Introduction
Ichthyosis is an umbrella term for a large
heterogeneous group of disorders of cornification, which are
usually monogenetic and mainly characterized by widespread
hyperkeratosis, xerosis and scaling of the skin; at times,
ichthyosis is also associated with syndromic features (1). Hundreds of genes, encoding for their
corresponding proteins, have a role in the normal differentiation
of keratinocytes and participate in the formation of a functional
epidermal barrier (2). Therefore,
inherited ichthyosis and related keratinization disorders may have
a complex etiology and overlapping manifestations; this poses a
huge challenge for clinical interpretation.
In general, four main types of nonsyndromic
ichthyosis have been reported, namely ichthyosis vulgaris (I.
vulgaris or IV), X-linked ichthyosis, autosomal recessive
congenital ichthyosis (ARCI) and keratinopathic ichthyosis
(1). Thus far, at least 67 genes
associated with various forms of inherited ichthyosis, either
syndromic or nonsyndromic, have been identified; these include the
filaggrin gene [FLG; online mendelian inheritance in man
(OMIM) no. 135940], steroid sulfatase (STS; OMIM no.
300747), keratin genes (KRT1/KRT10; OMIM no.
139350/148080) and member 12 of the ATP-binding cassette subfamily
A (OMIM no. 607800) (3). Following
advances in genetic diagnostic methods based on next-generation
sequencing (NGS), ~80–90% of inherited ichthyosis cases can be
resolved at present; this is beneficial not only for discovering
genetic causes and novel mutations but also for establishing
genotype-phenotype associations (4–7).
As the most common type of ichthyosis (prevalence,
~1/300), IV (OMIM no. 146700), with a typical feature of fine,
pale-grey scaling, is caused by autosomal, semi-dominant, inherited
loss-of-function mutations in the FLG gene (8). Patients with biallelic FLG
mutations, accounting for ~2/3 of all cases, tend to exhibit more
severe symptoms than those with a single heterozygous mutation
(9,10). To date, >175 FLG variants
that can cause IV, xerosis cutis or atopic dermatitis have been
detected (https://www.hgmd.cf.ac.uk/; pro
V2023.2). The second most common type, namely X-linked ichthyosis
(XLI), has an approximate prevalence of 1/2,000 in males, and it is
caused by STS deficiency (11);
this disorder is frequently associated with other clinical issues,
e.g., cryptorchidism or social communication deficits, such as
attention deficit or hyperactivity syndrome and autism, which
reflects the pleiotropy of the STS gene (12,13).
Another common group of ichthyosis is ARCI, with a prevalence of
~1/100,000; this disorder is associated with at least 10 genes
involved in the biosynthesis of acylceramide, lipid lamellae and
cornified lipid envelope (14).
Several other rare forms of ichthyosis frequently appear in
dermatological clinical settings, which increasingly rely on
genetic diagnosis for identification (15).
In the present study, seven patients with ichthyosis
or similar conditions were recruited and subjected to a genetic
analysis by whole-exome sequencing (WES). The detected variants
were distributed among several genes, thus reflecting the
heterogeneity and complexity of this disease. These variants
enriched the mutation spectrum of ichthyosis and provide strong
evidence for genetic counseling provided to the affected
families.
Patients and methods
Subjects
This study was approved by the Ethics Committee of
the First Hospital of Hebei Medical University (Shijiazhuang,
China; approval no. 20210095). Informed consent for genetic testing
and the results to be used in the study was obtained from all
participants. Furthermore, written informed consent was obtained
from the parent/legal guardian of the patients for the publication
of the details of their medical case and any accompanying images.
All procedures performed in the present study were in accordance
with the Declaration of Helsinki (1964) and its later amendments or
comparable ethical standards.
Between January 2018 and December 2021, patients
with hyperkeratosis, xerosis and scaling of the skin, particularly
those with a family history of the symptoms, were collected. The
principle of subject inclusion was based on the ‘receivables’
policy. The patients were clinically evaluated by physicians on the
basis of routine clinical examination and family surveys. Genomic
DNA was extracted from the peripheral blood specimens of the
patients and their parents by using the QIAamp DNA Midi Kit (Qiagen
GmbH) for further analysis.
