Introduction
Pyroptosis is a type of cell death that is
considered to be highly pro-inflammatory and different from
apoptosis (1–4). In contrast to apoptosis that without
an inflammatory response, pyroptosis has a unique cellular
morphology (i.e., nuclear integrity of pyroptotic cells is
maintained) and mechanism (1,2,4).
Pyroptosis was initially identified in macrophages (5–7) and
is characterized by membrane pore formation, cell membrane bubbling
and swelling and substantial release of pro-inflammatory mediators
or intracellular contents (2,8–11).
Nucleotide-binding oligomerization domain-like receptor family
pyrin domain containing 3 (NLRP3) inflammasome activation is
involved in triggering canonical macrophage pyroptosis (2,4,9).
When macrophages recognize pathogen-associated molecular patterns
derived from pathogenic infection and molecular patterns related to
damages induced by endogenous stress-associated signals, NLRP3
self-oligomerizes and binds to apoptosis-associated speck-like
protein, which contains a caspase recruitment domain. This
interaction recruits precursor of caspase-1 (pro-caspase-1) and
leads to its cleavage and activation (12). Active caspase-1 is a key enzyme
during pyroptosis that cleaves gasdermin D (GSDMD) and precursors
of interleukin (IL)-1β and IL-18 to generate their biologically
active forms (13,14). GSDMD serves as the central executor
of pyroptosis, and its cleavage produces an N-terminal p31 fragment
of GSDMD (GSDMD-N), which self-oligomerizes and creates membrane
pores in cells (15). When these
membrane pores form, intracellular substances including IL-1β and
IL-18 are released, along with an influx of water, causing cell
swelling and osmotic lysis (1,4,13,14).
Simultaneously, pro-inflammatory mediators (IL-1β and IL-18)
released by pyroptotic macrophages recruit inflammatory cells and
further amplify the inflammatory response (9,16).
A mounting body of evidence reports that excessive
macrophage pyroptosis exacerbates tissue damage and pathological
inflammation (2,14,17),
thereby contributing to initiation and progression of numerous
types of inflammatory disorder, such as sepsis and kidney,
neurodegenerative, liver and cardiovascular diseases (CVDs)
(18,19). Several studies have reported the
involvement of NLRP3/caspase-1/GSDMD-dependent macrophage
pyroptosis in the development of atherosclerosis (AS) (1,4,6,8,10,13).
Substantial evidence demonstrates that pyroptosis
pathway-associated proteins, such as NLRP3, GSDMD and caspase-1 are
prominently expressed in lesion macrophages both in animal and
human AS (1,9,20–22).
Moreover, risk factors associated with AS, including oxidized
low-density lipoprotein (ox-LDL), homocysteine, cholesterol crystal
and calcium phosphate can trigger NLRP3/caspase-1/GSDMD-dependent
pyroptosis in macrophages (13,14,22–24).
Pyroptosis of macrophages in early atherosclerotic lesions is
reported to be beneficial as the death of macrophage attenuates the
inflammatory response, scavenge cytotoxic lipoproteins and other
harmful substances and decrease the synthesis of matrix
metalloproteinases (4,25). However, sustained pyroptosis of
macrophages in advanced atherosclerotic plaques facilitates
formation of necrotic lipid cores and increases the instability of
atherosclerotic lesions, resulting in plaque rupture and arterial
thrombosis (25). The
aforementioned findings demonstrate that macrophage pyroptosis is
the predominant type of cell death in advanced atherosclerotic
plaques (6). Furthermore, a
growing body of evidence indicates that specific pharmacological
inhibition or genetic intervention targeting the
NLRP3/caspase-1/GSDMD axis can alleviate AS-associated risk
factor-induced pyroptosis in macrophages and plaque development in
apolipoprotein E (ApoE)−/− or low-density lipoprotein
receptor (Ldlr)−/− mice (22,26–32).
Therefore, elucidating the molecular mechanisms of pyroptosis in
macrophages may enhance current understanding of the pathogenesis
of CVDs, including AS (2) and pave
the way for developing promising therapeutic strategies for a broad
spectrum of inflammatory diseases, ranging from microbial infection
to AS (13,33).
Hydrogen sulfide (H2S), a gasotransmitter
with multiple biological actions, exerts a key regulatory function
in multiple physiological and pathophysiological events, including
cell death, differentiation, proliferation, hypertrophy,
metabolism, stress responses, inflammation, angiogenesis and
vasodilation (34–36). Evidence suggests that both
endogenous H2S and several exogenous H2S
donors exert notable vasodilatory, pro-angiogenic,
anti-hypertensive and anti-atherosclerotic effects by diverse
mechanisms including anti-oxidation, anti-inflammation and pro- and
anti-apoptosis (34,36–39).
Malfunction of endogenous H2S production is associated
with numerous CVDs (36,38). Exogenous H2S and
H2S derived from macrophages exert anti-inflammatory and
anti-pyroptotic effects in in vivo and in vitro
conditions as a response to pro-inflammatory or pro-atherogenic
stimuli. Previous studies have reported that this is achieved by
suppressing NLRP3 inflammasome activation (40–43),
however the underlying mechanisms by which H2S directly
regulates macrophage pyroptosis remains unclear. S-sulfhydration of
functional proteins induced by H2S is suggested as a key
mechanism responsible for the biological effects of H2S
(34,44). H2S mediated
S-sulfhydration of multiple critical target proteins (Liver kinase
B1, specificity protein 1, NF-κB, Parkin and Kelch-like
ECH-associated protein 1 and c-Jun) in numerous cellular signaling
pathways (AMP-activated protein kinase pathway, apoptotic pathway,
Ldlr protein pathway and NLRP3 inflammasome pathway) is involved in
cell differentiation, cell proliferation/hypertrophy, cell
metabolism, cell survival/death, inflammation and oxidative stress
(34,44). However, whether H2S
inhibits macrophage pyroptosis and exerts anti-inflammatory effects
through the S-sulfhydration of pyroptosis pathway-associated
proteins remains uncertain.
The present study used ox-LDL-stimulated THP-1
macrophages to establish an in vitro model that mimics
AS-induced macrophage pyroptosis according our previous study
(45). The impact of exogenous
H2S donors and inhibition of endogenous H2S
production on NLRP3/caspase-1/GSDMD-mediated macrophage pyroptosis
in response to ox-LDL stimulation was assessed. Pyroptosis-related
morphological features, propidium iodide (PI)-positive staining,
lactate dehydrogenase (LDH) release, caspase-1 activity and
expression and secretion of pyroptosis pathway-specific proteins
and pro-inflammatory cytokines were evaluated. Furthermore, the
mechanisms via which H2S serves its anti-pyroptotic role
in THP-1 macrophages through S-sulfhydrating caspase-1 were
assessed.
Materials and methods
Cell culture
Human THP-1 monocytic cells were purchased from
National Collection of Authenticated Cell Cultures (cat. no.
SCSP-567; The Chinese Academy of Sciences; https://cellbank.org.cn/search-detail.php?id=517).
THP-1 cells were cultured in RPMI-1640 medium (cat. no.
C11875500BT; Gibco; Thermo Fisher Scientific, Inc.) supplemented
with 10% fetal bovine serum (FBS; cat. no. 10270-106; Gibco; Thermo
Fisher Scientific, Inc.) and 1% penicillin/streptomycin (cat. no.
SV30010; HyClone; Cytiva). The culture was incubated at 37°C with
5% CO2.
Differentiation of THP-1 monocytes
into macrophages
For differentiation, THP-1 monocytes were plated in
6- (1×106 cells/well), 12-(1×105 cells/well)
or 96-well culture plates (1×104 cells/well) containing
RPMI-1640 medium, 10% FBS and 1% penicillin/streptomycin for
subsequent cultivation. THP-1 monocytes were incubated with
phorbol-12-myristate-13-acetate (PMA; cat. no. P6741; Beijing
Solarbio Science & Technology Co., Ltd.) for 24 h at 37°C.
Adherent differentiated THP-1 cells were rinsed with PBS and
incubated at 37°C for a further 24 h in RPMI-1640 complete media
without PMA.
Cell viability assay
THP-1 macrophages were seeded (1×105
cells/ml) in 96-well plates (100 µl/well) in a 37°C incubator.
After 24 h, the cells were treated with increasing concentrations
of NaHS (50, 100, 200 and 400 µM; cat. no. 161527; MilliporeSigma),
D,L-propargylglycine (PAG) (1.0, 2.5, 5.0 and 10.0 µM; cat. no.
P7888; MilliporeSigma) and ox-LDL (25, 50, 100 and 150 µg/ml; cat.
no. YB-002, http://www.yiyuanbiotech.com/PRO.asp?id=562#section2;
Guangzhou Yiyuan Biological Technology Co., Ltd.) for 24 h in a
37°C incubator. Cell viability was assessed using Cell Counting
Kit-8 (cat. no. K1018; APeXBIO Technology LLC) according to the
manufacturer's instructions. A total of 10 µl CCK-8 reagent was
added to each well and incubated for 2 h at 37°C. Optical density
(OD) was measured at 450 nm with a xMark microplate
spectrophotometer (Bio-Rad Laboratories, Inc.). For each treatment
group, cells were seeded in triplicate. Cell viability was
calculated according to the manufacturer's instructions.
Drug treatment
THP-1 macrophages were classified into six groups as
follows: Control, cells were cultured with RPMI-1640 for 24 h at
37°C; ox-LDL, cells were incubated with 100 µg/ml ox-LDL at 37°C
for 24 h to construct an in vitro model of pyroptosis or
activate the caspase-1-dependent pyroptosis signaling pathway; PAG,
cells were treated with 2.5 mM PAG supplemented in RPMI-1640 medium
for 24 h at 37°C; ox-LDL + NaHS, cells were pretreated with 200 µM
NaHS for 30 min before treatment with 100 µg/ml ox-LDL for 23.5 h
at 37°C; ox-LDL + PAG, the cells were pretreated with 2.5 mM PAG
for 30 min at 37°C prior to ox-LDL treatment for 23.5 h; and ox-LDL
+ PAG + NaHS, where cells were pretreated with 200 µM NaHS and 2.5
mM PAG for 30 min before treatment with ox-LDL for 23.5 h at
37°C.
In supplementary experiments containing NaHS and/or
lipopolysaccharide (LPS) + adenosine triphosphate (ATP) treatment,
THP-1 macrophages were classified into the following four groups:
Control, THP-1 cells were cultured with RPMI-1640 for 24 h at 37°C;
NaHS, cells were treated with 200 µM NaHS for 24 h at 37°C;
lipopolysaccharide (LPS; cat. no. L2880; MilliporeSigma) + ATP
(cat. no. 6419; MilliporeSigma), cells were first treated with 1
µg/ml LPS for 18 h and subsequently treated with 5 mM ATP at 37°C
for 6 h to construct an in vitro model of pyroptosis; and
LPS + ATP + NaHS, cells were pretreated with 200 µM NaHS for 30
min, then treated with 1 µg/ml LPS for 18 h at 37°C, before
pyroptosis was triggered by treatment with 5 mM ATP for 5.5 h.
To determine whether H2S inhibits
caspase-1 activity by sulfhydrating pro-caspase-1, THP-1 cells were
allocated into the following groups: Control, THP-1 cells were
treated with PBS for either 2 or 24 h at 37°C; NaHS, THP-1 cells
were exposed to 200 µM NaHS for 2 h at 37°C; dithiothreitol
(DTT)/ox-LDL, THP-1 cells were treated with 1 mM DTT (cat. no.
43816; MilliporeSigma) for 2 h or incubated with 100 µg/ml ox-LDL
for 24 h at 37°C; ox-LDL + NaHS, THP-1 cells were pretreated with
200 µM NaHS for 30 min, followed 100 µg/ml ox-LDL for an additional
23.5 h at 37°C; and DTT + NaHS, THP-1 cells were treated with 1 mM
DTT for 1.5 h at 37°C after the pretreatment with 200 µM NaHS for
30 min.
Measurement of H2S
concentration
H2S concentration in THP-1 cells was
assessed by a H2S detection kit (cat. no. A146-1-1;
Nanjing Jiancheng Bioengineering Institute), which uses the
classical methylene blue method, according to the manufacturer's
instructions. THP-1 cells (1×106 cells/well) were seeded
in a 60-mm culture dish and cultured overnight at 37°C. Cells were
incubated with the aforementioned treatments for 24 h and lysed
using 1 ml PBS (pH 7.4) via ultrasonication (ultrasonic treatment
five times, 8 sec at intervals of 10 sec, 1.5 min in total) on ice.
Subsequently, a 100 µl aliquot of the cell lysate was transferred
into a centrifuge tube with 1% w/v zinc acetate and 12% NaOH to
incubate for 90 min in a shaking metal bath at 37°C. Bradford
protein assay kit (cat. no. P0006C; Beyotime Institute of
Biotechnology) was used to ascertain the protein concentration in
the cell lysates. After adding 20 mM
N,N-dimethyl-p-phenylenediamine sulfate and 30 mM FeCl3
containing 1.2 M HCl at room temperature for 15 min, the reaction
was halted. Then, 10% trichloroacetic acid was added to precipitate
protein and the mixture was centrifuged for 5 min at 10,000 × g and
at room temperature. Using a xMark™ microplate
spectrophotometer (Bio-Rad Laboratories, Inc.), absorbance of the
resultant methylene blue in the supernatant was measured at 665 nm.
The H2S concentration in each sample was calculated as
follows: H2S (nmol/mg protein)=[(absorbance of the
sample-absorbance of the blank)/0.0044] × [total reaction
volume/(sample volume × protein concentration of the sample)].
Cell death assay
Micrographs of pyroptotic cell morphology were
captured under phase microscopy using a 40X objective prior to cell
harvesting. The proportion of pyroptosis-like cells was obtained by
the number of cells displaying pyroptotic morphology relative to
the total cell count in five randomly selected fields/well, each
containing ~50 cells. These experiments were performed at least
three times independently.
The morphological changes associated with pyroptosis
and pyroptotic cell death were assessed by Hoechst 33342/PI
staining using a fluorescent microscope (TH4-200; Olympus
Corporation) and LDH activity assay using a spectrophotometer
(xMark; Bio-Rad Laboratories, Inc.). Each experiment was
independently conducted at least three times. Specifically, for
Hoechst 33342/PI double staining, THP-1 cells (5×105
cells/well) were treated with the aforementioned drugs for 24 h.
