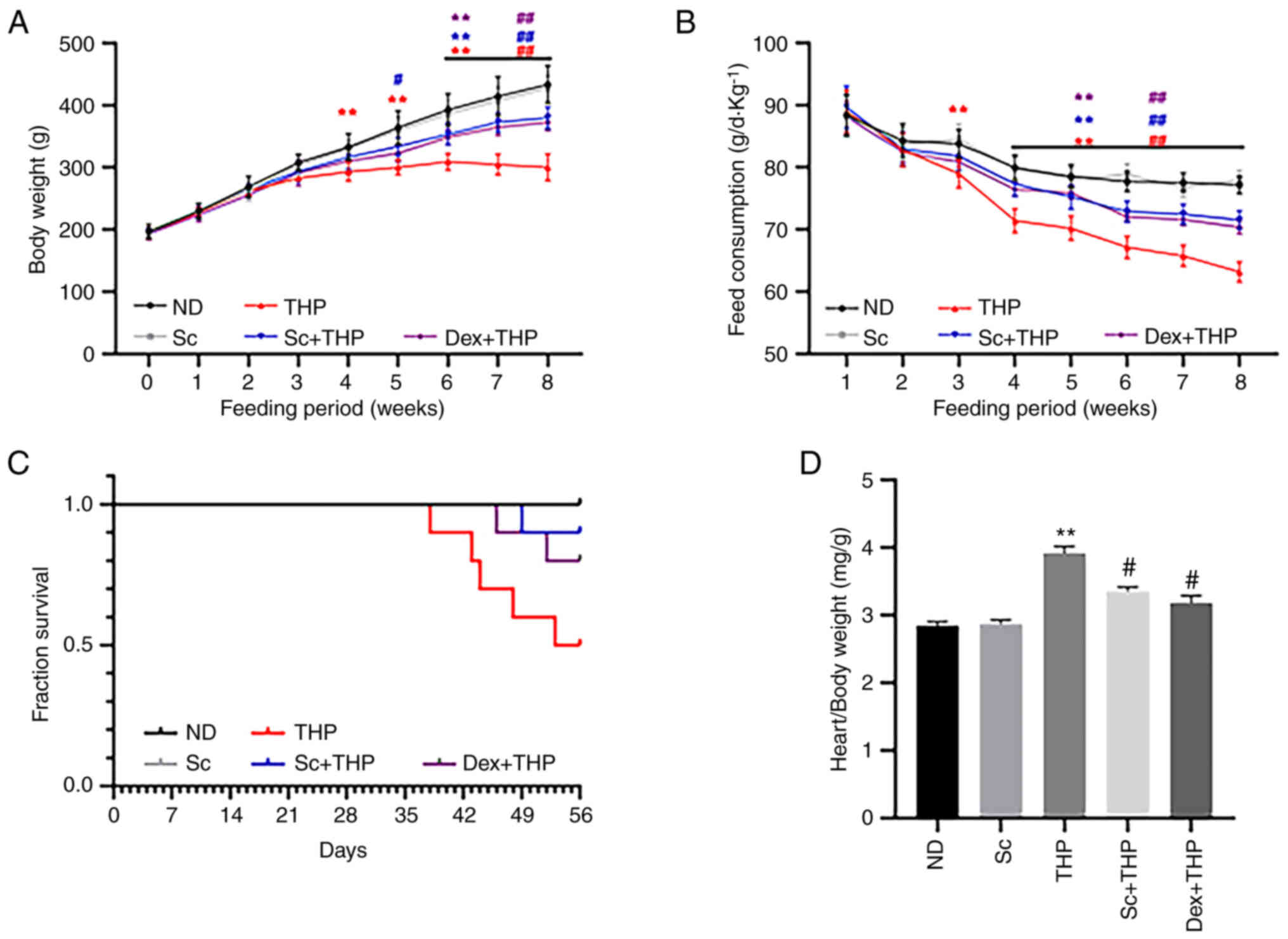
As shown in Fig. 1,
compared with the ND group, food intake was significantly reduced
from the third week in the THP group (THP vs. ND, P<0.01;
Fig. 1B). In addition, rat weight
was also notably decreased in the THP group at the fourth week
compared with the ND group (THP vs. ND, P<0.01; Fig. 1A). After eight weeks, the survival
rate of rats in the THP group was markedly reduced compared with
the ND group (Fig. 1C), while the
cardiac mass index was notably enhanced (THP vs. ND, P<0.01;
Fig. 1D). However, co-treatment of
rats with Sc and Dex significantly restored the aforementioned
changes (Sc + THP vs. THP: P<0.05, cardiac mass index and
P<0.01, body weight and food intake; Dex + THP vs. THP:
P<0.05, cardiac mass index and P<0.01, body weight and food
intake; Fig. 1A-D).
Sc effectively improves the
THP-induced abnormal changes in myocardial injury markers and
echocardiography in SD rats
As shown in Fig. 2,
after treatment of SD rats with THP for eight weeks, significant
changes were observed in the echocardiography parameters in the THP
group compared with the ND group (Fig.
2A), including decreased EF and FS (THP vs. ND, P<0.01;
Fig. 2B and C) and increased LVIDd
and LVIDs (THP vs. ND, P<0.01; Fig.
2D and E). At the same time, abnormal levels of the myocardial
injury-related markers, BNP, CK-MB, cTnT and LDH, were recorded in
the THP group (THP vs. ND, P<0.01; Fig. 2F-I). However, the aforementioned
changes were improved following treatment of SD rats with Sc and
Dex (Sc + THP vs. THP, P<0.05 for FS, LVIDd, LVIDs, cTnT and
LDH, and P<0.01 for EF, BNP, CK-MB; Dex + THP vs. THP, P<0.05
for EF, FS, LVIDd, LVIDs, BNP and LDH, and P<0.01 for CK-MB and
cTnT; Fig. 2A-I).
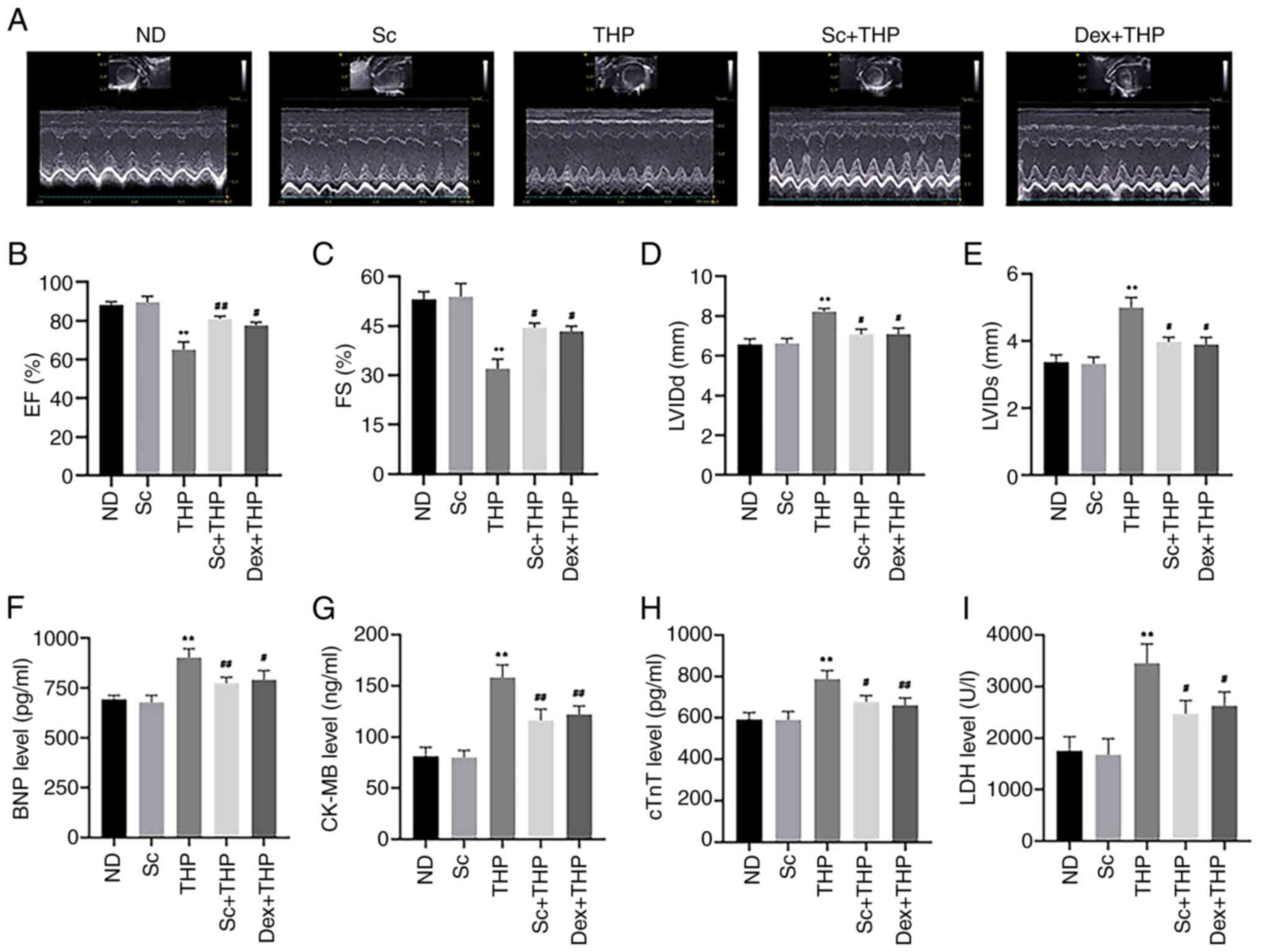 | Figure 2.Sc improves the effects of myocardial
injury markers and echocardiography results, induced by THP in SD
rats. (A) Results of echocardiography in various groups.