WES
WES was used to detect the sequence variants in the
probands' samples, as described in a previous study by our group
(16). In brief, target-region
sequence enrichment was performed using the Agilent Sure Select
Human Exon Sequence Capture Kit (Agilent Technologies, Inc.). DNA
libraries were tested by quantitative PCR (17), wherein the size, distribution and
concentration were determined using an Agilent Bioanalyzer 2100
(Agilent Technologies, Inc.). By utilizing ~150 bp pair-end reads,
the NovaSeq6000 platform (Illumina, Inc.) was used for DNA
sequencing with ~300 pM per sample with the NovaSeq Reagent kit
(Illumina, Inc.). Sequencing raw reads (quality level Q30>90%)
(17) were aligned to the human
reference genome (accession no. hg19/GRCh37) by using the Burrows
Wheeler Aligner tool (https://www.geneticsmr.com/keywords/burrow-wheeler-aligner-tool).
PCR duplicates were removed using Picardv1.57 (https://github.com/broadinstitute/picard). Variant
calling was conducted with the Verita Trekker® Variants Detection
system (v2.0; Berry Genomics) and the Genome Analysis Tool Kit
(https://software.broadinstitute.org/gatk/). The
variants were annotated and interpreted using ANNOVAR (v2.0)
(18) and the Enliven® Variants
Annotation Interpretation system(Berry Genomics)according to the
common guidelines issued by the American College of Medical
Genetics and Genomics (ACMG) (19). To accurately interpret variant
pathogenicity, we referred to three frequency databases (ExAC_EAS;
http://exac.broadinstitute.org;gnomAD_exome_EAS,
http://gnomad.broadinstitute.org; and
1000G_2015aug_eas, http://www.internationalgenome.org) and the Human Gene
Mutation Database (HGMD) pro V2021.10 (https://www.hgmd.cf.ac.uk/ac/index.php). Revel score
(a combined method of pathogenicity prediction, with a threshold
for damage of ≥0.700) (20) and
pLI score (which represents the tolerance for truncating variants)
were also used. Sanger sequencing (21) was performed on the suspected
variants as the validation method, with the 3730 DX Genetic
Analyzer (Applied Biosystems; Thermo Fisher Scientific, Inc.).
Conservation and structural analysis
of missense variants
The evolutionary conservation of all affected amino
acid residues by the corresponding missense variants was analyzed
using the online tool MEGA7 (http://www.megasoftware.net/.php; accessed on
September 25, 2022), with default parameters. Furthermore, the
SWISS-MODEL online program (https://swissmodel.expasy.org/) with default
parameters was used to generate models and compare the structures
of wild-type (WT) and mutant (MT) proteins with the missense
variants.
Results
Clinical manifestations
A total of seven patients with possible ichthyosis
were recruited at the First Hospital of Hebei Medical University
(Shijiazhuang, China; approval no. 20210095) between February 2018
and December 2021. All seven patients showed a dermatological
phenotype that was suspected to be associated with ichthyosis;
however, there were certain differences and each patient had their
own characteristics (Table I).
Case 1 was a 15-year-old female patient with ichthyosis vulgaris.
Case 2 was a 4-year-old male patient whose father and grandfather
were also affected by ichthyosis. Case 3 was a 17-year-old female
patient whose main clinical presentation was ichthyosis and atopic
dermatitis. Case 4 was a 2-month-old male with ichthyosis and
cryptorchidism as the main clinical symptoms. Case 5 was an
18-year-old female with the main clinical symptoms of ichthyosis
and attention deficit; this patient's father also had ichthyosis.
Case 6 was a 1-month-old male with the main clinical symptoms of
hyperkeratosis and scaly skin. Case 7 was a 16-year-old female with
the main clinical symptoms of palmoplantar hyperkeratosis and
acanthosis. Fig. S1 shows the
representative clinical features of several patients (based on the
discretion of patients or their guardians to disclose privacy,
clinical images of only part of the patients were included).
 | Table I.Clinical indications and genetic
variations of the seven cases in this study. |
Table I.