Cells were then washed with PBS and stained using 10 µl Hoechst
33342 staining solution from Hoechst 33342/PI dual staining kit
(cat. no. G023-1-1; Nanjing Jiancheng Bioengineering Institute) at
37°C for 10 min without light. Next, cells were stained using 5 µl
PI (cat. no. G023-1-1; Nanjing Jiancheng Bioengineering Institute)
at 25°C for 10 min without light. Cells were subsequently washed
three times with PBS and images of were captured under a TH4-200
fluorescent microscope (Olympus Corporation) at a magnification of
×200. Image J software (version 1.46r; National Institutes of
Health) was used to count PI-stained cells in five randomly
selected microscopic fields. Cells were quantified as follows:
PI-stained cells (%)=(number of red fluorescent cells/total blue
fluorescent cells) ×100.
LDH activity in the culture medium was measured
using an LDH activity assay kit (cat. no. A020-2-2; Nanjing
Jiancheng Bioengineering Institute). Cell culture supernatants (200
µl/sample) were harvested and centrifuged at 400 × g for 5 min at
4°C. Then, 20 µl supernatant was added to a 96-well test plate and
mixed with 5 µl coenzyme I or deionized water and 25 µl substrate
buffer. The mixture was then incubated at 37°C for 15 min. Then 25
µl 2,4-dinitrophenylhydrazine was added and samples incubated at
37°C for 15 min. Finally, 250 µl 0.4 M NaOH solution was added and
the mixture was incubated at 25°C for 5 min. The absorbance was
measured at 450 nm with a xMark microplate spectrophotometer
(Bio-Rad Laboratories, Inc.). The proportion of LDH release was
calculated according to the manufacturer's instructions.
Measurement of caspase-1 activity
THP-1 cells were incubated with the aforementioned
treatments for 2 or 24 h and caspase-1 activity was assessed using
a caspase-1 activity assay kit (cat. no. C1102; Beyotime Institute
of Biotechnology). The cells were lysed with the kit lysis buffer
on ice for 15 min and centrifuged at 18,000 × g for 15 min at 4°C.
Protein content in each sample was determined using a Bradford
protein assay kit (cat. no. P0006C; Beyotime Institute of
Biotechnology). Then, 40 reaction buffer and 10 µl 2 mM caspase-1
substrate were added to 50 µl supernatant, incubated at 37°C for 2
h and the amount of yellow formazan pNA was measured at 405 nm
using a xMark microplate spectrophotometer (Bio-Rad Laboratories,
Inc.). Caspase-1 activity was calculated according to the
manufacturer's instructions, which was represented as fold-change
in activity compared with the control.
Immunofluorescent staining
To determine if macrophages expressed active
caspase-1, immunofluorescence staining was performed. THP-1 cells
were treated with the aforementioned treatments for 24 h. THP-1
cells were fixed for 15 min at 25°C with 4% paraformaldehyde and
rinsed with PBS (three times at 5 min per time). The cells were
permeabilized for 3 min at room temperature using 0.2% Triton
X-100, and then blocked with 5% BSA (cat. no. SW3015; Beijing
Solarbio Science & Technology Co., Ltd.) at 37°C for 30 min in
a humidified incubating box. Then, cells were incubated with
antibodies against cleaved caspase-1 (1:200; cat. no. PA5-99390;
Thermo Fisher Scientific, Inc.) overnight at 4°C. Following PBS
washes (three times at 5 min per time), cells were incubated for
1.5 h at 37°C without exposure to light with a FITC-conjugated
secondary antibody (1:100; cat. no. ZF-0311; OriGene Technologies,
Inc.). Nuclei were counterstained with 1 µg/ml DAPI at room
temperature for 15 min without light. Images were captured at a
magnification of ×200 under a TH4-200 fluorescence microscope
(Olympus Corporation). The average fluorescence intensity (AFI)
value was calculated to assess the expression of active caspase-1
in THP-1 cells using Image J software (version 1.46r; National
Institutes of Health).
Immunoblotting
After incubating the THP-1 cells with the
aforementioned treatments for 24 h, total secreted proteins were
extracted from the culture medium using the methanol/chloroform
method, as previously described (46,47).
THP-1 cells were collected by centrifugation at 800 × g for 10 min
at 4°C and lysed in ice-cold RIPA lysis buffer (cat. no. R0010;
Beijing Solarbio Science & Technology Co., Ltd.) supplemented
with protease inhibitor cocktail (cat. no. P8340; MilliporeSigma)
and incubated at 4°C for 20 min. The supernatants from the cell
lysates were isolated by centrifugation at 12,000 × g for 15 min at
4°C and the protein concentration of the supernatants was examined
using a BCA protein assay kit (cat. no. 23227; Thermo Fisher
Scientific, Inc.). Protein pellets from cell-free media were
dissolved in 2X Laemmli buffer and the cell lysates of THP-1 cells
were mixed with 5X Laemmli buffer. Samples were denatured at 98°C
for 5 min. Equal amounts of protein (40 µg/lane) were loaded onto
10% (more than 60 kDa of molecular weight) or 12% (15–60 kDa of
molecular weight) SDS-PAGE gels and separated by electrophoresis.
The separated proteins were transferred to polyvinylidene fluoride
(PVDF) membranes (cat. nos. ISEQ00010 (0.22 µm), and IPVH00010
(0.45 µm); MilliporeSigma) with the pore size of 0.22 and 0.45 µm
using a wet-transfer system. The membranes were blocked at 25°C for
2 h using 5% skimmed milk, then incubated overnight at 4°C with
primary antibodies against NLRP3 (1: 1,000; cat. no. ab263899;
Abcam), pro-caspase-1 (1:1,000; cat. no. PA5-87536; Thermo Fisher
Scientific, Inc.), cleaved caspase-1 (1:1,000; cat. no. PA5-99390;
Thermo Fisher Scientific, Inc.), GSDMD (1:1,000; cat. no. ab210070;
Abcam), cleaved GSDMD (1:1,000; cat. no. 36425S; Cell Signaling
Technology, Inc.), pro-IL-1β (1:500; cat. no. ab315084; Abcam),
IL-1β (1:1,000; cat. no. 83186; Cell Signaling Technology, Inc.),
IL-18 (1:1,000; cat. no. A1115; ABclonal Biotech Co., Ltd.),
β-actin (1:1,000; cat. no. TA-09; OriGene Technologies, Inc.) and
GAPDH (1:1,000; cat. no. TA-08; OriGene Technologies, Inc.) in 5%
BSA buffer (cat. no. SW3015; Beijing Solarbio Science &
Technology Co., Ltd.). The membranes were then washed three times
with TBST (tris-buffered saline with 0.05% Tween-20) solution and
incubated for 2 h at 25°C with secondary antibody (1:10,000; cat.
nos. ZB-2301, and ZB-2305; OriGene Technologies, Inc.) conjugated
with horseradish peroxidase. After rinsing the membranes five times
with TBST (tris-buffered saline with 0.05% Tween-20), proteins
bands were visualized with an enhanced chemiluminescence detection
kit (cat. no. WBKLS0100; Merck KGaA) via X-ray film exposure or a
Tanon 5200 chemiluminescence imaging system (Tanon Science and
Technology Co., Ltd.) Image J software (version 1.46r; National
Institutes of Health) was used to assess and quantify the relative
integrated OD (IOD) of the protein bands as well as the relative
expression levels of the target proteins. To detect secreted active
caspase-1 and IL-1β, following the SDS-PAGE electrophoresis for
total secreted proteins extracted from the cell free media, the
upper part of the gel (>40 kDa) was stained by Coomassie
brilliant blue to indicate loading of lanes, and the lower part
(<40 kDa) was transferred to a PVDF membrane for the detection
of secreted caspase-1 and IL-1β by western blotting. Coomassie
brilliant blue staining from total secreted protein blots was
applied as a loading control for culture supernatant (48,49)
and GAPDH or β-actin acted as the loading control for the cell
lysates.
Quantification of NLRP3 and
pyroptosis-specific cytokines
To assess NLRP3 inflammasome activation and
secretion of pyroptosis-related pro-inflammatory cytokines,
PMA-differentiated THP-1 macrophages were seeded into 12-well
plates (5×105 cells/well) and incubated with 100 µg/ml
ox-LDL for 23.5 h at 37°C, in the presence or absence of 200 µM
NaHS and/or 2.5 mM PAG pretreatment for 30 min. THP-1 cells were
lysed in ice-cold RIPA lysis buffer (cat. no. R0010; Beijing
Solarbio Science & Technology Co., Ltd.) and incubated at 4°C
for 15 min. The supernatant obtained from the cell lysates was
isolated via centrifugation at 18,000 × g for 15 min at 4°C. The
protein concentration of the supernatants from THP-1 cell lysate
was determined using a BCA protein assay kit (cat. no. 23227;
Thermo Fisher Scientific, Inc.). Furthermore, cell-free culture
supernatant (500 µl/sample) was immediately collected by
centrifugation at 300 × g for 5 min at 4°C and frozen at −80°C.
ELISA kits were used according to the manufacturer's instructions
to quantify the concentrations of NLRP3 (cat. no. ab274401; Abcam)
in THP-1 cell lysate and the levels of IL-1β (cat. no. EK101B;
MultiSciences Biotech Co., Ltd.) and IL-18 (cat. no. EK118;
MultiSciences Biotech Co., Ltd.) in cell culture medium. Precoated
96-well plates and ELISA reagents were brought to room temperature.
Standards and 100 µl cell culture supernatants were added to each
well of the microplate, and precoated with biotinylated monoclonal
antibodies against IL-1β or IL-18 (1:100) for 10 min at 25°C. The
microplates were incubated with continuous shaking for 2 h at room
temperature, then rinsed with washing buffer for six times.
Subsequently, 100 µl HRP-conjugated streptavidin (1:100) was added
to each well and incubated in the dark at room temperature for 45
min with gentle shaking. The wells were washed six times and 100 µl
tetramethylbenzidine solution was added to each well and incubated
for 30 min at room temperature with no exposure to light. Finally,
100 µl stop solution was added to each well and the maximum
absorbance values at 450 nm or reference absorbance values at
570/630 nm were measured with a xMark microplate spectrophotometer
(Bio-Rad Laboratories, Inc.). The expression levels of NLRP3, IL-1β
and IL-18 in THP-1 cells or cell culture medium were determined
using the standard curve established from the kit standards.
Biotin switch assay for determination
S-sulfhydration of pro-caspase-1
The S-sulfhydration of pro-caspase-1 was assessed
using the biotin switch test described by Mustafa et al
(50), with minor adjustments.
THP-1 cells were harvested by centrifugation at 800 × g for 10 min
at 4°C and washed twice with ice-cold PBS following treatment with
or without ox-LDL, DTT or/and NaHS for 2 or 24 h as aforementioned,
The cells were then kept on ice and homogenized using
ultrasonication (sonicated three times, 15 sec each time, 1 min in
total) in an ice-cold HEN
[N-2-hydroxyethylpiperazine-N'-2-ethanesulfonic acid
(HEPES)/ethylene diamine tetraacetic acid (EDTA)/Neocuproine]
buffer containing protease inhibitor cocktail (cat. no. P1050;
Beyotime Institute of Biotechnology), 250.0 HEPES-NaOH, 1.0 EDTA
and 0.1 mM neocuproine and 100.0 µM deferoxamine. The cell lysate
was cleared by centrifugation at 14,000 × g at 4°C for 15 min and
protein concentration in the supernatant was measured using the
Pierce BCA Protein Assay kit (cat. no. 23227; Thermo Fisher
Scientific, Inc.). Cell lysate containing 800 µg total protein was
combined with 2 ml of HEN buffer [250.0 HEPES (pH 7.7), 1.0 EDTA
and 0.1 mM neocuproine and 100.0 µM deferoxamine], supplemented
with 2.5% SDS and 20 mM S-methyl methanethiosulfonate (MMTS) and
incubated at 50°C for 30 min with gentle and frequent vortexing to
block the free thiol (−SH). The blocked proteins were incubated
with 8 ml cooled acetone at −20°C for 30 min. The free MMTS plus
acetone were removed by centrifugation at 3,000 × g for 10 min at
4°C. The precipitated proteins were washed three times with 8 ml
ice-cold acetone and resuspended in 2 ml HEN buffer supplemented
with 1% SDS. The sample was then mixed with 0.4 mM
biotin-N-[6-(biotinamido) hexyl]-3′-(2′-pyridyldithio) propionamide
(cat. no. 21341; Thermo Fisher Scientific, Inc.) in DMSO and
incubated for 3 h at 25°C. After that, cooled acetone was added
into the biotin-labeled proteins to incubated for 40 min at −20°C.
The proteins were centrifuged at 3,000 × g for 10 min at 4°C and
rinsed with 2 ml ice-cold acetone and resuspended in 1 ml
neutralization buffer (25 HEPES-NaOH, 1 EDTA and 100 mM NaCl and
0.5% Triton X-100).
The samples were incubated with 20 µl
streptavidin-agarose beads (cat. no. S1638; MilliporeSigma) on a
rotator mixer overnight at 4°C to capture S-sulfhydrated proteins.
Samples were then centrifuged at 800 × g for 5 min at 4°C, and the
supernatant was discarded. The precipitated pellet of
streptavidin-agarose beads was then resuspended in 1 ml HEN buffer
containing 1% SDS and centrifuged for 5 min at 800 × g at 4°C.
S-sulfhydrated proteins were then washed five times with 1 ml HEN
buffer supplemented with 1% SDS, and spun down at 800 × g for 5 min
at 4°C. The S-sulfhydrated proteins of samples were eluted from the
streptavidin-agarose beads by 2X SDS-PAGE loading buffer (3% SDS,
1% β-mercaptoethanol, 62.5 mM tris-base, and 0.005% bromophenol
blue) at 37°C for 20 min on the rotator mixer. The S-sulfhydrated
proteins of samples were denatured at 98°C for 5 min and separated
on 12% SDS-PAGE gels, and transferred to a PVDF membrane (cat. no.