Quantitative analysis of the (B) EF, (C) FS, (D) LVIDd and (E)
LVIDs in each group. Quantitative analysis in each group of the
myocardial injury markers: (F) BNP, (G) CK-MB, (H) cTnT and (I)
LDH. Values are expressed as the means ± standard deviation.
**P<0.01 vs. CON. #P<0.05 and
##P<0.01 vs. THP. Sc, scutellarein; THP, pirarubicin;
EF, ejection fraction; FS, fractional shortening; LVIDd, left
ventricular end-diastolic diameter; LVIDs, left ventricular
end-systolic diameter; BNP, brain natriuretic peptide; CK-MB,
creatine kinase MB; cTnT, cardiac troponin T; LDH, lactate
dehydrogenase; ND, normal diet; Dex, dexrazoxane. |
Sc alleviates the THP-induced abnormal changes in
the oxidative stress- and ferroptosis-related indexes in blood and
myocardial tissues of SD rats. Compared with the ND group,
Fe2+, Fe3+ and total Fe levels were enhanced
in the THP group (THP vs. ND, P<0.05 for Fe3+ and
P<0.01 for Fe2+ and total Fe; Fig. 3A-C), while this effect was restored
by rat treatment with Sc and Dex (Sc + THP vs. THP, P<0.05 for
Fe2+ and total Fe; Dex + THP vs. THP, P<0.05 for
Fe2+ and total Fe; Fig. 3A
and C). In addition, the levels of CAT, GSH, GSH-Px, SOD and
T-AOC were reduced (THP vs. ND, P<0.01; Fig. 3D-H), while the level of MDA was
increased (THP vs. ND, P<0.01; Fig.
3I) in the THP group compared with the ND group, and these
effects were also restored following treatment of SD rats with Sc
and Dex (Sc + THP vs. THP, P<0.05 for CAT, GSH and SOD, and
P<0.01for GSH-Px, T-AOC and MDA; Dex + THP vs. THP, P<0.05
for GSH, SOD and T-AOC, and P<0.01 for CAT, GSH-Px and MDA;
Fig. 3D-I).
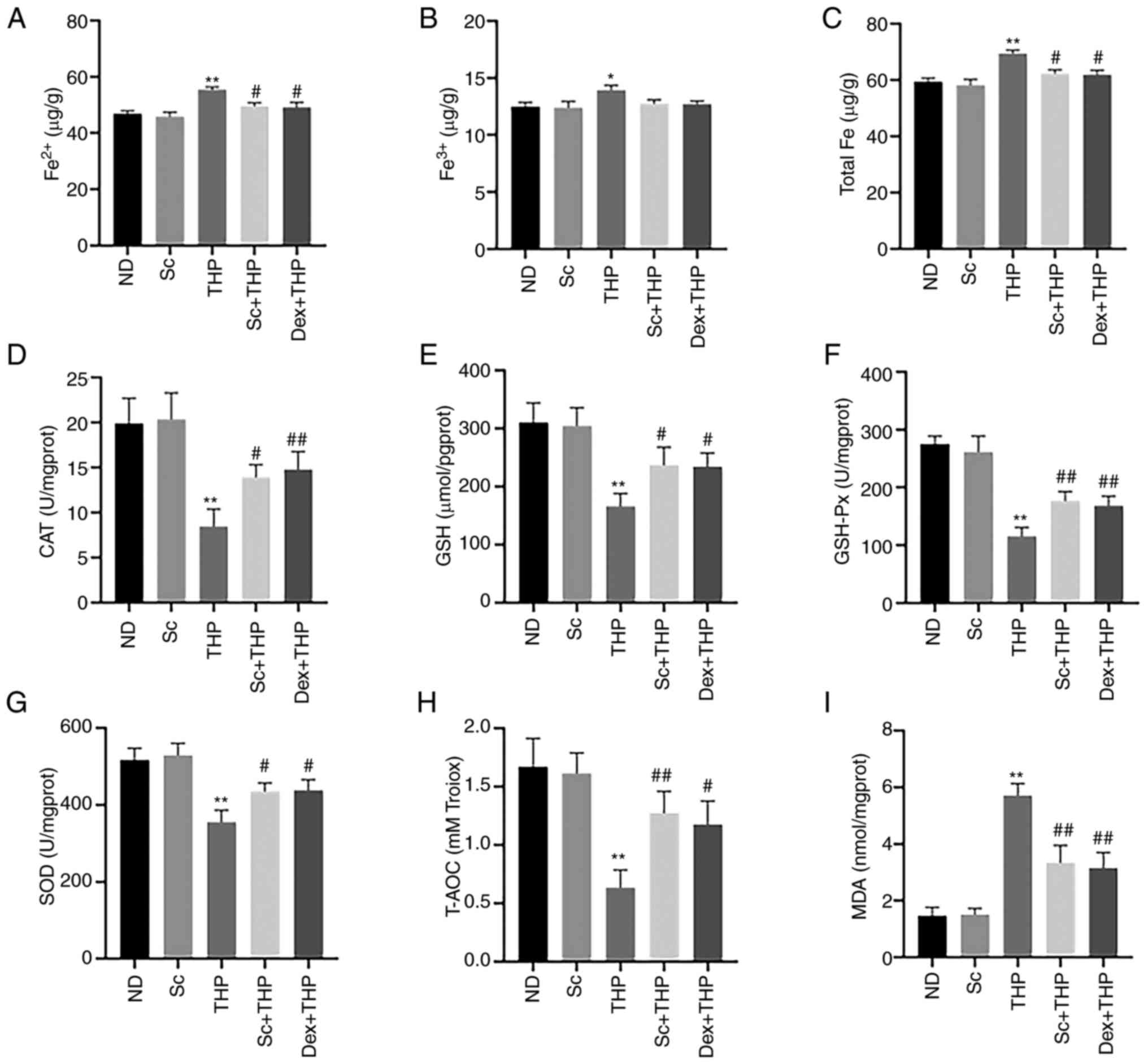 | Figure 3.Sc alleviates the THP-induced
aberrant effects of indexes related to oxidative stress and
ferroptosis in blood and myocardial tissue in vivo.
Quantitative analysis in each group of the indexes related to
oxidative stress and ferroptosis: (A) Fe2+, (B)
Fe3+, (C) total Fe, (D) CAT, (E) GSH, (F) GSH-Px, (G)
SOD, (H) T-AOC, and (I) MDA. Values are expressed as the means ±
standard deviation. *P<0.05 and **P<0.01 vs. CON.