Clinical indications and genetic
variations of the seven cases in this study.
| Case no. | Age | Sex | Main clinical
indication | Gene | Genomic
variation | Protein
variation | Frequency in 3
databasesa | HGMD pathogenicity
level | Revelb score | ACMG level
(evidence) | Allele frequencies
in gnomAD | CADD score |
|---|
| 1 | 15 years | F | Ichthyosis
vulgaris | FLG | c.7945delA | p.S2649Vfs*94 | 0.004; 0.0035;
0.0020 | DM | / | P
(PVS1+PP5+PM2) |
9.98×10−5 | / |
| 2 | 4 years | M | Ichthyosis
vulgaris | FLG | c.6950_6957del | p.S2317fs | 0.005; 0.0032;
0.0027 | DM | / | P
(PVS1+PP5+PM2) |
1.31×10−4 | / |
| 3 | 17 years | F | Ichthyosis
vulgaris; atopic dermatitis | FLG | c.3321delA | p.G1109Efs*13 | 0.0069; 0.0090;
0.0098 | DM | / | P (PVS1+PP5) |
2.32×10−4 | / |
| 4 | 2 months | M | Ichthyosis;
cryptorchidism | STS |
chrX:6968331_7894165del (0.93Mb) | Whole protein
absence | 0; 0; 0 | DM | / | P
(PVS1+PS4_Supporting+PM2+PP4) | 0 | / |
| 5 | 18 years | F | Ichthyosis;
attention deficit | STS | c.452C>T | p.P151L | 0; 0; 0 | / | 0.897 | LP
(PM1+PM2+PM5+PP4) | 0 | 0.453 |
| 6 | 1 month | M | Hyperkeratosis;
scaly skin; keratin clumping? | KRT10 | c.449T>C | p.M150T | 0; 0; 0 | DM | 0.954 | P
(PS3+PP3+PP5+PM1+PM5+PM2) | 0 | 4.387 |
| 7 | 16 years | F | Palmoplantar
hyperkeratosis; acanthosis |
SERPINB7 | c.796C>T | p.R266* | 0.0072;
0.0070;0.0119 | / | / | LP (PP5+PM2) |
2.50×10−4 | / |
|
|
|
|
|
| c.647_650del | p.L216fs | 0.0007; 0.0009;
0.003 | / | / | LP
(PVS1+PP5+PM2) |
6.16×10−6 | / |
Genetic findings
Table I summarizes
the detailed information of each genetic variation, including the
frequency in the three population-based databases, the index status
in the HGMD database, the Revel score and the pathogenicity level
according to the ACMG criteria. Fig.
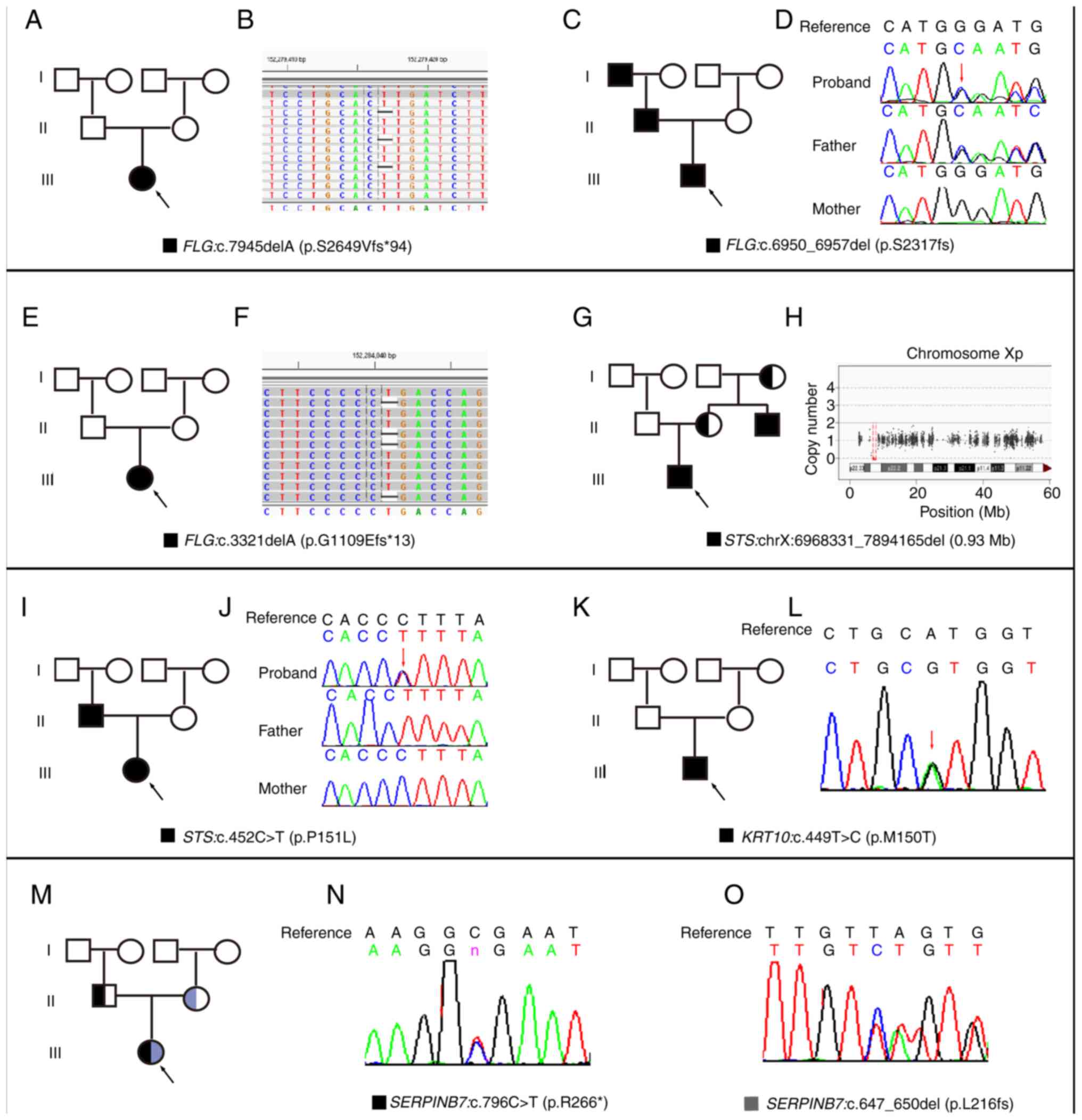
1 shows the results of NGS or Sanger sequencing verification,
together with the pedigree diagram and the variant carrying status