ISEQ00010; MilliporeSigma) with a pore size of 0.22 µm. The
membranes were blocked at 25°C for 2 h using 5% skimmed milk, and
S-sulfhydrated proteins underwent western blot analysis using a
pro-caspase-1-specific antibody (1:1,000; cat. no. ab207802;
Abcam). GAPDH (1:1,000; cat. no. TA-08; OriGene Technologies, Inc.)
acted as the loading control. The procedure of western blot
analysis for S-sulfhydrated pro-caspase-1 was the same as for
immunoblotting of pro-caspase-1 expression. Samples were run on
SDS-PAGE gels alongside the total target protein (‘input’) that had
not been subjected to the biotin switch assay procedure. The same
antibodies were used for western blotting to quantify
S-sulfhydrated proteins. The ratio of S-sulfhydrated pro-caspase-1
IOD relative to total pro-caspase-1 IOD was densitometrically
analyzed using Image J software (version 1.46r; National Institutes
of Health) and calculated to represent the relative S-sulfhydration
levels of pro-caspase-1.
Statistical analysis
GraphPad Prism 8 (version 8.0.2; Dotmatics) was used
to analyze data. All data are presented as the mean ± SEM of >3
independent experiments performed under identical conditions. The
statistical significance between two groups was assessed using the
unpaired two-tailed Student's t test in cell viability assay and
comparisons between multiple groups was analyzed by one-way ANOVA
followed by Bonferroni's post hoc test. P<0.05 was considered to
indicate a statistically significant difference.
Results
NaHS increases cell viability or
H2S concentration in THP-1 macrophages with or without
ox-LDL stimulation, while PAG and ox-LDL reduces them
Cytotoxicity of NaHS, PAG and ox-LDL toward THP-1
macrophages was assessed by the CCK-8 assay. THP-1 macrophages were
treated with NaHS, PAG and ox-LDL for 24 h. Compared with the
control group, 100, 200 and 400 µM NaHS significantly increased the
viability of THP-1 macrophages (Fig.
1A), whereas treatment with PAG (1, 2.5, 5 or 10 mM) (Fig. 1B) and ox-LDL (50, 100 and 150
µg/ml) (Fig. 1C) for 24 h resulted
in a significant decrease in cell viability of THP-1 macrophages.
Concentrations of 200 µM NaHS, 2.5 mM PAG and 100 µg/ml ox-LDL were
used in subsequent assays.
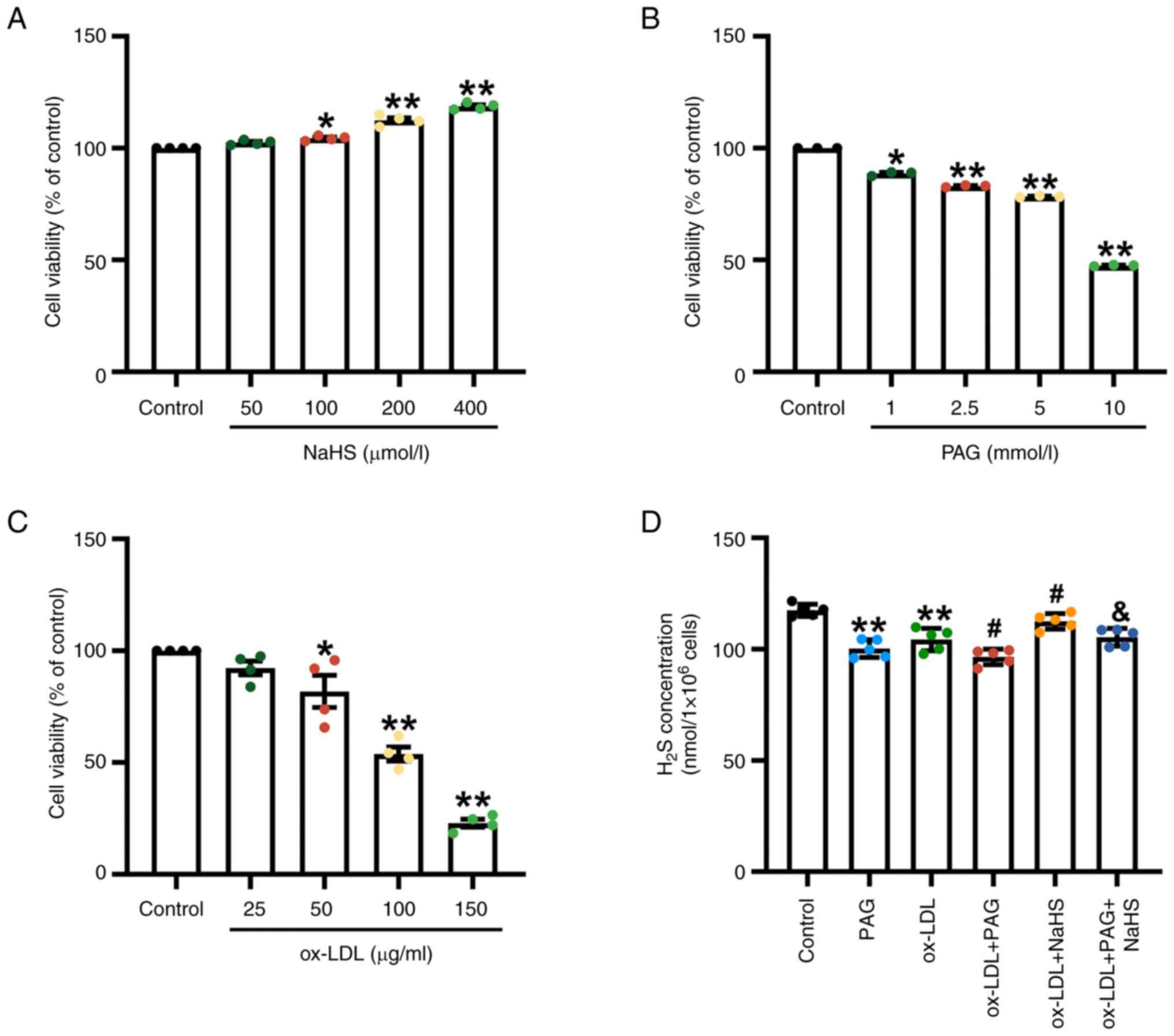 | Figure 1.Effect of NaHS, PAG and ox-LDL on
cytotoxicity and H2S levels in PMA-differentiated THP-1
macrophages. (A) The different concentrations of NaHS (50, 100, 200
and 400 µM) treatment alone for 24 h increases viability of THP-1
macrophages (n=4). (B) Cell viability was determined in THP-1 cells
treated with PAG at different concentrations of 1.0, 2.5, 5.0 and
10.0 µM for 24 h (n=3). (C) The effects of ox-LDL on the viability
of THP-1 macrophages. THP-1 cells were incubated with the different
concentrations of ox-LDL (25, 50, 100 and 150 µg/ml) for 24 h
(n=4). Cell Counting Kit-8 was used to measure the cell viability
of the macrophages. The viability of the control was normalised to
100%. (D) NaHS increases H2S concentration in THP-1
macrophages treated by ox-LDL or ox-LDL + PAG. THP-1 macrophages
were stimulated with 100 µg/ml ox-LDL for 23.5 h in the presence or
absence of pretreatment with 200 µM NaHS and/or 2.5 mM PAG for 30
min. H2S levels were assessed using a methylene blue
method-based H2S detection kit in THP-1 macrophages
(n=5). *P<0.05 and **P<0.01 vs. control,
#P<0.05 vs. ox-LDL, &P<0.05 vs.
ox-LDL + PAG. PAG, D,L-propargylglycine; ox-LDL, oxidized
low-density lipoprotein; PMA, phorbol-12-myristate-13-acetate. |
The main enzyme synthesizing endogenous
H2S in macrophages is cystathionine-γ-lyase (CSE)
(42,51,52).
Dysfunction in the CSE/H2S pathway induced by ox-LDL is
associated with ox-LDL-induced inflammation in macrophage (51,53).
Therefore, ox-LDL and the CSE inhibitor PAG were used in the
present study. To assess whether exogenous H2S donors
and the inhibition of endogenous H2S synthesis impact
H2S production in ox-LDL-stimulated THP-1 macrophages,
H2S concentration was measured using methylene blue
assay. Compared with the control group, ox-LDL and PAG treatment
alone significantly inhibited endogenous H2S production
in THP-1 cells (Fig. 1D). ox-LDL +
PAG treatment significantly reduced H2S production
compared with the ox-LDL group. However, pretreatment with an
exogenous H2S donor significantly increased
H2S concentrations compared with the ox-LDL group
(Fig. 1D). Similarly, NaHS
pretreatment also led to an increased H2S content in the
presence of a combination of ox-LDL and PAG compared with the
ox-LDL + PAG group (Fig. 1D).
H2S signaling suppresses
pyroptotic cell death in THP-1 macrophages exposed to ox-LDL and
LPS + ATP
Although the impact of H2S on pyroptosis
in numerous types of cell has been partially elucidated in previous
studies (41,54–56),
its morphological and biological influence on pyroptosis in
macrophages remains unclear. To assess the role of exogenous and
endogenous H2S in pyroptosis induced by ox-LDL and LPS +
ATP in macrophages, THP-1 macrophages were pretreated with 200 µM
NaHS and/or 2.5 mM PAG for 30 min, then treated with 100 µg/ml
ox-LDL or 1 µg/ml LPS + 5 mM ATP for 23.5 h. Changes in cell
morphology associated with pyroptosis were assessed using an
optical microscope. THP-1 cells in the control group exhibited
normal morphology (Figs. S1A and
2A). NaHS (200 µM) or PAG (2.5 mM)
treatment alone under the same concentration showed no significant
effect on morphology of macrophages compared with the control group
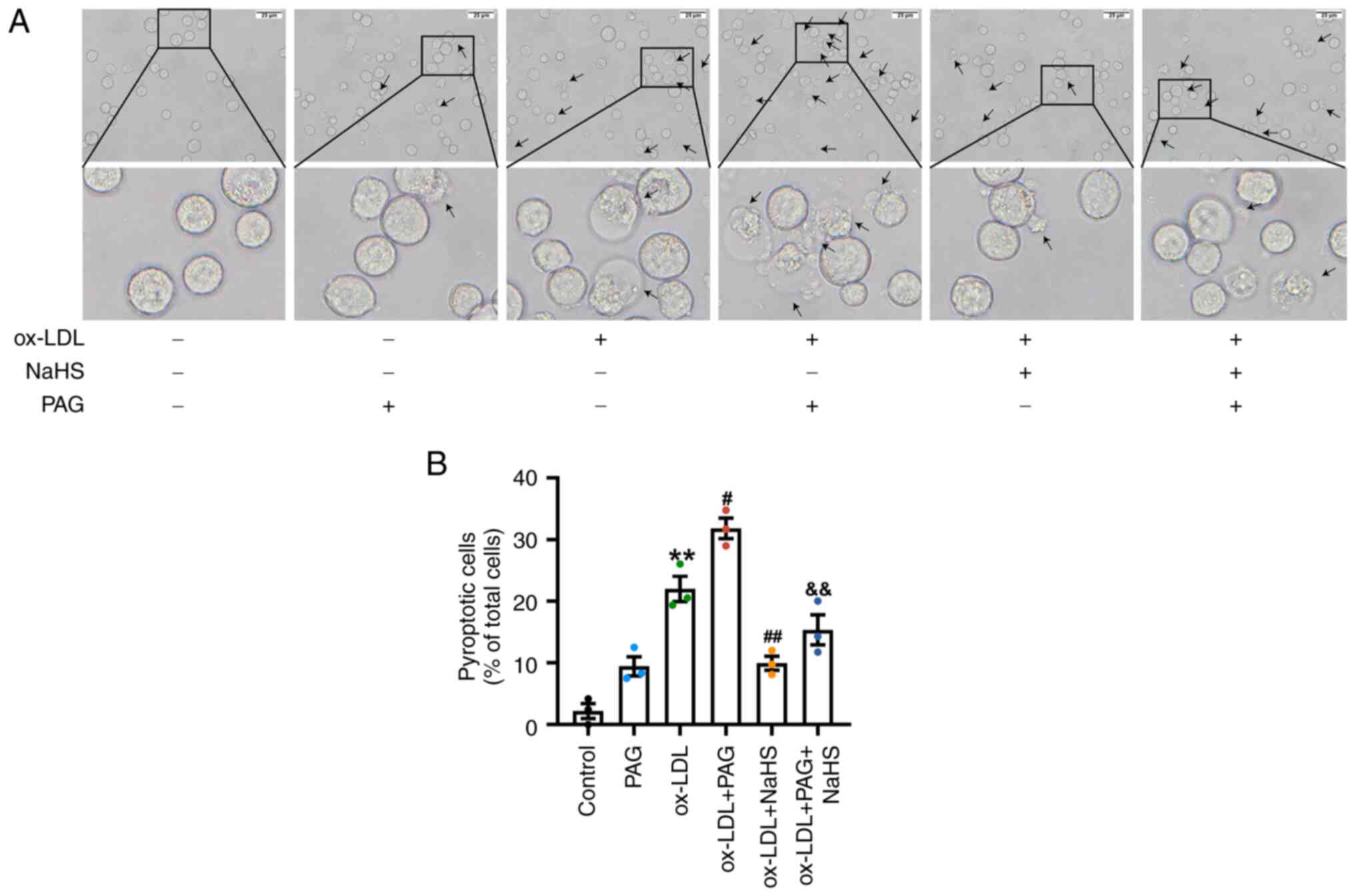
(Figs. S1A and 2A). By contrast, ox-LDL and LPS + ATP
induced a significant increase in the typical pyroptotic death of
THP-1 cells compared with the control group (Figs. S1 and 2), characterized by pronounced cell
swelling or membrane rupture and balloon-shaped bubbles extending
from the plasma membrane (Figs.
S1A and 2A). Meanwhile, THP-1
cells from ox-LDL + PAG group exhibited more obvious cell
morphology associated with pyroptosis and a more percentage of
pyroptotic THP-1 cells compared with the ox-LDL group (Fig. 2). However, pretreatment with NaHS
significantly decreased ox-LDL and LPS + ATP induced morphological
changes associated with pyroptosis and reduced the percentage of
pyroptotic THP-1 cells (Figs. S1
and 2). In addition, the
pretreatment of NaHS also effectively improved cell morphology
associated with pyroptosis, and reduced the percentage of
pyroptotic THP-1 cells in the presence of a combination of ox-LDL
and PAG compared with those in the ox-LDL + PAG group (Fig. 2).