#P<0.05 and ##P<0.01 vs. THP. Sc,
scutellarein; THP, pirarubicin; CAT, catalase; GSH, glutathione;
GSH-Px, glutathione peroxidase; SOD, superoxide dismutase; T-AOC,
total antioxidant capacity; MDA, malondialdehyde; ND, normal diet;
Dex, dexrazoxane. |
Effects of Sc and THP on the expression of oxidative
stress-, ferroptosis- and apoptosis-related proteins in the
myocardium of SD rats. The results of western blot analysis showed
that THP increased the expression of oxidative stress-related
proteins, such as NOX2 and NOX4, and decreased those of NRF2, in
the myocardial tissues of SD rats (THP vs. ND, P<0.01 for NOX2
and NRF2, and P<0.001 for NOX4; Fig. 4A-D). Additionally, THP
downregulated GPX4, a ferroptosis-related protein (THP vs. ND,
P<0.01; Fig. 4E). In terms of
apoptosis, THP notably enhanced the Bax/Bcl-2 ratio, the cleaved
caspase 3/total caspase 3 and cleaved caspase 9/total caspase 9
ratio (THP vs. ND, P<0.001 for Bax/Bcl-2, cleaved caspase
3/total caspase 3 and cleaved caspase 9/total caspase 9; Fig. 4A and F-H), while these were
restored by Sc and Dex (Sc + THP vs. THP, P<0.05 for NOX2, NRF2,
GPX4, cleaved caspase 3/total caspase 3 and cleaved caspase 9/total
caspase 9, and P<0.01 for NOX4, Bax/Bcl-2; Dex + THP vs. THP,
P<0.05 for NOX2, NRF2, GPX4, Bax/Bcl-2, cleaved caspase 3/total
caspase 3 and cleaved caspase 9/total caspase 9, and P<0.01 for
NOX4; Fig. 4B-H).
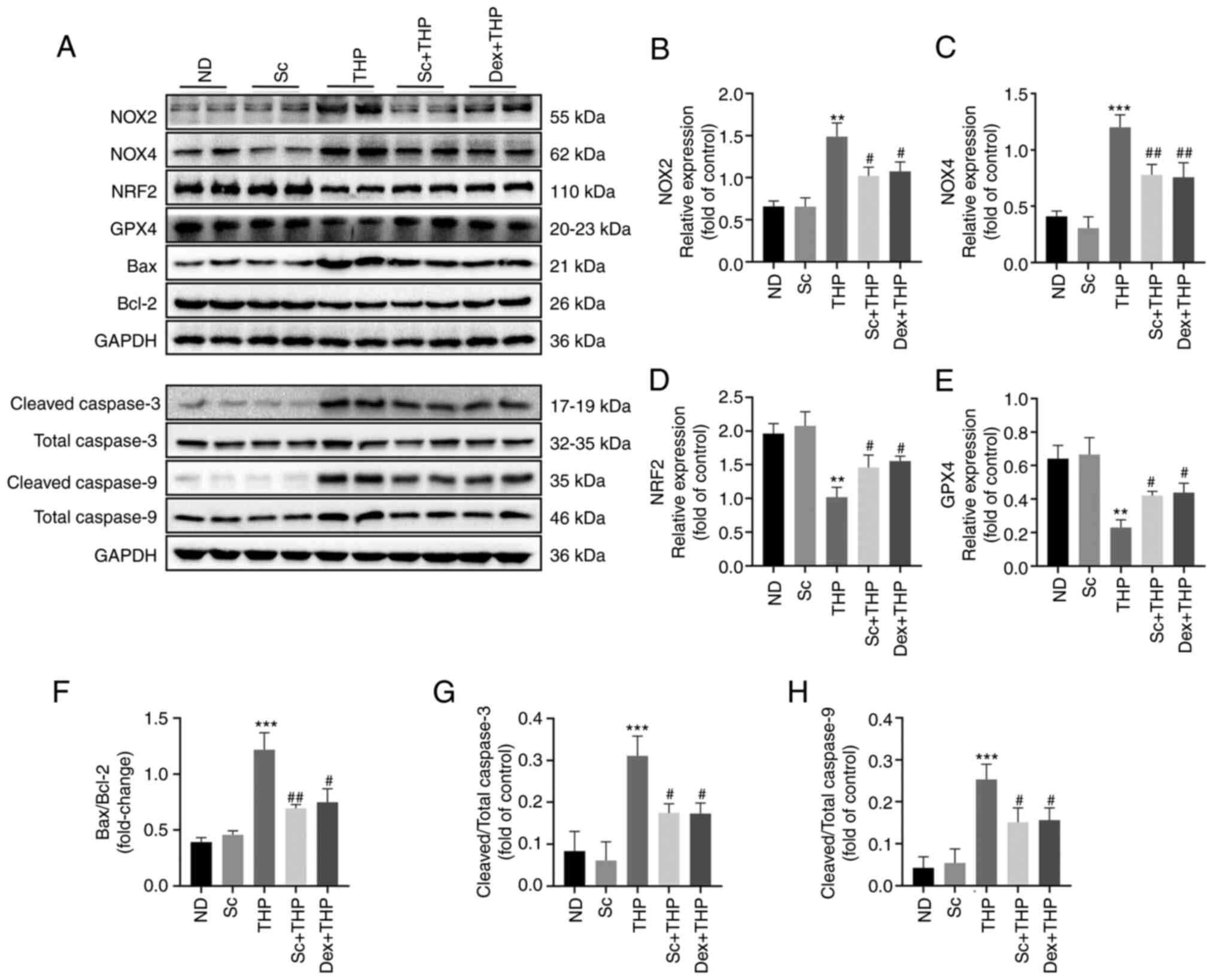 | Figure 4.Sc improves the oxidative stress,
ferroptosis, and apoptosis-related protein effects induced by THP
in the myocardium of SD rats. (A) Western blots. Semi-quantitative
analysis of the protein expression of (B) NOX2, (C) NOX4, (D) NRF2,
(E) GPX4, (F) Bax/Bcl-2, (G) cleaved caspase 3/total caspase 3 and
(H) cleaved caspase 9/total-caspase 9 in each group. Values are
expressed as the means ± standard deviation. **P<0.01 and
***P<0.001 vs. ND. #P<0.05 and
##P<0.01 vs. THP. Sc, scutellarein; THP, pirarubicin;
NOX2, NADPH oxidase 2; NOX4, NADPH oxidase 4; NRF2, erythroid
2-related factor 2; GPX4, glutathione peroxidase 4; ND, normal
diet; Dex, dexrazoxane. |
Sc ameliorates the THP-mediated
decrease in H9c2 myocardial cell viability
In vitro experiments using CCK-8 assays,
showed that 5 µmol/l THP and 100 µmol/l Sc were the optimal
concentrations to treat cells (5 µmol/l THP vs. CON, P<0.001;
100 µmol/l Sc + THP vs. THP, P<0.01; Fig. 5A and B). CCK-8 assays showed that
THP significantly reduced the viability of H9c2 cells, which was
improved by cell treatment with Sc, GSK and Fer-1 (THP vs. CON,
P<0.01; Sc + THP vs. THP, P<0.05; GSK + THP vs. THP,
P<0.05; and Fer-1 + THP vs. THP, P<0.05; Fig. 5C-E). Furthermore, erastin had a
similar effect with THP on cell viability, which was also
alleviated by Sc (erastin vs. CON, P<0.01; and erastin + Sc vs.
erastin, P<0.01; Fig. 5E).