of each family.
In all seven patients, the diagnostic variations
were identified in ichthyosis-associated causative genes. Cases 1–3
were associated with IV by known FLG variants reported by
previous studies (21–23); Case 1 (Fig. 1A and B) and Case 3 (Fig. 1E and F) were de novo, and
Case 2 was familial (Fig. 1C and
D). Cases 4 and 5 were XLI due to STS variations; Case 4
had a previously reported variant with deletion of the entire
STS gene (24,25), inherited from his heterozygous
carrier mother (Fig. 1G and H),
while Case 5 carried a novel variant, namely
STS:c.452C>T(p.P151L), inherited from her symptomatic
father (Fig. 1I and J). Case 6,
with a de novo known heterozygous KRT10:
c.449T>C(p.M150T) variant (26), was determined to have epidermolytic
hyperkeratosis (OMIM no. 113800; Fig.
1K and L). Case 7 had a compound heterozygous variation in
SERPINB7, which comprised c.796C>T(p.R266*) and
c.647_650del(p.L216fs); thus, this patient was determined to have
palmoplantar keratoderma, Nagashima-type (OMIM no. 615598). These
two variants were inherited from her parents (Fig. 1M-O).
Conservation and structural analysis
of the associated missense variants
Among the variants, there were two missense
variants, namely STS: c.452C>T(p.P151L) and KRT10:
c.449T>C(p.M150T). Conservation analysis for the amino acid
residues affected by each of them indicated that these residues
were highly conserved across species (Fig. 2A and B). The protein databank (PDB;
(https://www.rcsb.org/structure/1P49 for
STS:p.P151L) and alpha fold (AF; AF-P13645-F1; https://alphafold.ebi.ac.uk/entry/P13645
for KRT10:p.M150T) were used for prediction and structural analysis
of these two missense variants. Of note, the results showed that
neither mutant significantly affected the protein structure but
altered only the length of the local hydrogen bonds, which may
affect the stability of the protein structure (Fig. 2C and D).