To further assess the impact of H2S
signaling on pyroptosis in macrophages induced by ox-LDL and LPS +
ATP, Hoechst 33342/PI double staining in THP-1 cells and LDH
activity in the culture supernatant was assessed. PI enters the
cell via the ruptured membrane (57). ox-LDL significantly increased the
percentage of PI-positive THP-1 cells (Fig. 3A and B) and LDH release (Fig. 3C) compared with the control. The
percentage of PI-positive cells was significantly increased in the
ox-LDL + PAG group compared with the ox-LDL group (Fig. 3A and B). Likewise, LDH release was
also significantly increased in the ox-LDL + PAG compared with the
ox-LDL group (Fig. 3C).
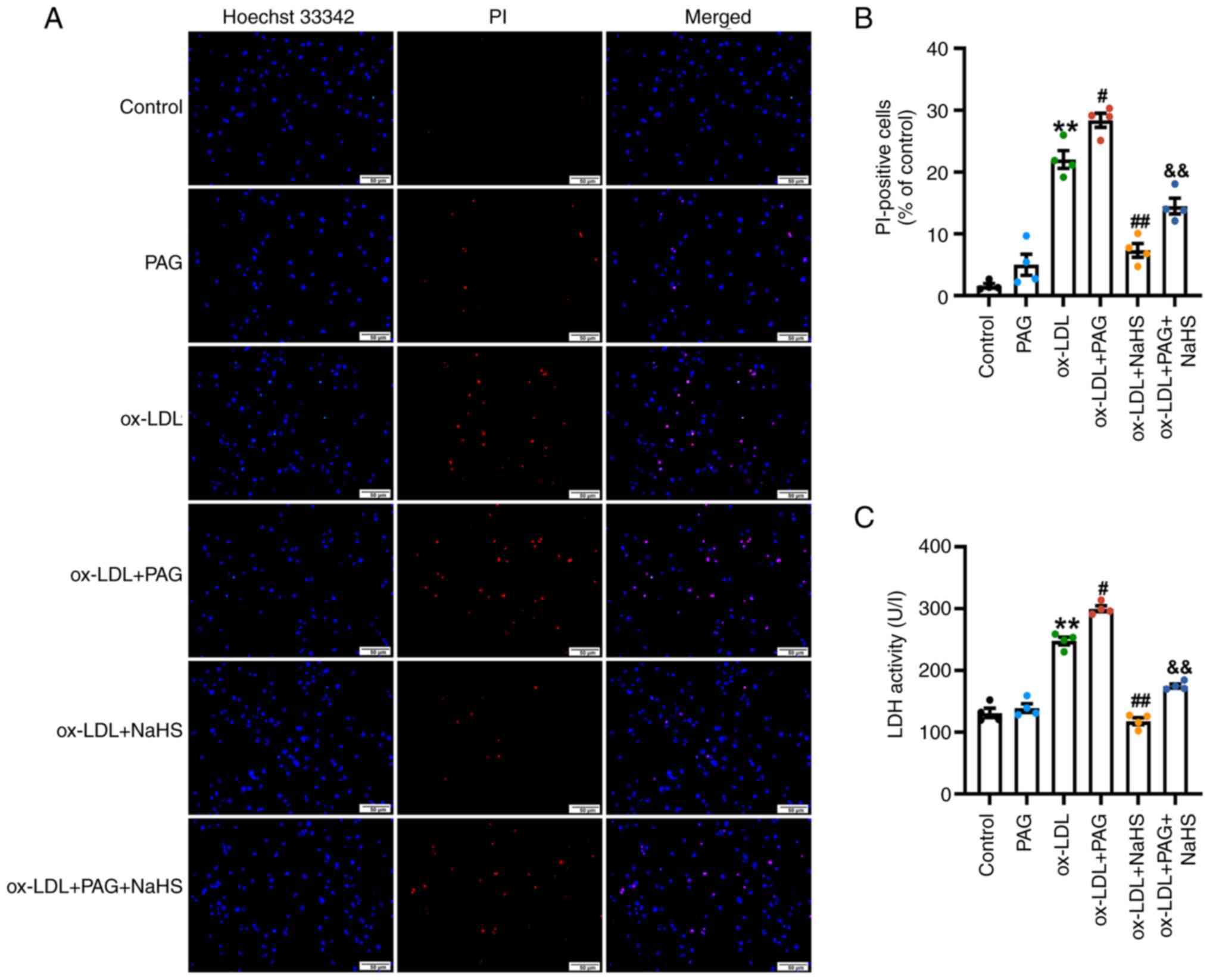 | Figure 3.H2S signaling inhibits
ox-LDL-induced pyroptosis in THP-1 macrophages. PMA-differentiated
THP-1 macrophages were stimulated with 100 µg/ml ox-LDL for 23.5 h
in the presence or absence of pretreatment with 200 µM NaHS and/or
2.5 mM PAG for 30 min. (A) NaHS inhibits ox-LDL or ox-LDL + PAG
-induced increase in the percentage of PI-positive cells.
Pyroptotic death in THP-1 cells was evaluated by Hoechst 33342/PI
double staining. Images were observed using fluorescence
microscopy. Magnification, ×200; scale bar, 50 µm. (B) Proportion
of pyroptotic cells was determined by PI-positive dead cells
relative to the total number of live cells exhibiting blue
fluorescence, demonstrated by Hoechst 33342 staining. n=4. (C) NaHS
inhibits LDH release in THP-1 macrophages treated by ox-LDL or
ox-LDL + PAG. Pyroptosis was assessed by measuring LDH activity in
the cell free media. n=4. **P<0.01 vs. control,
#P<0.05, ##P<0.01 vs. ox-LDL and
&&P<0.01 vs. ox-LDL + PAG. ox-LDL, oxidized
low-density lipoprotein; PMA, phorbol-12-myristate-13-acetate; PAG,
D,L-propargylglycine; PI, propidium iodide; LDH, lactate
dehydrogenase. |
NaHS (200 µM; Fig.
S2) or PAG treatment alone (2.5 mM; Fig. 3) under the same concentration did
not affect the percentage of PI-positive cells or LDH release.
These results suggested that ox-LDL induced pyroptosis in
macrophages derived from THP-1, consistent with a prior study that
reported that differentiated THP-1 macrophages exhibit increased
pyroptosis in response to different ox-LDL concentrations (50, 100
and 150 µg/ml) (45).
NaHS lowered the proportion of PI-positive cells
(Fig. 3A and B) and LDH release
(Fig. 3C) in ox-LDL + NaHS group
compared with those of the ox-LDL group in THP-1 cells.
Additionally, compared with ox-LDL + PAG treatment group, NaHS
pretreatment significantly inhibited the increase in the proportion
of PI-positive cells (Fig. 3A and
B) and LDH release (Fig. 3C)
in THP-1 cells from ox-LDL + PAG + NaHS group. Similarly, the
percentage of pyroptotic cells and LDH release in the LPS + ATP +
NaHS group was significantly lower compared with the LPS + ATP
group, suggesting LPS + ATP-induced macrophage pyroptosis could be
attenuated by NaHS pretreatment (Figs. S1 and S2). In summary, these results suggested
that H2S signaling protected macrophages from pyroptotic
cell death induced by ox-LDL.
H2S signaling inhibits
caspase-1 expression and activity in ox-LDL and LPS + ATP
stimulated THP-1 macrophages
Caspase-1 is a key enzyme in the canonical
pyroptotic pathway. Caspase-1 cleaves GSDMD, generating the active
GSDMD-N form, which forms pores in the cell membrane, initiating
pyroptosis (27). To validate
whether H2S directly modulates caspase-1, the role of
H2S signaling in intracellular localization of cleaved
caspase-1 as well as caspase-1 activity in THP-1 macrophages was
assessed.
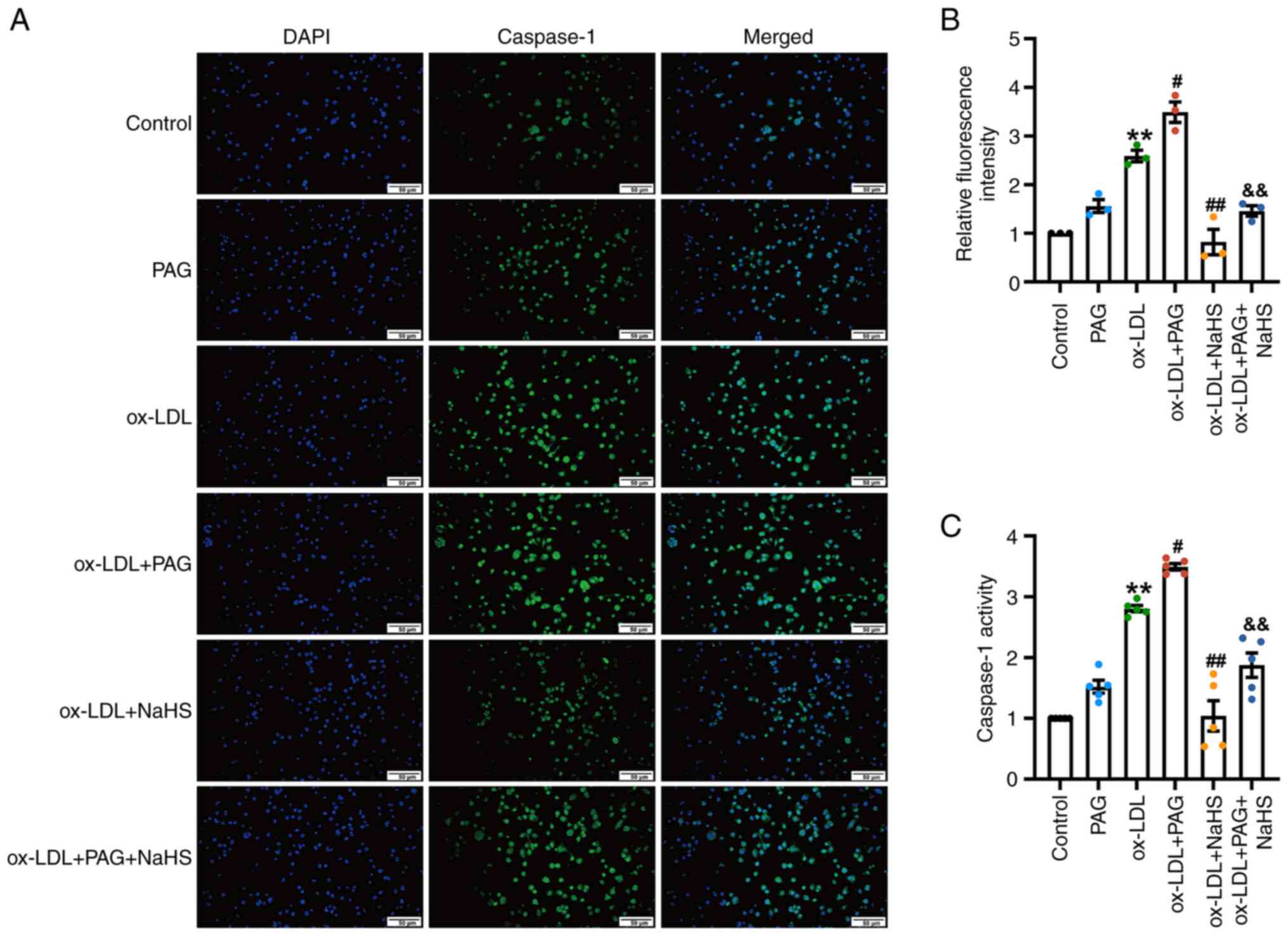
Immunofluorescence microscopy demonstrated that
ox-LDL significantly increased the fluorescence intensity of active
caspase-1 in THP-1 cells (Fig. 4A and
B) compared with the control. Co-treatment of PAG with ox-LDL
significantly increased the fluorescence intensity of caspase-1
compared with ox-LDL alone, whereas PAG treatment alone had no
significant impact on the fluorescence intensity of active
caspase-1 compared with the control. However, fluorescence
intensity of cleaved caspase-1 in THP-1 cells from the ox-LDL +
NaHS group significantly decreased compared with the ox-LDL group
(Fig. 4A and B). Fluorescence
intensity of caspase-1 in the ox-LDL + PAG + NaHS group was
significantly lower compared with the ox-LDL+ PAG group (Fig. 4A and B).
Likewise, ox-LDL significantly increased caspase-1
activity in THP-1 cells compared with the control (Fig. 4C). Furthermore, caspase-1 activity
of the ox-LDL + PAG group was significantly higher compared with
the ox-LDL group (Fig. 4C) while
NaHS pretreatment significantly decreased caspase-1 activity
compared with the ox-LDL group (Fig.
4C). ox-LDL + PAG + NaHS group had a significantly lower
caspase-1 activity compared with ox-LDL + PAG (Fig. 4C).
Moreover, the LPS + ATP-induced increase in
caspase-1 activity was also significantly decreased by NaHS
pretreatment (Fig. S3), while
treatment with PAG (Fig. 4C) or
NaHS (Fig. S3) alone did not
significantly affect caspase-1 activity. These results suggested
that H2S signaling ameliorated ox-LDL-induced pyroptosis
in THP-1 macrophages by suppressing active caspase-1 generation and
activity of caspase-1.
H2S signaling inhibits
ox-LDL- and LPS + ATP-induced activation of the canonical
pyroptosis signaling pathway
To further assess the effect of endogenous
H2S signaling or exogenous H2S donors on the
canonical pyroptosis signaling pathway. In the present study
western blotting assessed the protein expression of canonical
pyroptosis specific markers following treatment of THP-1 cells with
ox-LDL with or without pretreatment with NaHS or PAG. Treatment
with ox-LDL alone significantly increased protein expression levels
of NLRP3, GSDMD, GSDMD-N, pro-caspase-1, caspase-1 and IL-18
compared with the control. ox-LDL + PAG treatment significantly
increased protein expression levels of NLRP3, GSDMD-N, caspase-1
and IL-18 compared with the ox-LDL group (Fig. 5). However, NaHS (Fig. S4) or PAG alone (Fig. 5) did not significantly promote
protein expression of pyroptosis specific markers in THP-1 cells,
and ox-LDL + PAG treatment did not significantly increase GSDMD
expression compared with ox-LDL treatment alone (Fig. 5).