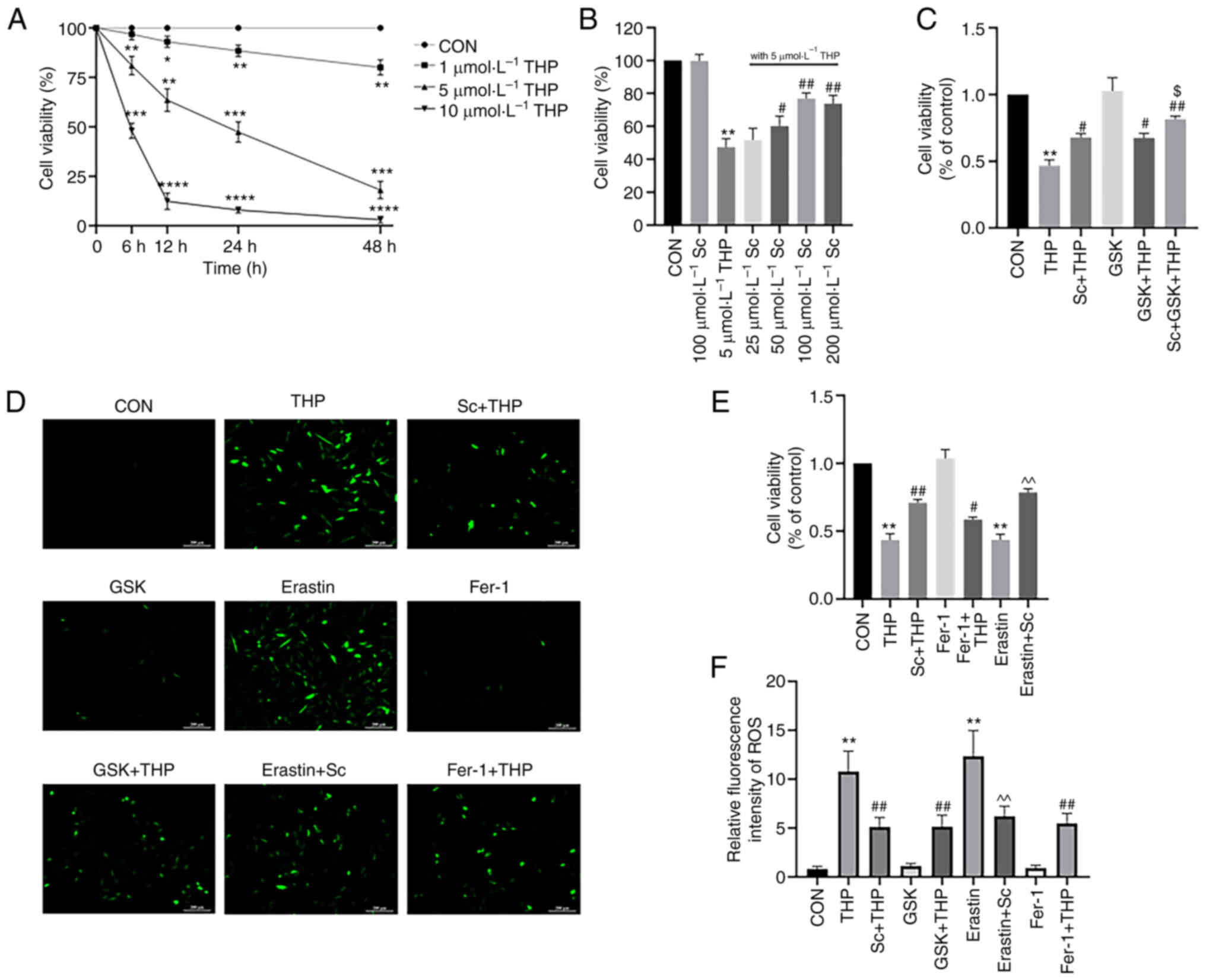 | Figure 5.Effects of Sc, THP, GSK, Fer-1, and
erastin on the cell viability and the production of ROS in H9c2
cells. According to CCK-8 assays, it was determined that the
treatment concentration of THP was (A) 5 µmol/l and that of (B) Sc
was 100 µmol/l. (C and E) Quantitative analysis of the CCK-8 assays
in each group receiving corresponding treatment. (D) ROS staining.
(F) Semi-quantitative analysis of ROS in each group. Values are
expressed as the means ± standard deviation. *P<0.05,
**P<0.01, ***P<0.001 and ****P<0.0001 vs. CON.
#P<0.05 and ##P<0.01 vs. THP.
$P<0.05 vs. GSK + THP. ^^P<0.01 vs.
erastin. Sc, scutellarein; THP, pirarubicin; GSK, GSK2795039;
Fer-1, ferrostatin-1; ROS, reactive oxygen species; CCK-8, Cell
Counting Kit-8; CON, control. |
Effects of Sc, THP, GSK, Fer-1 and
erastin on ROS generation in H9c2 cardiomyocytes
As shown in Fig. 5D and
F, THP enhanced ROS production in H9c2 cells, while Sc and GSK
antagonized this effect (THP vs. CON, P<0.01; Sc + THP vs. THP,
P<0.01; and GSK + THP vs. THP, P<0.01; Fig. 5F). Furthermore, erastin also
increased ROS production in H9c2 cells, which was alleviated by Sc
(erastin vs. CON, P<0.01; erastin + Sc vs. erastin, P<0.01;
Fig. 5F). Consistently, Fer-1 also
improved the THP-mediated increase in ROS production (Fer-1 + THP
vs. THP, P<0.01; Fig. 5F).
Effects of Sc, THP, GSK, Fer-1 and
erastin on H9c2 cardiomyocyte apoptosis
TUNEL staining results indicated that THP promoted
H9c2 cell apoptosis (THP vs. CON, P<0.01; Fig. 6A and B). This effect was abrogated
by cell treatment with GSK, Fer-1 and Sc (Sc + THP vs. THP,
P<0.01; GSK + THP vs. THP, P<0.01; and Fer-1 + THP vs. THP,
P<0.01; Fig. 6A and B). In
addition, erastin also enhanced H9c2 cell apoptosis, which was also
improved by Sc (erastin vs. CON, P<0.01; erastin + Sc vs.
erastin, P<0.01; Fig. 6A and
B).
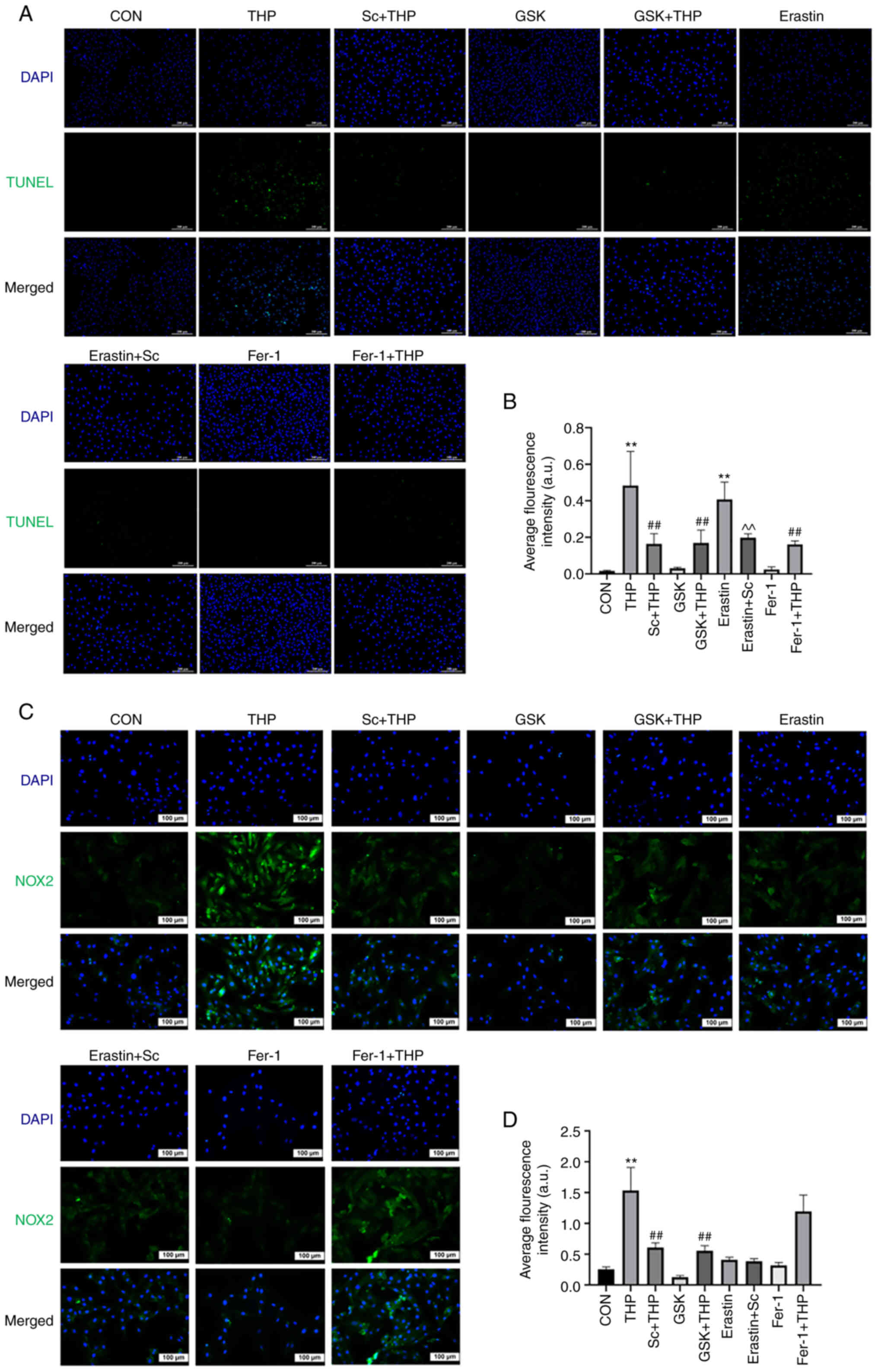 | Figure 6.Effects of Sc, THP, GSK, Fer-1, and
erastin on the apoptosis of H9c2 cardiomyocytes and the expression
of NOX2. (A) TUNEL assay. (B) Semi-quantitative analysis of the
average fluorescence intensity of the results of the TUNEL assay.