Discussion
Inherited ichthyosis and keratodermas include a
phenotypically heterogeneous group of disorders; these conditions
are associated with several genes involved with the structural
components of the epidermis, epidermal lipid metabolism, cell-cell
adhesion or keratinocyte differentiation (3). Patients with these disorders
generally manifest with dry and scaly skin, with considerable
phenotypic variation. Among the traditional diagnostic methods,
electron microscopy and tissue immunofluorescence can enable the
identification of specific types; however, the cost, invasiveness
and tediousness of these methods limit their application (27,28).
NGS, with its advantages of high-throughput, efficiency and
accuracy, is gradually becoming the first-line technology for
diagnosing inherited dermal disorders (5,6,9,16).
The Xp22.31 microdeletion containing the STS
gene accounts for >80% of the pathogenesis of XLI (13). In Case 5 in the present study, the
proband carried a paternally inherited heterozygous STS
missense variant and the proband's father also showed typical
ichthyosis symptoms. This situation is rare, because XLI is usually
considered as an X-linked recessive disease, and female carriers
with Xp22.31 duplication tend to manifest this disease with dermal
symptoms such as blistering/desquamation (29). Hence, it is interesting to further
investigate how this missense variant affects the function of STS
and causes indications in this female carrier in Case 5. The
results of the conservation analysis were supportive of the
pathogenicity of this variant.
Keratinopathic ichthyosis (KI) is caused by
mutations in the keratin genes, namely KRT1, KRT10 and
KRT2, with mutations in KRT10 accounting for >50%
of cases (15). The de novo
missense variant carried by Case 6 of the present study was
originally detected by Paller et al (26) as early as 1994 and was found to be
associated with epidermolytic hyperkeratosis, a severe subtype of
KI; this finding indicates that this variant is valuable as a
screening target for patients with KI. The amino acid affected by
this variant remains highly conserved across species, further
supporting its pathogenicity. The results of the structural
analysis of this case and the missense variant in Case 5 were not
unequivocally strongly supportive; however, more detailed analyses,
such as molecular dynamics simulations and in vitro
functional experiments, are required to clarify the mechanism of
specific variations (30).
Case 7, which was initially a confusing case, was
finally determined to not be associated with typical ichthyosis by
genetic diagnosis. The patient had Nagashima-type palmoplantar
keratosis (NPPK; OMIM no. 615598). NPPK was initially thought to be
caused by biallelic putative loss-of-function mutations in
SERPINB7 (OMIM no. 603357), and the c.796C>T(p.R266*)
mutation was considered a major founder mutation in the East Asian
population (31). To date, >20
causative variants have been identified by several studies [STS:
c.452C>T(p.P151L) (https://www.hgmd.cf.ac.uk/) (32–36)]. Our finding of Case 7 not only
confirmed the widespread prevalence of this c.796C>T variant but
also contributed one novel variant, c.647_650del(p.L216fs), to
extend the mutation spectrum of the SERPINB7 gene. A
noteworthy finding is that the proband's other siblings exhibited a
25% likelihood of developing this disease; hence, appropriate
genetic testing is recommended.
Patients with hyperkeratosis, xerosis and scaling of
the skin were recruited into the present study. A total of seven
patients with suspected ichthyosis were recruited in our centers.
Based on the genetic findings, appropriate counseling was offered
to their families and it was recommended that the patients'
relatives should also undergo targeted testing. For the families
with a familial inheritance pattern, such as Cases 2, 4, 5 and 7,
it was recommended that they consider prenatal diagnosis of
subsequent pregnancies to determine the course of pregnancy or to
plan for adequate caretaking of the newborns. For the families with
the de novo pattern (Cases 1, 3 and 6), they were reminded
that the risk of future recurrence is not extremely low because of
the possibility of gonad mosaicism (37). As different variants lead to
different phenotypes in these seven patients, the
genotype-phenotype correlation of ichthyosis requires further
elucidation with more genetic data.