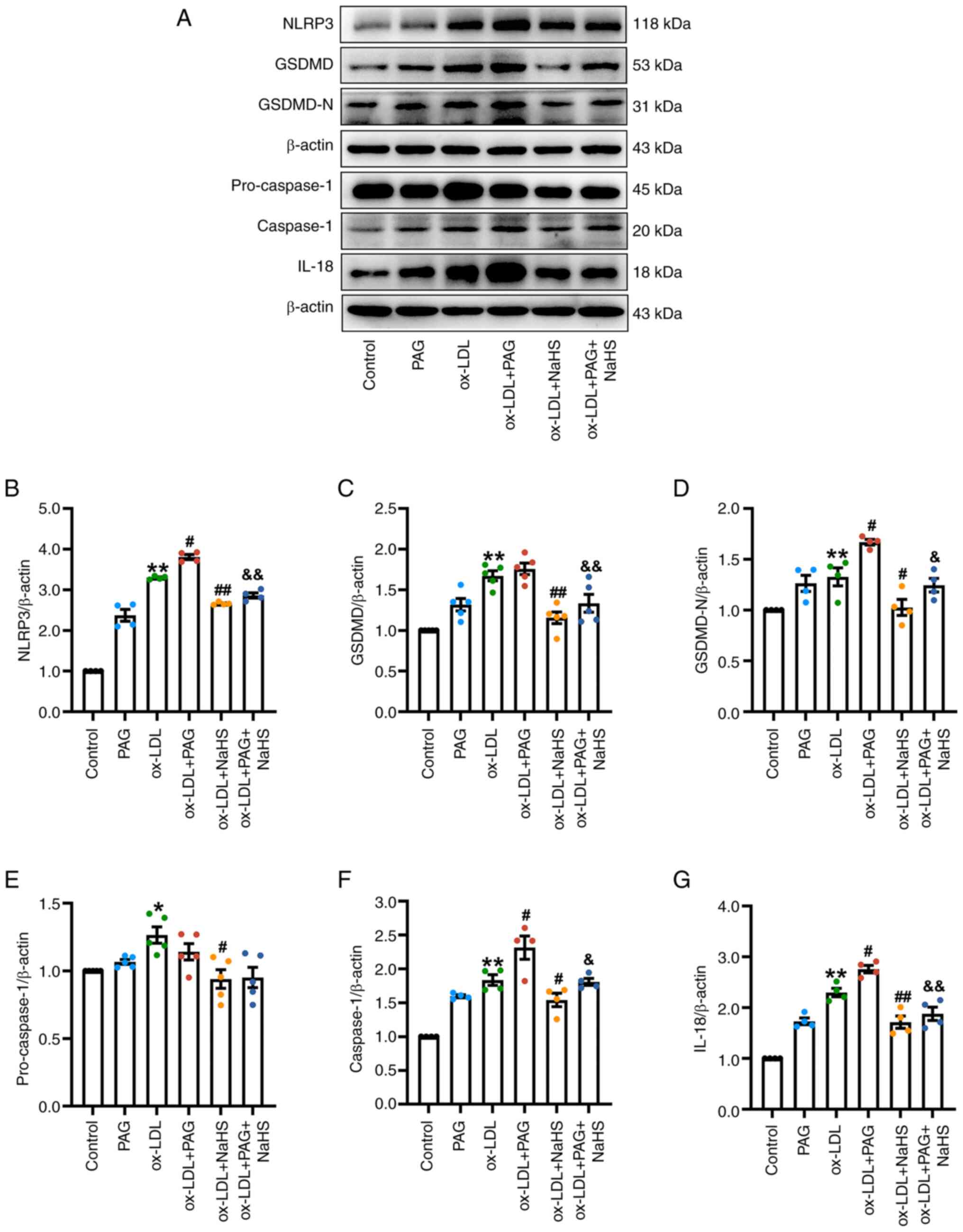 | Figure 5.H2S signaling inhibits
ox-LDL-induced activation of the canonical pyroptotic pathway.
Immunoblotting was performed to determine the protein levels of
NLRP3, pro-caspase-1, caspase-1, GSDMD, GSDMD-N and IL-18 in THP-1
macrophages treated with 100 µg/ml ox-LDL for 23.5 h in the
presence or absence of pretreatment with 200 µM NaHS and/or 2.5 mM
PAG for 30 min. (A) NaHS reduces the expression of pyroptosis
specific markers in THP-1 macrophages treated by ox-LDL or ox-LDL +
PAG. Representative immunoblots of pyroptotic indicators in the
extracts of THP-1 cells. β-actin served as an internal reference
protein. The relative protein levels were quantified based on band
intensity. The relative IOD of (B) NLRP3 (n=4), (C) GSDMD (n=5),
(D) GSDMD-N (n=4), (E) pro-caspase-1 (n=5), (F) caspase-1 and (G)
IL-18 (both n=4) was examined using Image J software. The relative
expression levels of these pyroptotic proteins in control group
were normalized to 1. *P<0.05, **P<0.01 vs. control,
#P<0.05, ##P<0.01 vs. ox-LDL,
&P<0.05 and &&P<0.01 vs.
ox-LDL + PAG. NLRP3, nucleotide-binding oligomerization domain-like
receptor family pyrin domain containing 3; GSDMD-N, N-terminal
fragment of gasdermin D; ox-LDL, oxidized low-density lipoprotein;
PAG, D,L-propargylglycine; IOD, integrated optical density. |
Pretreatment with NaHS significantly attenuated the
protein expressions of pyroptosis specific markers (NLRP3, GSDMD,
GSDMD-N, caspase-1 and IL-18) induced by ox-LDL, ox-LDL + PAG and
LPS + ATP in THP-1 cells compared with the ox-LDL group, ox-LDL +
PAG group and LPS + ATP group, respectively (Figs. 5 and S4). Moreover, pretreatment with NaHS did
not affect pro-caspase-1 expression (Fig. 5A and E) in the ox-LDL + PAG + NaHS
group compared with the ox-LDL + PAG group.
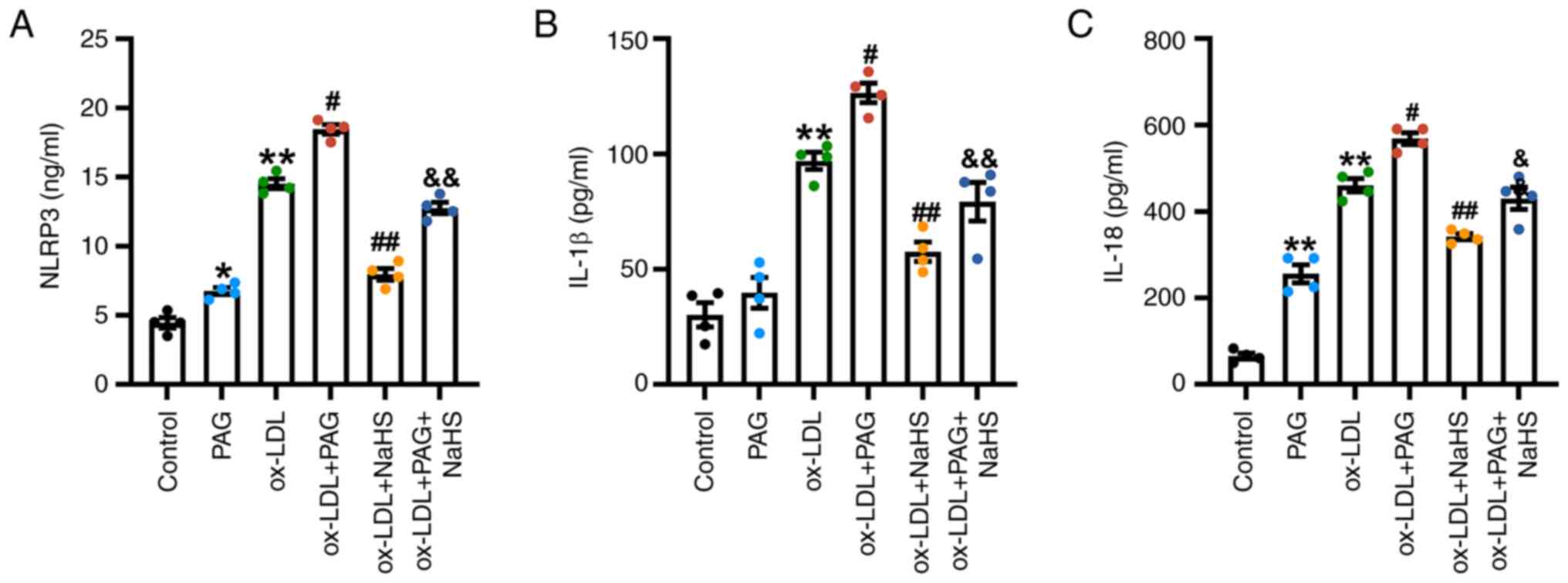
To evaluate the impact of H2S signaling
on the pyroptosis pathway activation, ELISA was used to assess the
concentrations of NLRP3 in THP-1 cell extract, and IL-1β and IL-18
secretion in cell-free media. ox-LDL and ox-LDL + PAG significantly
increased the levels of NLRP3 in THP-1 cell extract and IL-1β and
IL-18 in cell culture supernatants compared with the control and
ox-LDL groups, respectively (Fig.
6). NaHS pretreatment significantly decreased levels of NLRP3
in THP-1 cell extract and IL-1β and IL-18 secretion induced by
ox-LDL and ox-LDL + PAG in the cell culture supernatant compared
with the ox-LDL and ox-LDL + PAG groups, respectively (Fig. 6). However, no significant change in
IL-1β levels was demonstrated in the PAG compared with the control
group (Fig. 6B). Notably, PAG
alone increased the levels of NLRP3 in THP-1 cell extract and IL-18
secretion in the cell-free media (Fig.
6A and C).
H2S inhibits activity of
caspase-1 and reduces ox-LDL-induced caspase-1 dependent pyroptosis
via S-sulfhydration of pro-caspase-1
S-sulfhydration is the H2S mediated
post-translational modification (58,59)
that regulates the structure and functionality of proteins
(60). To evaluate whether
H2S mediated S-sulfhydration affects caspase-1 mediated
pyroptosis, the level of sulfhydrated pro-caspase-1, release of
caspase-1 and IL-1β and caspase-1 activity were measured in THP-1
cells treated with ox-LDL and DTT(S-sulfhydration modification
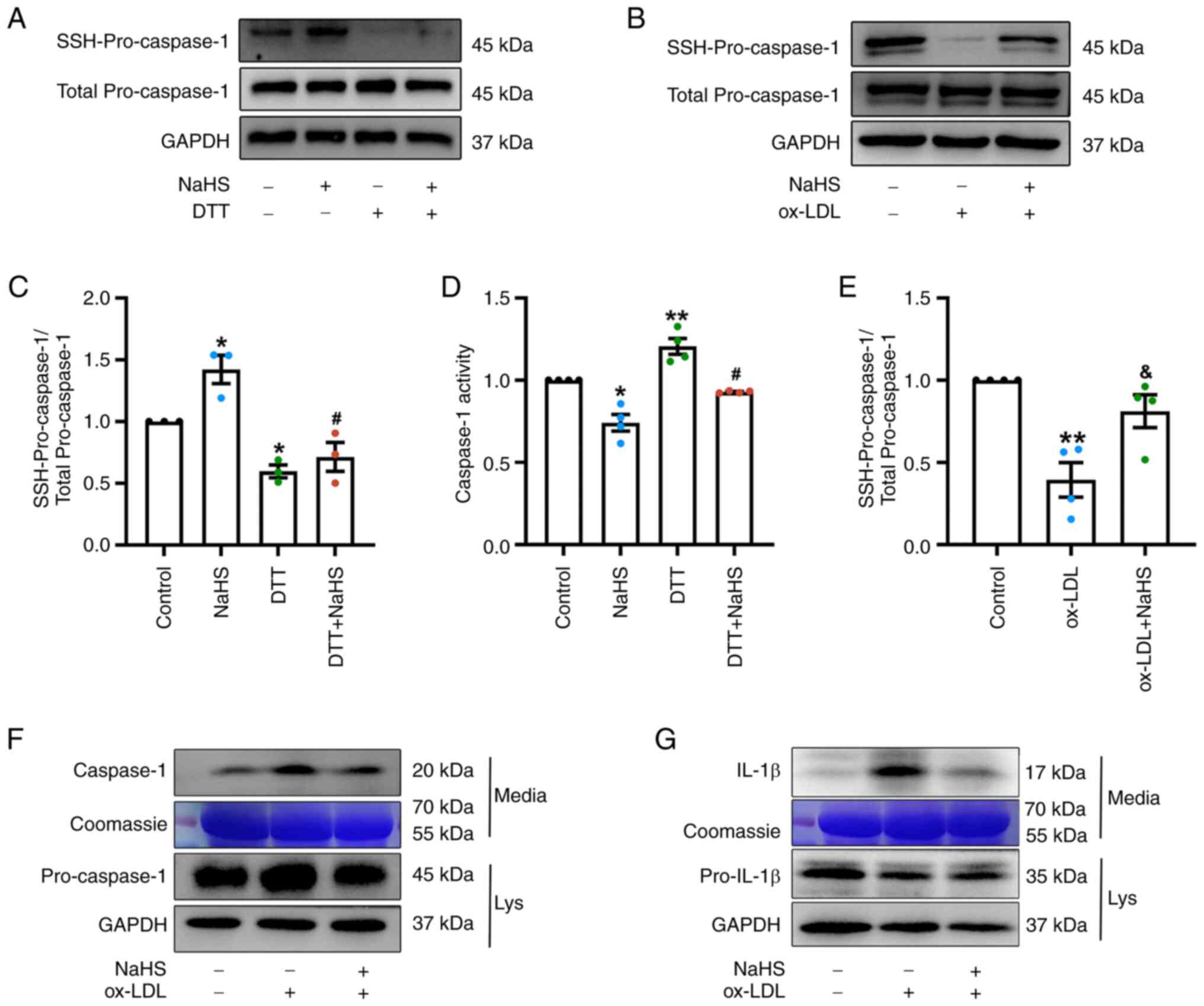
remover) in the presence or absence of NaHS pretreatment. In the
control group, pro-caspase-1 underwent S-sulfhydration even in the
absence of exogenous H2S. Moreover, treatment with NaHS
alone for 2 h increased the relative levels of pro-caspase-1
S-sulfhydration in THP-1 cells compared with the control group
(Fig. 7A and C). By contrast,
incubation with DTT induced a significant reduction in the
S-sulfhydration levels of pro-caspase-1 in THP-1 cells treated with
DTT or DTT + NaHS compared with the control group and the NaHS
group, respectively (Fig. 7A and
C).