(C) Immunofluorescence staining of NOX2. (D) Semi-quantitative
analysis of the average fluorescence intensity of NOX2. Values are
expressed as means ± standard deviation. **P<0.01 vs. CON.
##P<0.01 vs. THP. ^^P<0.01 vs. erastin.
Sc, scutellarein; THP, pirarubicin; GSK, GSK2795039; Fer-1,
ferrostatin-1; NOX2, NADPH oxidase 2; CON, control. |
Effects of Sc, THP, GSK, Fer-1 and erastin on the
expression of oxidative stress-, ferroptosis- and apoptosis-related
proteins in H9c2 cardiomyocytes. The in vitro immunofluorescence
results shown in Fig. 6C and D,
revealed that compared with the CON group, NOX2 was upregulated in
the THP group (THP vs. CON, P<0.01; Fig. 6C and D). The protein expression
levels of NOX2 were restored following cell treatment with Sc and
GSK (Sc + THP vs. THP, P<0.01; and GSK + THP vs. THP, P<0.01;
Fig. 6C and D). However, Fer-1 and
erastin did not significantly affect NOX2 expression. Furthermore,
the western blot results also demonstrated that THP markedly
upregulated NOX2, increased the Bax/Bcl-2, cleaved caspase 3/total
caspase 3, cleaved caspase 9/total caspase 9 ratio and
downregulated GPX4 (THP vs. CON, P<0.01 for cleaved caspase
3/total caspase 3; P<0.001 for GPX4, cleaved caspase 9/total
caspase 9; and P<0.0001 for NOX2 and Bax/Bcl-2; Fig. 7C). The aforementioned results were
restored by cell treatment with Sc (Sc + THP vs. THP, P<0.05 for
NOX2, GPX4, Bax/Bcl-2, cleaved caspase 3/total caspase 3 and
cleaved caspase 9/total caspase 9; Fig. 7C). At the same time, the
aforementioned effects were also improved by GSK treatment (GSK +
THP vs. THP, P<0.05 for NOX2, GPX4, Bax/Bcl-2, cleaved caspase
3/total caspase 3 and cleaved caspase 9/total caspase 9; Fig. 7C). Consistent with the
immunofluorescence results, erastin and Fer-1 had no effect on NOX2
expression (Fig. 7D). Notably,
H9c2 cell treatment with erastin increased the Bax/Bcl-2 ratio,
upregulated cleaved caspase 3/9, total caspase 3/9 and
downregulated GPX4 (erastin vs. CON, P<0.01 for cleaved caspase
3/total caspase 3, cleaved caspase 9/total caspase 9; and
P<0.001 for GPX4 and Bax/Bcl-2; Fig. 7D), while Sc improved some
aforementioned effects (erastin + Sc vs. erastin, P<0.05 for
cleaved caspase 3/total caspase 3 and cleaved caspase 9/total
caspase 9; and P<0.01 for GPX4 and Bax/Bcl-2; Fig. 7D). In addition, Fer-1 improved
aberrant protein effects in H9c2 cells induced by THP (Fer-1 + THP
vs. THP, P<0.05 for GPX4, cleaved caspase 3/total caspase 3 and
cleaved caspase 9/total caspase 9; and P<0.01 for Bax/Bcl-2;
Fig. 7D).
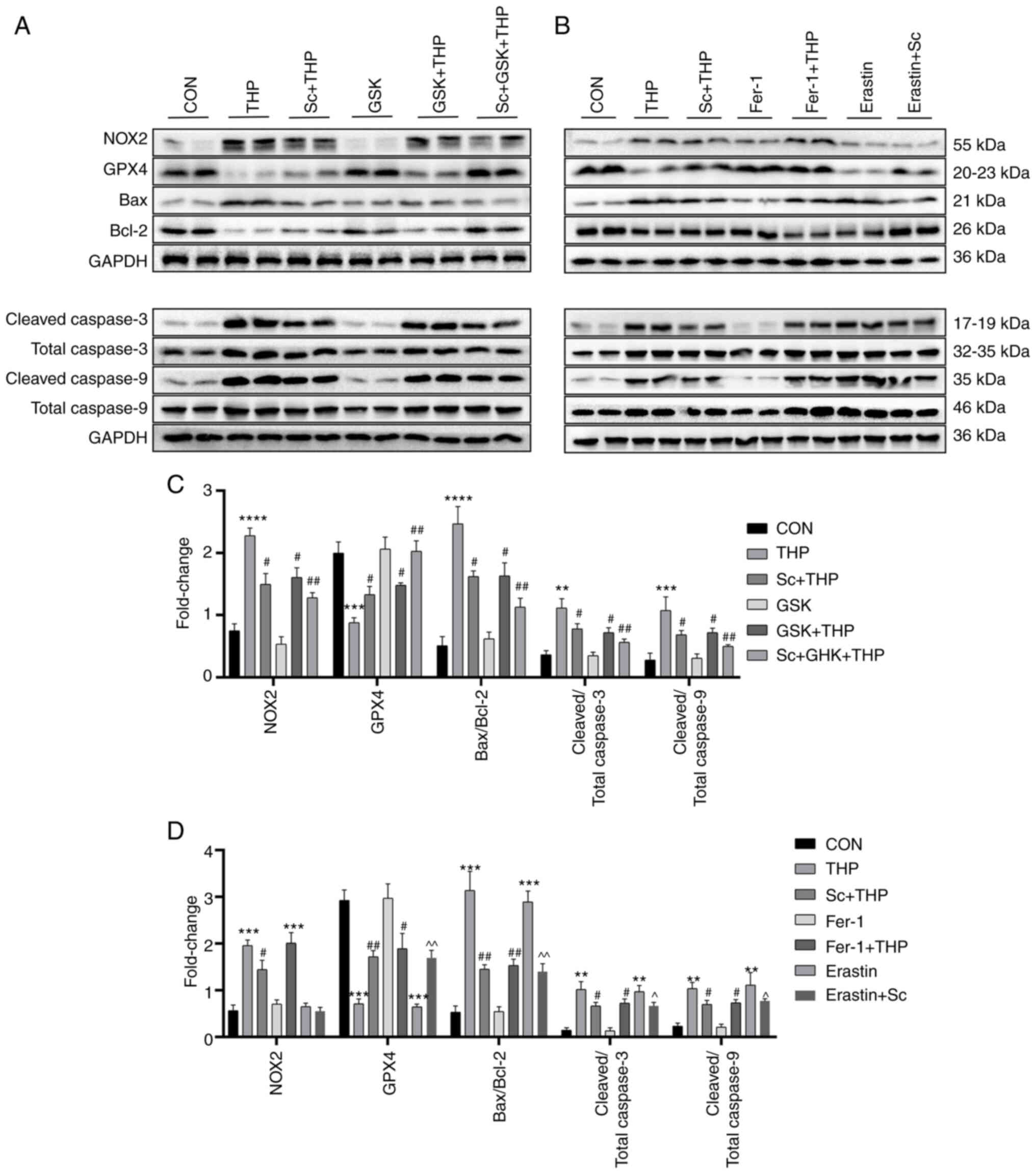 | Figure 7.Effects of Sc, THP, GSK, Fer-1, and
erastin on oxidative stress, ferroptosis, and apoptosis-related
proteins in H9c2 cells. (A and B) Western blots. (C and D)
Semi-quantitative analysis of the protein expression of GPX4, NOX2,
Bax/Bcl-2, cleaved caspase 3/total caspase 3 and cleaved caspase
9/total caspase 9 in each group. Values are expressed as the means
± standard deviation. **P<0.01, ***P<0.001 and
****P<0.0001 vs. CON. #P<0.05 and
##P<0.01 vs. THP. ^P<0.05 and
^^P<0.01 vs. erastin. Sc, scutellarein; THP,
pirarubicin; GSK, GSK2795039; Fer-1, ferrostatin-1; GPX4,
glutathione peroxidase 4; NOX2, NADPH oxidase 2. |
Discussion
With the increasing incidence of malignant tumors,
chemotherapy-induced myocardial toxicity has become a public health
problem that cannot be ignored. CTP is considered as a significant
component of the aforementioned problem (29,30).