In conclusion, in the present study, a comprehensive
clinical and genetic analysis of seven patients with ichthyosis and
NPPK was conducted and their respective diagnostic mutations were
detected. WES detected the diagnostic variants of the FLG,
STS and KRT10 genes in these seven patients. A total of
eight variants were detected and confirmed. Among all the variants,
two were identified for the first time, namely STS:
c.452C>T(p.P151L) and c.647_650del(p.L216fs). The current
findings further enrich the mutant spectrum of ichthyosis and a new
variant in the SERPINB7 gene was identified in a patient
with NPPK; this finding provides a basis for further investigation
of its pathogenesis.
Supplementary Material
Supporting Data
Acknowledgements
Not applicable.
Funding
Funding: No funding was received.
Availability of data and materials
The data that support the findings of this study are
available in the figshare repository at https://doi.org/10.6084/m9.figshare.22680832. Due
to patient self-determination, only 4/7 cases of data were publicly
released and the remaining data maybe requested from the
corresponding author.
Authors' contributions
GQZ initiated the project and was involved in the
conception and design of the study. JZ and YY collected cases and
performed clinical evaluation. YT, HYH and JZ performed
experimental validation. LXZ analyzed the data. JZ wrote the
manuscript. GQZ revised the manuscript. All authors have read and
approved the manuscript. YT and YY checked and confirmed the
authenticity of the raw data.
Ethics approval and consent to
participate
This study was approved by the Ethics Committee of
the First Hospital of Hebei Medical University (Shijiazhuang,
China; approval no. 20210095). All research participants or their
legal representatives signed informed consent forms for
participation in clinical and genetic testing. All methods were
carried out in accordance with relevant guidelines and
regulations.
Patient consent for publication
All study participants or their legal
representatives provided written consent to publish their clinical
details and images.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Vahlquist A and Torma H: Ichthyosis: A
road model for skin research. Acta Derm Venereol. 100:adv000972020.
View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Fischer J and Bourrat E: Genetics of
inherited ichthyoses and related diseases. Acta Derm Venereol.
100:adv000962020. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Uitto J, Youssefian L, Saeidian AH and
Vahidnezhad H: Molecular genetics of keratinization
disorders-what's new about ichthyosis. Acta Derm Venereol.
100:adv000952020. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Hotz A, Kopp J, Bourrat E, Oji V, Komlosi
K, Giehl K, Bouadjar B, Bygum A, Tantcheva-Poor I, Hellstrom Pigg
M, et al: Meta-Analysis of Mutations in ALOX12B or ALOXE3
Identified in a Large Cohort of 224 Patients. Genes (Basel).
12:802021. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Ballin N, Hotz A, Bourrat E, Küsel J, Oji
V, Bouadjar B, Brognoli D, Hickman G, Heinz L, Vabres P, et al:
Genetical, clinical, and functional analysis of a large
international cohort of patients with autosomal recessive
congenital ichthyosis due to mutations in NIPAL4. Hum Mutat.
40:2318–2333. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Abdel-Hamid MS, Issa MY, Elbendary HM,
Abdel-Ghafar SF, Rafaat K, Hosny H, Girgis M, Abdel-Salam GMH and
Zaki MS: Phenotypic and mutational spectrum of thirty-five patients
with Sjogren-Larsson syndrome: Identification of eleven novel
ALDH3A2 mutations and founder effects. J Hum Genet. 64:859–865.
2019. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Youssefian L, Touati A, Saeidian AH,
Zargari O, Zeinali S, Vahidnezhad H and Uitto J: A novel mutation
in ST14 at a functionally significant amino acid residue expands
the spectrum of ichthyosis-hypotrichosis syndrome. Orphanet J Rare
Dis. 12:1762017. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Smith FJ, Irvine AD, Terron-Kwiatkowski A,
Sandilands A, Campbell LE, Zhao Y, Liao H, Evans AT, Goudie DR,
Lewis-Jones S, et al: Loss-of-function mutations in the gene
encoding filaggrin cause ichthyosis vulgaris. Nat Genet.