Consistent with these observations, NaHS treatment
alone notably decreased activity of caspase-1, whereas treatment
with DTT significantly increased caspase-1 activity compared with
the control. Likewise, the DTT + NaHS significantly increased
caspase-1 activity compared with the NaHS group (Fig. 7D). These results suggest that
H2S may inhibit caspase-1 activity via S-sulfhydration
of caspase-1. Furthermore, it was demonstrated that ox-LDL
significantly decreased S-sulfhydration of pro-caspase-1 in THP-1
cells (Fig. 7B and E), and
increased levels of secreted active caspase-1 (Fig. 7F) and IL-1β (Fig. 7G) in the culture media when
compared with THP-1 cells and the culture media of the control
group. Similarly, LPS + ATP induced increase in the levels of
caspase-1 and IL-1β in cell culture supernatants (Fig. S5). However, NaHS pretreatment
restored the decreased S-sulfhydration of pro-caspase-1 induced by
ox-LDL with significantly increased levels of S-sulfhydrated
pro-caspase-1 compared with the ox-LDL group (Fig. 7B and E). ox-LDL + NaHS group and
LPS + ATP + NaHS group had decreased levels of secreted caspase-1
and IL-1β (Figs. S5, 7F and G) when compared with the culture
media of the ox-LDL group or LPS + ATP group, while NaHS treatment
alone did not increase the levels of secreted caspase-1 and IL-1β
(Fig. S5).
Discussion
Although previous studies have reported the role of
H2S in countering inflammation and pyroptosis (40–43,45,54–56,61),
the specific molecular mechanisms by which H2S regulates
the pyroptosis signaling pathway and inhibits macrophage pyroptosis
are largely unknown. The present study demonstrated a molecular
mechanism by which H2S signaling directly regulates
caspase-1-dependent macrophage pyroptosis via S-sulfhydration of
pro-caspase-1. To the best of our knowledge, this mechanism has not
been previously identified. ox-LDL was demonstrated to
significantly decrease S-sulfhydration of pro-caspase-1 by
inhibiting endogenous H2S production, consequently
inducing pyroptosis in THP-1 macrophages. This induction occurs via
activation of the canonical pyroptosis signaling pathway as well as
an increase in enzymatic activity of caspase-1, as reported by
previous studies (6,13,32,45).
Inhibition of endogenous H2S production by PAG enhanced
ox-LDL-induced expression and activity of pyroptosis-associated
proteins, leading to pyroptotic death in macrophages. The exogenous
H2S donor NaHS not only ameliorated ox-LDL-induced
macrophage pyroptosis but also mitigated the effect of co-treatment
with PAG and ox-LDL on pyroptosis. Finally, decreased levels of
pro-caspase-1 S-sulfhydration by DTT or ox-LDL increased caspase-1
activity and secretion of pyroptosis-related proteins (caspase-1,
IL-1β and IL-18). Conversely, increased S-sulfhydration of
pro-caspase-1 mediated by H2S decreased caspase-1
activity and secretion of pyroptosis-related proteins (caspase-1,
IL-1β and IL-18). These findings, in conjunction with a previous
study (45), provide evidence that
pro-caspase-1 was a target of H2S-mediated
S-sulfhydration in the modulation of pyroptosis. The present study
also demonstrated the role of H2S-induced
S-sulfhydration in the canonical pyroptosis signaling pathway.
These findings increase understanding of the molecular mechanisms
through which H2S exerts anti-inflammatory and
anti-pyroptotic effects. Furthermore, the findings of the present
study establish a foundation for the development of novel
H2S-releasing drugs and anti-atherosclerotic agents
targeting the pyroptosis signaling pathway.
Previous studies (1,2,4,6,8,11,13,23)
suggest that both excessive pyroptosis in macrophages and release
of pro-inflammatory molecules by pyroptotic macrophages contribute
to the progression of AS. This process enlarges the size of the
necrotic lipid core in atherosclerotic plaque, increases plaque
instability and increases the risk of arterial thrombosis (4,25,62,63).
ox-LDL is an independent pathogenic factor associated with
inflammatory responses and AS. ox-LDL has been reported to activate
the NLRP3 inflammasome and caspase-1 in macrophages (8,14,32).
Activated caspase-1 triggers pyroptosis by directly cleaving GSDMD,
inducing development of GSDMD-N formed cell membrane pores and
cleaving precursors of IL-1β and IL-18 into active forms (8,13,14,32).
Pro-inflammatory cytokines IL-1β and IL-18 are secreted during
pyroptosis via GSDMD-N membrane pores, and NLRP3 inflammasome or
caspase-1 activation is linked to the maturation and release of
these cytokines (1,4,8,11,13,14).
During pyroptosis, the plasma membrane is damaged, allowing water
influx causing cell swelling, osmotic lysis and release of
intracellular contents including pro-inflammatory mediators and
LDH, a hallmark of pyroptosis, into the extracellular space
(13,54).
The present study demonstrated that ox-LDL
decreased cell viability and induced pyroptotic cell death, marked
by pronounced cell swelling, membrane rupture and formation of
balloon-shaped bubbles extending from the plasma membrane. ox-LDL
also increased proportion of PI-stained cells, LDH activity and the
protein expression of NLRP3, caspase-1, GSDMD, GSDMD-N, IL-1β and
IL-18, which are related to the pyroptosis pathway in THP-1 cells.
Moreover, ox-LDL could promote the secretion of
pyroptosis-associated cytokines (IL-1β and IL-18) were secreted in
cell-free culture supernatant. The aforementioned results are in
line with a previous study that showed differentiated THP-1
macrophages can be stimulated to undergo increased pyroptosis by
ox-LDL at different ox-LDL concentrations (50, 100 and 150 µg/ml)
(45), suggesting that ox-LDL can
induce macrophage pyroptosis. Numerous studies have reported
increased expression of caspase-1 in lesion macrophages in animal
and human AS and in macrophages stimulated by atherosclerotic risk
factors (1,9,22,24,42).
This implies that caspase-1 is involved in formation of
atherosclerotic lesions mediated by macrophage pyroptosis.
Consistent with these studies, the present study also demonstrated
that ox-LDL increased caspase-1 activity in macrophages.
Furthermore, increasing evidence indicates that specific
pharmacological inhibition or genetic knockout of caspase-1
decreases macrophage pyroptosis induced by AS-associated risk
factors (ox-LDL, homocysteine and cholesterol) and plaque
development in ApoE−/− mice (22,24,26–28,31).
Therefore, inhibiting the caspase-1-dependent pyroptosis signaling
pathway and decreasing macrophage pyroptosis are promising
therapeutic strategies for the treatment of AS.
Studies have reported the involvement of
H2S in numerous physiological processes such as lipid
metabolism, blood vessel relaxation, blood pressure regulation and
neurotransmission (34,36,64,65).
H2S exhibits potent anti-inflammatory properties in
numerous types of inflammatory and immune disease including
atherosclerosis, multiple sclerosis, inflammatory bowel diseases,
ulcerative colitis, asthma and sepsis-induced cardiomyopathy
(38,40,41,66–68).
In particular, the anti-inflammatory properties of H2S
are associated with its anti-pyroptotic effects (40–43,55,68,69).
Administration or pretreatment with exogenous H2S donors
such as GYY4137 and NaHS have been reported to alleviate
inflammatory responses by suppressing activation of the NLRP3
inflammasome and the caspase-1-dependent canonical pyroptosis
pathway in numerous types of cell (40–43,45,55,68–72).
This causes a significant decrease in the inflammation of tissues
and organ damage induced by numerous pro-inflammatory pathological
factors (40–43,54–56,66,68,69).
However, the aforementioned studies have not demonstrated the
mechanism by which H2S inhibits macrophage pyroptosis.
The present study demonstrated that exogenous H2S can
inhibit the expression and secretion of pyroptosis-specific markers
(NLRP3, caspase-1, GSDMD, GSDMD-N, IL-1β and IL-18), and caspase-1
activity in response to ox-LDL treatment, thereby attenuating
morphological and biochemical changes associated with macrophage
pyroptosis induced by ox-LDL. Therefore, exogenous H2S
exerts both anti-inflammatory and anti-pyroptotic effects.
Furthermore, the present study demonstrated that
inhibition of endogenous H2S production increases the
pro-pyroptotic effects of ox-LDL in macrophages by increasing
activation of the canonical pyroptosis signaling pathway. Thus, the
results of the present study align with previous research
demonstrating the anti-pyroptotic effects of H2S in
countering NLRP3/caspase-1/GSDMD-mediated pyroptosis in cellular
and mammalian models of acute or chronic inflammation (40,70).
As Yue et al (71) and
Zheng et al (72) reported,
a potential link may exist between H2S metabolism and AS
via the NLRP3/caspase-1/IL-1β signaling pathway. Zheng et al
(72) reported that increased
activation of caspase-1 and expression of NLRP3 and mature IL-1β in
endothelial cells, induced by high glucose + ox-LDL stimulation, is
reversed by the addition of exogenous H2S. Furthermore,
the present study suggested that the effects of H2S
against ox-LDL-induced macrophage pyroptosis were mediated through
the inhibition of the NLRP3/caspase-1/GSDMD-dependent canonical
pyroptosis signaling pathway. Moreover, the effects of NaHS
treatment alone on macrophage pyroptosis were assessed in the
present study, which demonstrated that NaHS (200 µM) treatment
alone under the same concentration did not significantly promote
pyroptosis specific proteins and (NLRP3, GSDMD and GSDMD-N)
pyroptosis of THP-1-derived macrophages, while LPS + ATP-induced
macrophage pyroptosis was attenuated by NaHS pretreatment. Cell
viability assays in the present and a previous study (45) also demonstrated that NaHS treatment
alone has no adverse effect on THP-1 macrophages and increased
concentrations (50, 100 and 200 µM;) of NaHS significantly reduced
ox-LDL-induced pyroptosis in THP-1-derived macrophages. Based on
these results, it is suggested that NaHS administration alleviated
pyroptosis in ox-LDL-stimulated THP-1 macrophages by inhibiting the
activation of the canonical pyroptotic pathway, which exhibited a
positive correlation with increased levels of H2S in
THP-1 macrophages.
The dysfunction of H2S signaling is
implicated in development of AS and is associated with
ox-LDL-induced macrophage inflammation (38,51,73).
In mammalian tissue, H2S is primarily synthesized
endogenously by three key enzymes, CSE, 3-mercaptopyruvate sulfur
transferase and cystathionine β-synthetase (CBS), using L-cysteine
and homocysteine as primary substrates (34,67).
However, the expression and distribution of these three enzymes are
tissue- and cell-specific (52).
CSE is the main enzyme involved in endogenous H2S
production in multiple types of macrophage (42,51,52).
Previous studies also suggest that by suppressing expression and
function of CSE and CBS, ox-LDL attenuates production of
H2S in THP-1 cells as well as Raw264.7 macrophages
(51,73). ox-LDL can induce DNA
hypermethylation of the CSE promoter in macrophages, resulting in
suppression of H2S production and CSE transcription,
thereby exacerbating inflammatory responses (74). CSE deficiency or inhibition induces
NLRP3 inflammasome activation in inflammatory cellular and mouse
models with peritonitis, acute kidney injury, high choline-induced
cardiac dysfunction, diabetic cardiomyopathy and colitis (40,70,75–77).
Therefore, the present study investigated the impact of endogenous
and exogenous H2S on macrophage pyroptosis following
ox-LDL stimulation. The experiment in Fig. 1D assessed the alterations in
endogenous H2S levels in macrophages treated with
numerous drugs. It was demonstrated that ox-LDL and/or PAG
decreased H2S synthesis by inhibiting CSE, while NaHS
increased H2S levels in ox-LDL- and/or PAG-treated THP-1
cells, consistent with previous findings (51). Notably, inhibiting endogenous
generation of H2S with PAG promoted pyroptosis in
ox-LDL-stimulated THP-1 macrophages by exacerbating the activation
of the pyroptosis signaling pathway. This was observed through
morphological changes associated with pyroptosis, increased
PI-positive cell counts, LDH release, accumulation of cleaved
caspase-1, caspase-1 activity and upregulation of
pyroptosis-related proteins in THP-1 cells, compared with those
stimulated by ox-LDL alone. This exacerbation of pyroptotic effects
by co-treatment with PAG with ox-LDL was attenuated by pretreatment
with NaHS.
Numerous studies have reported that CSE
deficiency/silencing/inhibition by PAG in monosodium urate and
dextran sulphate sodium stimulated bone marrow-derived macrophages,
THP-1 cells, RAW264.7 cells murine peritoneal macrophages
stimulated with LPS (40,71,77–79)
increase the protein expression levels of NLRP3, active caspase-1,
GSDMD and IL-1β, which are associated with the pyroptosis pathway.
Conversely, treatment with exogenous H2S donors can
suppress expression of NLRP3, GSDMD-N, cleaved caspase-1 and IL-1β
in CSE-deficient mice and macrophages (40,77,79).
These findings align with the present study, demonstrating that the
suppression of endogenous H2S production by PAG
exacerbated the pro-pyroptotic role of ox-LDL in macrophages
derived from THP-1, while supplementation with exogenous
H2S via NaHS inhibited macrophage pyroptosis by
downregulating the canonical pyroptosis pathway. This suggests that
H2S signaling served an anti-pyroptotic role in
ox-LDL-induced macrophage inflammation. Moreover, endogenously
produced H2S by CSE may be a critical negative regulator
of the canonical pyroptosis pathway.