It has been reported that oxidative stress and ROS, a key product
of oxidative stress, are closely associated with the onset of CTP
in myocardial cells (8).
Therefore, regulating oxidative stress can be a significant entry
point for the prevention and treatment of CTP (31). In the present study, the results
demonstrated that THP notably inhibited the growth of SD rats,
reduced their survival rate and severely impaired cardiac function,
as verified by the abnormal elevation of the myocardial
injury-related markers, BNP, CK-MB, cTnT and LDH, and the changes
in cardiac echocardiography. The aforementioned findings verified
that THP could successfully induce myocardial toxicity in SD rats.
In addition, THP promoted abnormal changes in oxidative
stress-related indexes in the blood and myocardium of SD rats, thus
further supporting that oxidative stress may play a significant
role in THP-induced myocardial toxicity. Notably, the in
vivo experiments also revealed that THP promoted aberrant
changes in the expression of apoptosis-related indicators, such as
Bax, Bcl-2, cleaved caspase 3 and cleaved caspase 9, and
ferroptosis-related indicators, including Fe2+, total Fe
and GPX4, in SD rats. Therefore, it was suggested that CTP may be
associated with oxidative stress, ferroptosis and apoptosis.
Sc has strong antioxidant properties and is widely
used in the medical and food industries (20–22).
Therefore, herein, to explore the effect of Sc on CTP, SD rats were
subjected to food therapy with Sc. Currently, oxidative stress is
considered the central mechanism of anthracycline-induced
myocardial toxicity (8,32). Different from other cells,
myocardial cells have high energy demands and therefore are rich in
mitochondria, where ROS-producing enzymes, such as NOX2, are
located. Therefore, the majority of ROS is produced in mitochondria
(33–35). When cells are induced, NOX2 is
activated to produce ROS via delivering electrons from NADPH to
oxygen through the transmembrane (36). Previous studies showed that
anthracycline chemotherapeutic drugs aggravated oxidative stress in
myocardial cells and promoted the production of ROS, thus
suggesting that myocardial cells are vulnerable to anthracycline
drugs (8,37). Consistent with the aforementioned
finding, in the present study, treatment of myocardial cells with
THP promoted oxidative stress and ROS overproduction. Furthermore,
cell co-treatment with Sc improved the THP-mediated NOX2
upregulation, thus further improving the increase of ROS and
alleviating the THP-induced oxidative stress.
Mitochondria are a significant regulatory target of
apoptosis, while ROS is one of the triggering factors of
mitochondrial-mediated apoptosis (38,39).
Low levels of ROS are critical for cell proliferation, signal
transduction and other physiological processes, while its enhanced
levels are associated with cytotoxicity, which promotes DNA damage,
mitochondrial dysfunction, reduced protein synthesis and
destruction of intracellular calcium homeostasis, eventually
leading to cardiomyocyte apoptosis (39–41).
The results of the present study indicated that THP increased the
expression of apoptosis-related proteins, namely Bax/Bcl-2, cleaved
caspase 3 and cleaved caspase 9, in cardiomyocytes. However,
treatment with Sc and GSK reversed these effects, thus suggesting
that regulating oxidative stress could improve THP-induced
cardiomyocyte apoptosis.
On the other hand, THP also promoted changes in the
expression of the key ferroptosis-related protein, GPX4, thus
supporting that in addition to oxidative stress and apoptosis,
ferroptosis may be also involved in CTP. As aforementioned, THP
induced oxidative stress and promoted ROS production in myocardial
cells. A previous study showed that the excessive production of ROS
induced lipid peroxidation, thus indicating that ferroptosis could
be considered as a newly discovered unique method of cell death
driven by iron-dependent lipid peroxidation (19). It has been reported that
ferroptosis is regulated by several cellular metabolic pathways,
such as iron homeostasis, redox homeostasis, mitochondrial activity
and various disease-related signal transduction pathways (19,42,43).
A previous study demonstrated that NOX4 promoted ferroptosis in
astrocytes through oxidative stress-induced lipid peroxidation
(44). GPX4, also known as
phospholipid hydrogen glutathione peroxidase, prevented ferroptosis
via converting lipid hydroperoxide into non-toxic lipid alcohols
(42–44). Furthermore, astragaloside IV
attenuated ferroptosis in myocardial cells via promoting the
expression of GPX4 through activating the NRF2 signaling pathway
and regulating oxidative stress (45).
In the present study, ferroptosis was induced and
inhibited following cell treatment with erastin and Fer-1,
respectively. Erastin is a ferroptosis inducer, which functions
through ROS and iron-dependent signaling (46,47).
A previous study showed that erastin inhibited voltage-dependent
anion channels 2/3 and accelerated oxidation, thus leading to the
endogenous accumulation of ROS, which in turn induced lipid
peroxidation, ultimately promoting ferroptosis (48,49).
Additionally, Fer-1, as a radical-trapping antioxidant, attenuated
the accumulation of lipid hydroperoxides via a reduction mechanism,
thereby inhibiting ferroptosis (50,51).
Notably, in addition to ferroptosis the effects of erastin and
Fer-1 were also explored on apoptosis through in vitro
experiments. The in vitro experiments indicated that erastin
promoted apoptosis of H9c2 cells, while Fer-1 reduced myocardial
cell apoptosis induced by THP. In addition, erastin promoted the
expression of apoptosis-related proteins in H9c2 cells (Bax/Bcl-2
and cleaved caspase 3), while Fer-1 improved the aberrant
expression of apoptosis-related proteins induced by THP. In
addition, research has shown that erastin-induced increases in Bax
and cytochrome c levels were counteracted by ferrostatin-1
pretreatment (52). It was
therefore hypothesized that the effects on apoptosis may be
associated with the mechanisms of erastin and Fer-1 on regulating
ferroptosis via intensifying/inhibiting oxidative stress. Of note,
the findings of the present study indicated that the regulation of
erastin and Fer-1 for oxidative stress appeared to be independent
of NOX2. As aforementioned, oxidative stress is closely associated
with apoptosis, and the results revealed that THP downregulated
GPX4, thus indicating that THP promoted ferroptosis in myocardial
cells. In addition, GSK improved the THP-induced reduction of GPX4
expression, while Sc improved the erastin- and THP-mediated GPX4
downregulation. Furthermore, the combined treatment of myocardial
cells with Sc and GSK further improved the THP-mediated reduction
of GPX4 expression. The aforementioned findings indicated that
regulation of oxidative stress improved the THP-induced ferroptosis
in myocardial cells.
The toxic effect of anthracycline drugs on
myocardium cannot be ignored, since it is a major public health
problem that needs to be urgently solved. Herein, in vivo
studies demonstrated that CTP was closely associated with oxidative
stress, apoptosis and ferroptosis. Therefore, improving CTP via
regulation of oxidative stress to inhibit myocardial cell apoptosis
and ferroptosis appears to be a feasible strategy. Further
experiments verified that food therapy with Sc inhibited
cardiomyocyte apoptosis and ferroptosis via regulation of oxidative
stress, thereby improving CTP. The aforementioned findings may
provide novel insights into the clinical application of Sc and a
significant theoretical basis for the implementation of Sc or even
all anthracycline antineoplastic drugs in preventing and treating
THP-induced cardiotoxicity.
However, the present study has some limitations.
Although the current study investigated the protective effect of Sc
on THP-induced myocardial injury and its association with oxidative
stress, apoptosis and ferroptosis based on existing literature, its
specific underlying mechanism remains unclear. In addition, the
in vivo molecular mechanism underlying the protective effect
of Sc on CTP was not explored. Furthermore, clinical trials on the
effectiveness of Sc are still lacking. Therefore, further studies
and clinical trials on this subject should be carried out in the
future.