38:337–342. 2006. View
Article : Google Scholar : PubMed/NCBI
|
|
9
|
Chen S, Kong X, Wei X, Sun Y, Yin D, Zhang
Q, Du L, Man J, Mao L, Li H, et al: Targeted next-generation
sequencing identifies nine novel filaggrin gene variants in Chinese
Han patients with ichthyosis vulgaris. Br J Dermatol.
177:e202–e203. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Oji V, Seller N, Sandilands A, Gruber R,
Gerss J, Huffmeier U, Hamm H, Emmert S, Aufenvenne K, Metze D, et
al: Ichthyosis vulgaris: Novel FLG mutations in the German
population and high presence of CD1a+ cells in the epidermis of the
atopic subgroup. Br J Dermatol. 160:771–781. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Webster D, France JT, Shapiro LJ and Weiss
R: X-linked ichthyosis due to steroid-sulphatase deficiency.
Lancet. 1:70–72. 1978.PubMed/NCBI
|
|
12
|
Kent L, Emerton J, Bhadravathi V,
Weisblatt E, Pasco G, Willatt LR, McMahon R and Yates JR: X-linked
ichthyosis (steroid sulfatase deficiency) is associated with
increased risk of attention deficit hyperactivity disorder, autism
and social communication deficits. J Med Genet. 45:519–524. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Diociaiuti A, Angioni A, Pisaneschi E,
Alesi V, Zambruno G, Novelli A and El Hachem M: X-linked
ichthyosis: Clinical and molecular findings in 35 Italian patients.
Exp Dermatol. 28:1156–1163. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Richard G: Autosomal Recessive Congenital
Ichthyosis. GeneReviews®. Adam MP, Everman DB, Mirzaa GM, et al:
University of Washington; Seattle, WA: pp. 1993–2022
|
|
15
|
Vahlquist A, Fischer J and Torma H:
Inherited nonsyndromic ichthyoses: An Update on pathophysiology,
diagnosis and treatment. Am J Clin Dermatol. 19:51–66. 2018.
View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Yao Y, Yang K, Qi KY, Zeng LX and Zhang
GQ: Diverse clinical and genetic characteristics of six cases of
inherited epidermolysis bullosa. Exp Ther Med. 24:7272022.
View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Arslan S, Garcia FJ, Guo M, Kellinger MW,
Kruglyak S, LeVieux JA, Mah AH, Wang H, Zhao J, Zhou C, et al:
Sequencing by avidity enables high accuracy with low reagent
consumption. Nat Biotechnol. 42:132–138. 2024. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Wang K, Li M and Hakonarson H: ANNOVAR:
Functional annotation of genetic variants from next-generation
sequencing data. Nucleic Acids Res. 38:e1642010. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Richards S, Aziz N, Bale S, Bick D, Das S,
Gastier-Foster J, Grody WW, Hegde M, Lyon E, Spector E, et al:
Standards and guidelines for the interpretation of sequence
variants: A joint consensus recommendation of the American college
of medical genetics and genomics and the association for molecular
pathology. Genet Med. 17:405–424. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Ioannidis NM, Rothstein JH, Pejaver V,
Middha S, McDonnell SK, Baheti S, Musolf A, Li Q, Holzinger E,
Karyadi D, et al: REVEL: An ensemble method for predicting the
pathogenicity of rare missense variants. Am J Hum Genet.
99:877–885. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Chen H, Ho JCC, Sandilands A, Chan YC,
Giam YC, Evans AT, Lane EB and McLean WHI: Unique and Recurrent
mutations in the filaggrin gene in singaporean Chinese patients
with ichthyosis vulgaris. J Invest Dermatol. 128:1669–1675. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Zhang H, Guo Y, Wang W, Shi M, Chen X and
Yao Z: Mutations in the filaggrin gene in Han Chinese patients with
atopic dermatitis. Allergy. 66:420–427. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Nomura T, Sandilands A, Akiyama M, Liao H,
Evans AT, Sakai K, Ota M, Sugiura H, Yamamoto K, Sato H, et al:
Unique mutations in the filaggrin gene in Japanese patients with
ichthyosis vulgaris and atopic dermatitis. J Allergy Clin Immunol.