Although increasing evidence suggests a role for
H2S signaling in the inhibition of NLRP3 inflammasome
activation and caspase-1-dependent canonical pyroptosis (40–43,45,54–56,61),
there is a lack of understanding regarding the molecular mechanisms
by which H2S signaling inhibits macrophage pyroptosis.
H2S-mediated protein post-translational modification
targeting sulfhydryl groups, also known as S-sulfhydration, is a
key molecular mechanism by which H2S regulates structure
and function of target proteins (34,44,60).
Numerous target proteins undergo S-sulfhydration and there is
growing evidence that these proteins are involved in
anti-inflammatory responses and anti-atherosclerotic effects
(34,42,59,73,80).
A previous study reported that by S-sulfhydrating c-Jun in THP-1
macrophages, H2S decreases activation of the NLRP3
inflammasome induced by H2O2 (42). However, to date there is no direct
evidence for H2S mediated S-sulfhydration directly
regulates pyroptosis specific proteins in the protection against
macrophage pyroptosis.
Caspase-1 and caspase-3 both belong to the caspase
family; activated caspase-3 can induce macrophage pyroptosis by
cleaving GSDME, a member of the gasdermin family, into GSDME-N
(2,16). Pro-caspase-3 is constitutively
S-sulfhydrated in unstimulated cells and H2S can inhibit
activity of caspase-3 and cell apoptosis by enhancing the
S-sulfhydration levels of caspase-3 (60,64).
Based on these findings, along with the suppressive effects of
H2S on caspase-1 activity and accumulation demonstrated
in the present study, it was hypothesized that caspase-1 serves as
a target protein for H2S-mediated S-sulfhydration and
that H2S inhibits activation or activity of caspase-1
and macrophage pyroptosis through the S-sulfhydration of caspase-1.
To validate this hypothesis, the desulfhydration reagent DTT and
NaHS were used to assess the effect of S-sulfhydration induced by
H2S on caspase-1 activity. The biotin switch assay used
is an internationally recognized measurement technique for
detecting protein S-sulfhydration (34,44,50).
Specifically, the results demonstrated the existence of basal
S-sulfhydrated pro-caspase-1. S-sulfhydration of pro-caspase-1 was
increased following treatment with NaHS, resulting in a notable
decrease in the activity of caspase-1 in NaHS treated THP-1 cells.
Notably, both the basal S-sulfhydration of pro-caspase-1 and the
NaHS-induced S-sulfhydration of pro-caspase-1 were significantly
decreased by the desulfhydration reagent DTT, which increased
caspase-1 activity or mitigated the inhibitory effect of NaHS on
caspase-1 activity in macrophages. These findings suggested that
H2S-induced suppression of caspase-1 activity and
macrophage pyroptosis may be attributed to reversible
S-sulfhydration of protein thiols within pro-caspase-1.
To validate the inhibitory effects of
H2S-mediated caspase-1 S-sulfhydration on macrophage
pyroptosis induced by ox-LDL, the impact of the S-sulfhydration of
pro-caspase-1 on pyroptosis-induced secretion of caspase-1 and
IL-1β, mediated by GSDMD-N-formed membrane pores in THP-1 cells
exposed to ox-LDL was assessed. This demonstrated a notable
reduction in the basal S-sulfhydration of pro-caspase-1 in THP-1
cells treated with ox-LDL. Attenuation of S-sulfhydration of
pro-caspase-1 induced by ox-LDL was mitigated by NaHS pretreatment.
Meanwhile, NaHS pretreatment could reduce macrophage
pyroptosis-induced secretion of activated caspase-1 and mature
IL-1β. Therefore, these results suggested that pro-caspase-1 can be
S-sulfhydrated by H2S, which inhibited the generation of
active caspase-1, and led to the deactivation of downstream
pyroptotic signaling pathways. The present study demonstrated that
ox-LDL can induce inflammation-mediated macrophage pyroptosis by
upregulating the NLRP3/caspase-1/GSDMD-dependent canonical
pyroptosis signaling pathway. ox-LDL can also inhibit endogenous
H2S synthesis, thereby diminishing the anti-inflammatory
and anti-pyroptotic effects of endogenous H2S by
decreasing the constitutive S-sulfhydration of pro-caspase-1
mediated by endogenous H2S, leading to increased
caspase-1 activity and activation of the caspase-1-dependent
pyroptosis pathway. By contrast, pretreatment with exogenous
H2S increased intracellular H2S
concentrations, thereby exerting an anti-pyroptotic effect by
directly inhibiting ox-LDL-induced activation of the
NLRP3/caspase-1/GSDMD axis. The increased intracellular
H2S resulting from exogenous H2S pretreatment
increased the S-sulfhydration of pro-caspase-1. It was plausible to
hypothesize that H2S inhibits caspase-1 activity by
S-sulfhydrating pro-caspase-1, thereby preventing cleavage of
GSDMD/IL-1β, and decreasing macrophage pyroptosis.
There are several limitations to the present study.
First, only the function of the CSE/H2S pathway in
pyroptosis of macrophages was assessed. However, as the
downregulated endogenous CBS/H2S pathway is implicated
in ox-LDL-induced macrophage inflammation (73), further study is required to
elucidate the role of the CBS/H2S pathway in preventing
macrophage pyroptosis. Second, the present study did not identify
specific residues that are S-sulfhydrated in pro-caspase-1.
Although the present study represented a preliminary investigation
of the relationship between S-sulfhydration of pro-caspase-1 and
canonical caspase-1-dependent pyroptosis, before implementing it,
it would be profitable to identify the specific or crucial cysteine
residues in caspase-1 that are S-sulfhydrated. Liquid
chromatography with tandem mass spectrometry can determine
post-translational modifications of proteins; to the best of our
knowledge, however, there are few studies (50,64,81)
detecting the specific cysteine sites of S-sulfhydration
modification using mass spectrometry due to this technique has a
propensity to identify false positive cysteine sites (82,83).
Therefore, mass spectrometry results identifying S-sulfhydrated Cys
in pro-caspase-1 would require verification through mutation
studies to exclude the possibility of false positive S-sulfhydrated
sites (84,85). In future studies, these techniques
should be used to identify the key Cys residues that are
S-sulfhydrated in pro-caspase-1. Likewise, mutation studies should
be used to clarify the specific effects of Cys site, as well as
potential synergistic effects of multiple Cys site mutations on
bioactivity of caspase-1 and the interactions between
NLRP3/caspase-1/GSDMD signaling and macrophage pyroptosis.
Previous studies (86,87)
have reported key interplay and competition between nitric
oxide-induced S-nitrosylation and H2S-induced
S-sulfhydration. These post-translational modifications can occur
at the same Cys residues of target proteins but exert opposing
effects. This suggests that these post-translational modifications
may maintain normal function of target proteins (44,82).
Additional studies are required to assess the S-nitrosylation
status of pro-caspase-1 under normal conditions and in response to
pro-inflammatory stimuli. Moreover, the role of homeostasis between
S-nitrosylation and S-sulfhydration of caspase-1 at key Cys sites
in macrophage pyroptosis warrants further investigation. Lastly, in
addition to modulating the function and activity of target
proteins, S-sulfhydration can also regulate subcellular
localization, protein-protein interactions and protein stability.
Thus, immunoprecipitation experiments should be performed to assess
how S-nitrosylation and S-sulfhydration at different Cys residues
of caspase-1 affects interactions with other pyroptosis-specific
proteins.
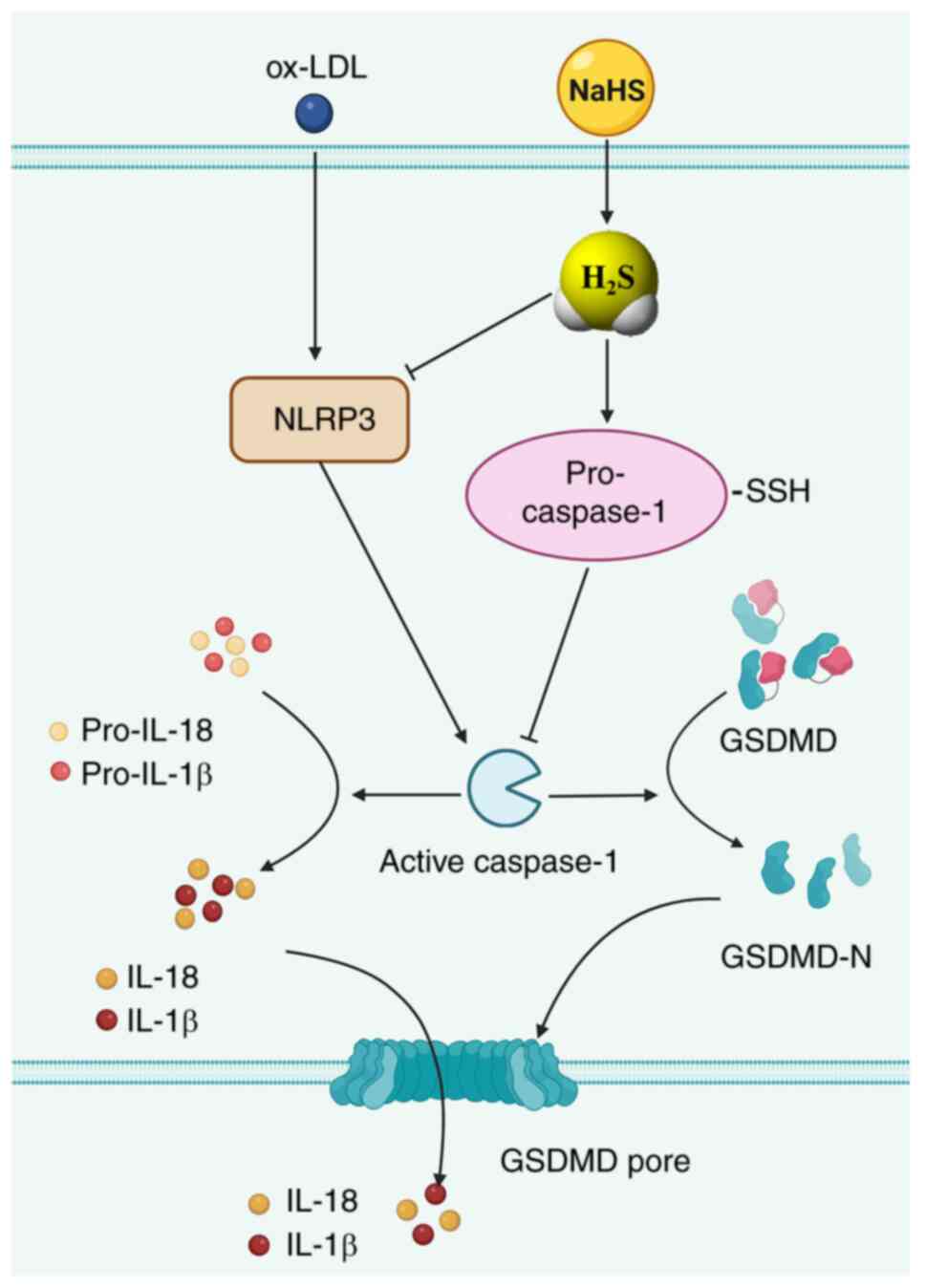
In conclusion, it was hypothesized that endogenous
pro-caspase-1 undergoes constitutive S-sulfhydration in macrophages
under normal conditions. Furthermore, decreased generation of
H2S triggered by ox-LDL stimulation in macrophages
decreases basal S-sulfhydration of pro-caspase-1. This increases
the activation of caspase-1, thereby initiating a downstream events
in the caspase-1-dependent canonical pyroptosis, including GSDMD
cleavage, GSDMD-N-mediated development of membrane pores and
release of activated caspase-1 and pro-inflammatory cytokines via
GSDMD-N membrane pores. Consequently, macrophage pyroptosis is
induced (Fig. 8). Finally, the
present study demonstrated that exogenous H2S donors
mitigates ox-LDL-induced macrophage pyroptosis by suppressing
expression of the NLRP3 inflammasome, the generation of functional
caspase-1, caspase-1 activity, production of the pyroptosis
executioner (GSDMD-N) and the secretion of mature pro-inflammatory
cytokines (IL-1β and IL-18). This mitigation occurs by elevating
H2S levels and S-sulfhydration of pro-caspase-1
(Fig. 8). Such modifications may
account for deactivation of downstream signaling pathways
associated with caspase-1, including the generation of active
caspase-1, the suppression of GSDMD cleavage and formation of
membrane pores mediated by GSDMD-N. Furthermore, this leads to a
decrease in the release of pro-inflammatory mediators through
GSDMD-N-formed membrane pores.
The present study demonstrated a novel molecular
mechanism by which H2S directly regulates the
caspase-1-dependent pyroptotic pathway, thereby ameliorating
macrophage pyroptosis induced by ox-LDL stimulation. Furthermore,
the present results provide novel insights into the involvement of
S-sulfhydration in functional regulation of caspase-1. The results
of the present study supported the development of novel
H2S-releasing drugs specifically target
pyroptosis-specific proteins and the S-sulfhydration of
caspase-1.
Supplementary Material
Supporting Data
Acknowledgements
The authors would like to thank Professor Wenjing
Xu (Department of Pathology, School of Basic Medical Science, Xi'an
Medical University, Xi'an, Shanxi, China.) for technical assistance
with biotin switch assay and Miss Yan Wang (Ministry of Education
Key Laboratory of Xinjiang Endemic and Ethnic Diseases, Shihezi
University, Shihezi, Xinjiang, China) for providing technical
assistance with immunofluorescent staining.
Funding
The present study was supported by National Natural Science
Foundation of China (grant no. 81600325), Project of Science and
Technology Innovation from Xinjiang Production and Construction
Corps (grant no. 2022CB002-10), Non-profit Central Research
Institute Fund of Chinese Academy of Medical Sciences (grant no.
2020-PT330-003), Open Research Fund of NHC Key Laboratory of
Prevention and Treatment of Central Asia High Incidence Diseases
(grant no. KF202107) and Science and Technology Project of Shihezi
University (grant nos. CXBJ201906 and ZZZC202135).
Availability of data and materials
The data generated in the present study may be
requested from the corresponding author.