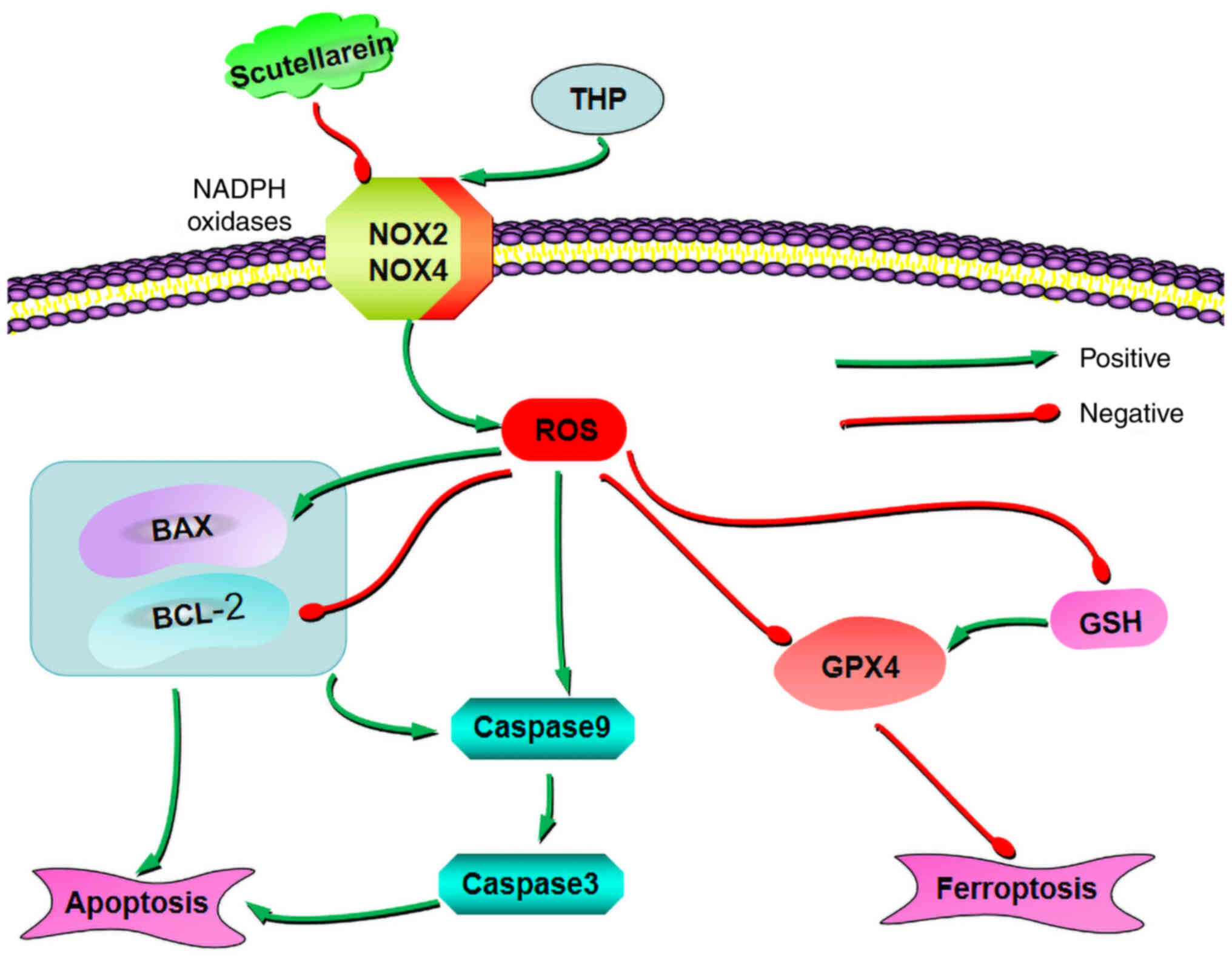
In conclusion, the present study indicated that Sc
had antioxidant, anti-apoptotic, and anti-ferroptosis effects in
CTP. In addition, the results suggested that Sc could further
inhibit cell apoptosis and ferroptosis via negatively regulating
the oxidative stress-related axis, NOX2/ROS, thereby improving the
THP-induced cardiotoxicity (Fig.
8).
Acknowledgements
Not applicable.
Funding
This study was supported by the National Key R&D Program of
China (grant nos. 2018YFC1311400 and 2018YFC1311404).
Availability of data and materials
The data generated in the present study are not
publicly available due the fact that elements of the current basic
and clinical research remain uncompleted and ongoing, and as a
patent application will be filed. However, data may be requested
from the corresponding author.
Authors' contributions
YL and FT confirm the authenticity of all the raw
data. YL and FT performed the experiments. HT and PP analyzed and
interpreted the data. QH and LD verified the results. YL and FT
wrote the manuscript. QH and LD contributed to the conception,
design and supervision of the study. All authors reviewed and
approved the final version of the manuscript.
Ethics approval and consent to
participate
This study was approved by the Animal Ethics
Committee of the First Affiliated Hospital of Chongqing Medical
University (approval no. IACUC-CQMU-2022-0127, Chongqing,
China).
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Mullard A: Addressing cancer's grand
challenges. Nat Rev Drug Discov. 19:825–826. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Hait WN: Anticancer drug development: The
grand challenges. Nat Rev Drug Discov. 9:253–254. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
von Minckwitz G and Loibl S: Evolution of
adjuvant chemotherapy for breast cancer. Lancet. 385:1812–1814.
2015. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Shen SJ and Liu CM: Chemotherapy for
early-stage breast cancer: The more the better? Lancet.
401:1243–1245. 2023. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Gabizon AA, Patil Y and La-Beck NM: New
insights and evolving role of pegylated liposomal doxorubicin in
cancer therapy. Drug Resist Updat. 29:90–106. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Pugazhendhi A, Edison TNJI, Velmurugan BK,
Jacob JA and Karuppusamy I: Toxicity of Doxorubicin (Dox) to
different experimental organ systems. Life Sci. 200:26–30. 2018.
View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Yu J, Wang C, Kong Q, Wu X, Lu JJ and Chen
X: Recent progress in doxorubicin-induced cardiotoxicity and
protective potential of natural products. Phytomedicine.
40:125–139. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Kong CY, Guo Z, Song P, Zhang X, Yuan YP,
Teng T, Yan L and Tang QZ: Underlying the mechanisms of
doxorubicin-induced acute cardiotoxicity: Oxidative stress and cell
death. Int J Biol Sci. 18:760–770. 2022. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Tocchetti CG, Carpi A, Coppola C,
Quintavalle C, Rea D, Campesan M, Arcari A, Piscopo G, Cipresso C,
Monti MG, et al: Ranolazine protects from doxorubicin-induced
oxidative stress and cardiac dysfunction. Eur J Heart Fail.
16:358–366. 2014. View
Article : Google Scholar : PubMed/NCBI
|
|
10
|
Zhao L, Tao X, Qi Y, Xu L, Yin L and Peng
J: Protective effect of dioscin against doxorubicin-induced
cardiotoxicity via adjusting microRNA-140-5p-mediated myocardial
oxidative stress. Redox Biol. 16:189–198. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
McLaughlin D, Zhao Y, O'Neill KM, Edgar
KS, Dunne PD, Kearney AM, Grieve DJ and McDermott BJ: Signalling
mechanisms underlying doxorubicin and Nox2 NADPH oxidase-induced
cardiomyopathy: Involvement of mitofusin-2. Br J Pharmacol.
174:3677–3695. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Li Q, Qin M, Tan Q, Li T, Gu Z, Huang P
and Ren L: MicroRNA-129-1-3p protects cardiomyocytes from
pirarubicin-induced apoptosis by down-regulating the
GRIN2D-mediated Ca2+ signalling pathway. J Cell Mol Med.
24:2260–2271. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Han D, Wang Y, Wang Y, Dai X, Zhou T, Chen
J, Tao B, Zhang J and Cao F: The tumor-suppressive human circular
RNA CircITCH sponges miR-330-5p to ameliorate doxorubicin-induced
cardiotoxicity through upregulating SIRT6, survivin, and SERCA2a.
Circ Res. 127:e108–e125. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Chen YH, Chen ZW, Li HM, Yan XF and Feng
B: AGE/RAGE-Induced EMP release via the NOX-Derived ROS pathway. J
Diabetes Res. 2018:68230582018. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Zhang J, Wang X, Vikash V, Ye Q, Wu D, Liu
Y and Dong W: ROS and ROS-Mediated cellular signaling. Oxid Med
Cell Longev. 2016:43509652016. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Drummond GR and Sobey CG: Endothelial
NADPH oxidases: Which NOX to target in vascular disease? Trends
Endocrinol Metab. 25:452–463. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Prosser BL, Ward CW and Lederer WJ: X-ROS
signaling: Rapid mechano-chemo transduction in heart. Science.
333:1440–1445. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Orrenius S, Gogvadze V and Zhivotovsky B:
Mitochondrial oxidative stress: Implications for cell death. Annu
Rev Pharmacol Toxicol. 47:143–183. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Stockwell BR: Ferroptosis turns 10:
Emerging mechanisms, physiological functions, and therapeutic
applications. Cell. 185:2401–2421. 2022. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Spiegel M, Marino T, Prejanò M and Russo
N: On the scavenging ability of scutellarein against the OOH
radical in water and Lipid-like environments: A Theoretical study.