119:434–440. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Valdes-Flores M, Kofman-Alfaro SH, Vaca AL
and Cuevas-Covarrubias SA: Mutation report: A novel partial
deletion of exons 2–10 of the STS gene in recessive X-linked
ichthyosis. J Invest Dermatol. 114:591–593. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Matsukura H, Fuchizawa T, Ohtsuki A,
Higashiyama H, Higuchi O, Higuchi A and Miyawaki T: End-stage renal
failure in a child with X-linked ichthyosis. Pediatr Nephrol.
18:297–300. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Paller AS, Syder AJ, Chan YM, Yu QC,
Hutton E, Tadini G and Fuchs E: Genetic and clinical mosaicism in a
type of epidermal nevus. N Engl J Med. 331:1408–1415. 1994.
View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Dahlqvist J, Klar J, Hausser I,
Anton-Lamprecht I, Pigg MH, Gedde-Dahl T Jr, Gånemo A, Vahlquist A
and Dahl N: Congenital ichthyosis: Mutations in ichthyin are
associated with specific structural abnormalities in the granular
layer of epidermis. J Med Genet. 44:615–620. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Diociaiuti A, Fortugno P, El Hachem M,
Angelo C, Proto V, De Luca N, Martinelli D, Boldrini R, Castiglia D
and Zambruno G: Early immunopathological diagnosis of ichthyosis
with confetti in two sporadic cases with new mutations in keratin
10. Acta Derm Venereol. 94:579–582. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Gubb SJA, Brcic L, Underwood JFG, Kendall
KM, Caseras X, Kirov G and Davies W: Medical and neurobehavioural
phenotypes in male and female carriers of Xp22.31 duplications in
the UK Biobank. Hum Mol Genet. 29:2872–2881. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Yang K, Xu YC, Hu HY, Li YZ, Li Q, Luan
YY, Liu Y, Sun YQ, Feng ZK, Yan YS and Yin CH: Investigation of a
Novel NTRK1 variation causing congenital insensitivity to pain with
anhidrosis. Front Genet. 12:7634672021. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Kubo A, Shiohama A, Sasaki T, Nakabayashi
K, Kawasaki H, Atsugi T, Sato S, Shimizu A, Mikami S, Tanizaki H,
et al: Mutations in SERPINB7, encoding a member of the serine
protease inhibitor superfamily, cause Nagashima-type palmoplantar
keratosis. Am J Hum Genet. 93:945–956. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Yin J, Xu G, Wang H, Zhao J, Duo L, Cao X,
Tang Z, Lin Z and Yang Y: New and recurrent SERPINB7 mutations in
seven Chinese patients with Nagashima-type palmoplantar keratosis.
J Invest Dermatol. 134:2269–2272. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Zhao J, Yang Z, Xiang X and Ma L: SERPINB7
novel mutation in Chinese patients with Nagashima-type palmoplantar
keratosis and cases associated with atopic dermatitis. Int J
Dermatol. 59:e320–e322. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Li Q, Zhu X, Wang C, Meng J, Chen D and
Kong X: Identification of a rare case with nagashima-type
palmoplantar keratoderma and 18q deletion syndrome via exome
sequencing and low-coverage whole-genome sequencing. Front Genet.
12:7074112021. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Xiao T, Liu Y, Wang T, Ren J, Xia Y and
Wang X: Two novel mutations of SERPINB7 in eight cases of
Nagashima-type palmoplantar keratosis in the Chinese population. J
Dermatol. 49:539–544. 2022. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Liu J, Chen Z, Hu L, Song Z, Mo R, Tsang
LS, Liu Y, Huang X, Gong Z, Lin Z and Yang Y: Investigation of
Nagashima-type palmoplantar keratoderma in China: A cross-sectional
study of 234 patients. J Dermatol. 50:375–382. 2023. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
van der Meulen MA, van der Meulen MJ and
te Meerman GJ: Recurrence risk for germinal mosaics revisited. J
Med Genet. 32:102–104. 1995. View Article : Google Scholar : PubMed/NCBI
|