Antioxidants (Basel). 11:2242022. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Lin Y, Lin Y, Ren N, Li S, Chen M and Pu
P: Novel anti-obesity effect of scutellarein and potential
underlying mechanism of actions. Biomed Pharmacother.
117:1090422019. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Chagas MDSS, Behrens MD, Moragas-Tellis
CJ, Penedo GXM, Silva AR and Gonçalves-de-Albuquerque CF: Flavonols
and flavones as potential anti-inflammatory, antioxidant, and
antibacterial compounds. Oxid Med Cell Longev. 2022:99667502022.
View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Gao L, Tang H, Zeng Q, Tang T, Chen M and
Pu P: The anti-insulin resistance effect of scutellarin may be
related to antioxidant stress and AMPKα activation in diabetic
mice. Obes Res Clin Pract. 14:368–374. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Mei X, Zhang T, Ouyang H, Lu B, Wang Z and
Ji L: Scutellarin alleviates blood-retina-barrier oxidative stress
injury initiated by activated microglia cells during the
development of diabetic retinopathy. Biochem Pharmacol. 159:82–95.
2019. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Shi H, Tang H, Ai W, Zeng Q, Yang H, Zhu
F, Wei Y, Feng R, Wen L, Pu P and He Q: Schisandrin B antagonizes
cardiotoxicity induced by pirarubicin by inhibiting mitochondrial
permeability transition pore (mPTP) opening and decreasing
cardiomyocyte apoptosis. Front Pharmacol. 12:7338052021. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Chai Y, Cao Z, Yu R, Liu Y, Yuan D and Lei
L: Dexmedetomidine attenuates LPS-Induced Monocyte-Endothelial
adherence via inhibiting Cx43/PKC-α/NOX2/ROS signaling pathway in
monocytes. Oxid Med Cell Longev. 2020:29304632020. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Zhang H, Wang Z, Liu Z, Du K and Lu X:
Protective effects of dexazoxane on rat ferroptosis in
Doxorubicin-Induced cardiomyopathy through regulating HMGB1. Front
Cardiovasc Med. 8:6854342021. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Li S, Lei Z, Yang X, Zhao M, Hou Y, Wang
D, Tang S, Li J and Yu J: Propofol protects myocardium from
ischemia/reperfusion injury by inhibiting ferroptosis through the
AKT/p53 signaling pathway. Front Pharmacol. 13:8414102022.
View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Lim GB: Circular RNA prevents
doxorubicin-induced cardiotoxicity. Nat Rev Cardiol. 19:5742022.
View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Gianni L, Herman EH, Lipshultz SE, Minotti
G, Sarvazyan N and Sawyer DB: Anthracycline cardiotoxicity: From
bench to bedside. J Clin Oncol. 26:3777–3784. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Han XZ, Gao S, Cheng YN, Sun YZ, Liu W,
Tang LL and Ren DM: Protective effect of naringenin-7-O-glucoside
against oxidative stress induced by doxorubicin in H9c2
cardiomyocytes. Biosci Trends. 6:19–25. 2012.PubMed/NCBI
|
|
32
|
Alanazi AM, Fadda L, Alhusaini A, Ahmad R,
Hasan IH and Mahmoud AM: Liposomal resveratrol and/or carvedilol
attenuate doxorubicin-induced cardiotoxicity by modulating
inflammation, oxidative stress and S100A1 in rats. Antioxidants
(Basel). 9:1592020. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Rosca MG and Hoppel CL: Mitochondria in
heart failure. Cardiovasc Res. 88:40–50. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Hausenloy DJ and Ruiz-Meana M: Not just
the powerhouse of the cell: Emerging roles for mitochondria in the
heart. Cardiovasc Res. 88:5–6. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
ChenY R and Zweier JL: Cardiac
mitochondria and reactive oxygen species generation. Circ Res.
114:524–537. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Bedard K and Krause KH: The NOX family of
ROS-generating NADPH oxidases: physiology and pathophysiology.
Physiol Rev. 87:245–313. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Li D, Yang Y, Wang S, He X, Liu M, Bai B,
Tian C, Sun R, Yu T and Chu X: Role of acetylation in
doxorubicin-induced cardiotoxicity. Redox Biol. 46:1020892021.
View Article : Google Scholar : PubMed/NCBI
|
|
38
|
D'Autreaux B and Toledano MB: ROS as
signalling molecules: Mechanisms that generate specificity in ROS
homeostasis. Nat Rev Mol Cell Biol. 8:813–824. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Stockwell BR: A powerful cell-protection
system prevents cell death by ferroptosis. Nature. 575:597–598.
2019. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Tang D, Chen X, Kang R and Kroemer G:
Ferroptosis: Molecular mechanisms and health implications. Cell
Res. 31:107–125. 2021. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Park MW, Cha HW, Kim J, Kim JH, Yang H,
Yoon S, Boonpraman N, Yi SS, Yoo ID and Moon JS: NOX4 promotes
ferroptosis of astrocytes by oxidative stress-induced lipid
peroxidation via the impairment of mitochondrial metabolism in
Alzheimer's diseases. Redox Biol. 41:1019472021. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Bersuker K, Hendricks JM, Li Z, Magtanong
L, Ford B, Tang PH, Roberts MA, Tong B, Maimone TJ, Zoncu R, et al:
The CoQ oxidoreductase FSP1 acts parallel to GPX4 to inhibit
ferroptosis. Nature. 575:688–692. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Jiang X, Stockwell BR and Conrad M:
Ferroptosis: Mechanisms, biology and role in disease. Nat Rev Mol
Cell Biol. 22:266–282. 2021. View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Conrad M and Pratt DA: The chemical basis
of ferroptosis. Nat Chem Biol. 15:1137–1147. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Liu Z, Zhou Z, Ai P, Zhang C, Chen J and
Wang Y: Astragaloside IV attenuates ferroptosis after subarachnoid
hemorrhage via Nrf2/HO-1 signaling pathway. Front Pharmacol.
13:9248262022. View Article : Google Scholar : PubMed/NCBI
|
|
46
|
Dixon SJ, Lemberg KM, Lamprecht MR, Skouta
R, Zaitsev EM, Gleason CE, Patel DN, Bauer AJ, Cantley AM, Yang WS,
et al: Ferroptosis: An iron-dependent form of nonapoptotic cell
death. Cell. 149:1060–1072. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
47
|
Yan R, Xie E, Li Y, Li J, Zhang Y, Chi X,
Hu X, Xu L, Hou T, Stockwell BR, et al: The structure of
erastin-bound xCT-4F2hc complex reveals molecular mechanisms
underlying erastin-induced ferroptosis. Cell Res. 32:687–690. 2022.
View Article : Google Scholar : PubMed/NCBI
|
|
48
|
Gan B: How erastin assassinates cells by
ferroptosis revealed. Protein Cell. 14:84–86. 2023.PubMed/NCBI
|
|
49
|
Yang Y, Luo M, Zhang K, Zhang J, Gao T,
Connell DO, Yao F, Mu C, Cai B, Shang Y and Chen W: Nedd4
ubiquitylates VDAC2/3 to suppress erastin-induced ferroptosis in
melanoma. Nat Commun. 11:4332020. View Article : Google Scholar : PubMed/NCBI
|
|
50
|
Zilka O, Shah R, Li B, Friedmann Angeli
JP, Griesser M, Conrad M and Pratt DA: On the mechanism of
cytoprotection by Ferrostatin-1 and Liproxstatin-1 and the role of
lipid peroxidation in ferroptotic cell death. ACS Cent Sci.
3:232–243. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
51
|
Miotto G, Rossetto M, Di Paolo ML, Orian
L, Venerando R, Roveri A, Vučković AM, Bosello Travain V, Zaccarin
M, Zennaro L, et al: Insight into the mechanism of ferroptosis
inhibition by ferrostatin-1. Redox Biol. 28:1013282020. View Article : Google Scholar : PubMed/NCBI
|
|
52
|
Park JS, Kim DH, Choi HI, Kim CS, Bae EH,
Ma SK and Kim SW: 3-Carboxy-4-methyl-5-propyl-2-furanpropanoic acid
(CMPF) induces cell death through ferroptosis and acts as a trigger
of apoptosis in kidney cells. Cell Death Dis. 14:782023. View Article : Google Scholar : PubMed/NCBI
|