Authors' contributions
ZJ, XZ, ZL and HY performed H2S
detection, Hoechst 33342/PI double staining, caspase-1 activity
measurement, immunofluorescent staining, immunoblotting, ELISA and
analyzed/interpreted data. ZJ and XZ conducted biotin switch assay.
CL analyzed the data of biotin switch assay. XT and KM analyzed and
interpreted data of Hoechst 33342/PI double staining,
immunofluorescent staining and ELISA. LZ and LL designed the
experiments in this study and drafted/revised the manuscript. ZJ,
XZ, LZ and LL confirm the authenticity of all the raw data. All
authors have read and approved the final manuscript.
Ethics approval and consent to
participate
Not applicable.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Xu YJ, Zheng L, Hu YW and Wang Q:
Pyroptosis and its relationship to atherosclerosis. Clin Chim Acta.
476:28–37. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Wang Q, Wu J, Zeng Y, Chen K, Wang C, Yang
S, Sun N, Chen H, Duan K and Zeng G: Pyroptosis: A pro-inflammatory
type of cell death in cardiovascular disease. Clin Chim Acta.
510:62–72. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Miao EA, Rajan JV and Aderem A:
Caspase-1-induced pyroptotic cell death. Immunol Rev. 243:206–214.
2011. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
He X, Fan X, Bai B, Lu N, Zhang S and
Zhang L: Pyroptosis is a critical immune-inflammatory response
involved in atherosclerosis. Pharmacol Res. 165:1054472021.
View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Zychlinsky A, Prevost MC and Sansonetti
PJ: Shigella flexneri induces apoptosis in infected macrophages.
Nature. 358:167–169. 1992. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Peng X, Chen H, Li Y, Huang D, Huang B and
Sun D: Effects of NIX-mediated mitophagy on ox-LDL-induced
macrophage pyroptosis in atherosclerosis. Cell Biol Int.
44:1481–1490. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Hilbi H, Moss JE, Hersh D, Chen Y, Arondel
J, Banerjee S, Flavell RA, Yuan J, Sansonetti PJ and Zychlinsky A:
Shigella-induced apoptosis is dependent on caspase-1 which binds to
IpaB. J Biol Chem. 273:32895–32900. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
He B, Nie Q, Wang F, Han Y, Yang B, Sun M,
Fan X, Ye Z, Liu P and Wen J: Role of pyroptosis in atherosclerosis
and its therapeutic implications. J Cell Physiol. 36:7159–7175.
2021. View Article : Google Scholar
|
|
9
|
Lin L, Zhang MX, Zhang L, Zhang D, Li C
and Li YL: Autophagy, pyroptosis, and ferroptosis: New regulatory
mechanisms for atherosclerosis. Front Cell Dev Biol. 9:8099552022.
View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Liu JR, Wang C, Li J, Yu Y, Liu Y, Liu H,
Peng Q and Guan X: Autophagy blockage promotes the pyroptosis of
ox-LDL-treated macrophages by modulating the p62/Nrf2/ARE axis. J
Physiol Biochem. 77:419–429. 2021. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Xu XD, Chen JX, Zhu L, Xu ST, Jiang J and
Ren K: The emerging role of pyroptosis-related inflammasome pathway
in atherosclerosis. Mol Med. 28:1602022. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Taabazuing CY, Okondo MC and Bachovchin
DA: Pyroptosis and apoptosis pathways engage in bidirectional
crosstalk in monocytes and macrophages. Cell Chem Biol.
24:507–514.e4. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Martinet W, Coornaert I, Puylaert P and De
Meyer GRY: Macrophage death as a pharmacological target in
atherosclerosis. Front Pharmacol. 10:3062019. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Zeng C, Wang R and Tan H: Role of
pyroptosis in cardiovascular diseases and its therapeutic
implications. Int J Biol Sci. 15:1345–1357. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Humphries F, Shmuel-Galia L,
Ketelut-Carneiro N, Li S, Wang B, Nemmara VV, Wilson R, Jiang Z,
Khalighinejad F, Muneeruddin K, et al: Succination inactivates
gasdermin D and blocks pyroptosis. Science. 369:1633–1637. 2020.
View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Ji N, Qi Z, Wang Y, Yang X, Yan Z, Li M,
Ge Q and Zhang J: Pyroptosis: A new regulating mechanism in
cardiovascular disease. J Inflamm Res. 14:2647–2666. 2021.
View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Huang SS, Guo DY, Jia BB, Cai GL, Yan J,
Lu Y and Yang ZX: Dimethyl itaconate alleviates the pyroptosis of
macrophages through oxidative stress. BMC Immunol. 22:722021.
View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Wang K, Sun Q, Zhong X, Zeng M, Zeng H,
Shi X, Li Z, Wang Y, Zhao Q, Shao F and Ding J: Structural
mechanism for GSDMD targeting by autoprocessed caspases in
pyroptosis. Cell. 180:941–955.e20. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Yuan YY, Xie KX, Wang SL and Yuan LW:
Inflammatory caspase-related pyroptosis: Mechanism, regulation and
therapeutic potential for inflammatory bowel disease. Gastroenterol
Rep (Oxf). 6:167–176. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Jiang M, Sun X, Liu S, Tang Y, Shi Y, Bai
Y, Wang Y, Yang Q, Yang Q, Jiang W, et al: Caspase-11-gasdermin
D-mediated pyroptosis is involved in the pathogenesis of
atherosclerosis. Front Pharmacol. 12:6574862021. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Puylaert P, Van Praet M, Vaes F, Neutel
CHG, Roth L, Guns PJ, De Meyer GRY and Martinet W: Gasdermin D
deficiency limits the transition of atherosclerotic plaques to an
inflammatory phenotype in ApoE knock-out mice. Biomedicines.
10:11712022. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Wang R, Wang Y, Mu N, Lou X, Li W, Chen Y,
Fan D and Tan H: Activation of NLRP3 inflammasomes contributes to
hyperhomocysteinemia-aggravated inflammation and atherosclerosis in
apoE-deficient mice. Lab Invest. 97:922–934. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Liu C, Jiang Z, Pan Z and Yang L: The
function, regulation and mechanism of programmed cell death of
macrophages in atherosclerosis. Front Cell Dev Biol. 9:8095162022.
View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Liu S, Tao J, Duan F, Li H and Tan H: HHcy
induces pyroptosis and atherosclerosis via the lipid raft-mediated
NOX-ROS-NLRP3 inflammasome pathway in apoE−/− mice.
Cells. 11:24382022. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Zeng Z, Li G, Wu S and Wang Z: Role of
pyroptosis in cardiovascular disease. Cell Prolif. 52:e125632019.
View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Hendrikx T, Jeurissen MLJ, van Gorp PJ,
Gijbels MJ, Walenbergh SMA, Houben T, van Gorp R, Pöttgens CC,
Stienstra R, Netea MG, et al: Bone marrow-specific caspase-1/11
deficiency inhibits atherosclerosis development in Ldlr(−/-) mice.
FEBS J. 282:2327–2338. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Jin Y, Liu Y, Xu L, Xu J, Xiong Y, Peng Y,
Ding K, Zheng S, Yang N, Zhang Z, et al: Novel role for caspase 1
inhibitor VX765 in suppressing NLRP3 inflammasome assembly and
atherosclerosis via promoting mitophagy and efferocytosis. Cell
Death Dis. 13:5122022. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Liu S, Xu DS, Ma JL, Huang P, Wu D and Ren
LQ: LncRNA H19 mitigates oxidized low-density lipoprotein induced
pyroptosis via caspase-1 in Raw 264.7 Cells. Inflammation.
44:2407–2418. 2021. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Opoku E, Traughber CA, Zhang D, Iacano AJ,
Khan M, Han J, Smith JD and Gulshan K: Gasdermin D mediates
inflammation-induced defects in reverse cholesterol transport and
promotes atherosclerosis. Front Cell Dev Biol. 9:7152112021.
View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Rathkey JK, Zhao J, Liu Z, Chen Y, Yang J,
Kondolf HC, Benson BL, Chirieleison SM, Huang AY, Dubyak GR, et al:
Chemical disruption of the pyroptotic pore-forming protein
gasdermin D inhibits inflammatory cell death and sepsis. Sci
Immunol. 3:eaat27382018. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Westerterp M, Fotakis P, Ouimet M, Bochem
AE, Zhang H, Molusky MM, Wang W, Abramowicz S, la Bastide-van
Gemert S, Wang N, et al: Cholesterol efflux pathways suppress
inflammasome activation, NETosis, and atherogenesis. Circulation.
138:898–912. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Zeng W, Wu D, Sun Y, Suo Y, Yu Q, Zeng M,
Gao Q, Yu B, Jiang X and Wang Y: The selective NLRP3 inhibitor
MCC950 hinders atherosclerosis development by attenuating
inflammation and pyroptosis in macrophages. Sci Rep. 11:193052021.
View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Robinson N, Ganesan R, Hegedűs C, Kovács
K, Kufer TA and Virág L: Programmed necrotic cell death of
macrophages: Focus on pyroptosis, necroptosis, and parthanatos.
Redox Biol. 26:1012392019. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Gupta R, Sahu M, Tripathi R, Ambasta RK
and Kumar P: Protein S-sulfhydration: Unraveling the prospective of
hydrogen sulfide in the brain, vasculature and neurological
manifestations. Ageing Res Rev. 76:1015792022. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Pang PP, Zhang HY, Zhang DC, Tang JX, Gong
Y, Guo YC and Zheng CB: Investigating the impact of protein
S-sulfhydration modification on vascular diseases: A comprehensive
review. Eur J Pharmacol. 966:1763452024. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Cirino G, Szabo C and Papapetropoulos A:
Physiological roles of hydrogen sulfide in mammalian cells,
tissues, and organs. Physiol Rev. 103:31–276. 2023. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Bibli SI, Hu J, Leisegang MS, Wittig J,
Zukunft S, Kapasakalidi A, Fisslthaler B, Tsilimigras D, Zografos
G, Filis K, et al: Shear stress regulates cystathionine γ lyase
expression to preserve endothelial redox balance and reduce
membrane lipid peroxidation. Redox Biol. 28:1013792020. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Zhang H, Bai Z, Zhu L, Liang Y, Fan X, Li
J, Wen H, Shi T, Zhao Q and Wang Z: Hydrogen sulfide donors:
Therapeutic potential in anti-atherosclerosis. Eur J Med Chem.
205:1126652020. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Zhang L, Wang Y, Li Y, Li L, Xu S, Feng X
and Liu S: Hydrogen sulfide (H2S)-releasing compounds:
Therapeutic potential in cardiovascular diseases. Front Pharmacol.
9:10662018. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Castelblanco M, Lugrin J, Ehirchiou D,
Ishii I, So A, Martinon F and Busso N: Hydrogen sulfide inhibits
NLRP3 inflammasome activation and reduces cytokine production both
in vitro and in a mouse model of inflammation. J Biol Chem.
293:2546–2557. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Li J, Ma J, Li M, Tao J, Chen J, Yao C and
Yao S: GYY4137 alleviates sepsis-induced acute lung injury in mice
by inhibiting the PDGFRβ/Akt/NF-κB/NLRP3 pathway. Life Sci.
271:1191922021. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Lin Z, Altaf N, Li C, Chen M, Pan L, Wang
D, Xie L, Zheng Y, Fu H, Han Y and Ji Y: Hydrogen sulfide
attenuates oxidative stress-induced NLRP3 inflammasome activation
via S-sulfhydrating c-Jun at Cys269 in macrophages. Biochim Biophys
Acta Mol Basis Dis. 1864:2890–2900. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Luo ZL, Ren JD, Huang Z, Wang T, Xiang K,
Cheng L and Tang LJ: The role of exogenous hydrogen sulfide in free
fatty acids induced inflammation in macrophages. Cell Physiol
Biochem. 42:1635–1644. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Zhang D, Du J, Tang C, Huang Y and Jin H:
H2S-induced sulfhydration: Biological function and
detection methodology. Front Pharmacol. 8:6082017. View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Zhang XL, Tian XQ, Jia ZL, Liu BW, Liang
GY, Zhang ZC, Li L and Zhang L: Sodium hydrosulfide attenuates
pyroptosis of macrophages by inhibiting classical pyroptosis
signaling pathway. Chin J Pathophysiol. 38:1015–1023. 2022.
|
|
46
|
Chiu HW, Li LH, Hsieh CY, Rao YK, Chen FH,
Chen A, Ka SM and Hua KF: Glucosamine inhibits IL-1β expression by
preserving mitochondrial integrity and disrupting assembly of the
NLRP3 inflammasome. Sci Rep. 9:56032019. View Article : Google Scholar : PubMed/NCBI
|
|
47
|
Mayes-Hopfinger L, Enache A, Xie J, Huang
CL, Köchl R, Tybulewicz VLJ, Fernandes-Alnemri T and Alnemri ES:
Chloride sensing by WNK1 regulates NLRP3 inflammasome activation
and pyroptosis. Nat Commun. 12:45462021. View Article : Google Scholar : PubMed/NCBI
|
|
48
|
Xu G, Fu S, Zhan X, Wang Z, Zhang P, Shi
W, Qin N, Chen Y, Wang C, Niu M, et al: Echinatin effectively
protects against NLRP3 inflammasome-driven diseases by targeting
HSP90. JCI Insight. 6:e1346012021. View Article : Google Scholar : PubMed/NCBI
|
|
49
|
Chen Y, Li R, Wang Z, Hou X, Wang C, Ai Y,
Shi W, Zhan X, Wang JB, Xiao X, et al: Dehydrocostus lactone
inhibits NLRP3 inflammasome activation by blocking ASC
oligomerization and prevents LPS-mediated inflammation in vivo.
Cell Immunol. 349:1040462020. View Article : Google Scholar : PubMed/NCBI
|
|
50
|
Mustafa AK, Gadalla MM, Sen N, Kim S, Mu
W, Gazi SK, Barrow RK, Yang G, Wang R and Snyder SH: H2S
signals through protein S-sulfhydration. Sci Signal. 2:ra722009.
View Article : Google Scholar : PubMed/NCBI
|
|
51
|
Wang XH, Wang F, You SJ, Cao YJ, Cao LD,
Han Q, Liu CF and Hu LF: Dysregulation of cystathionine γ-lyase
(CSE)/hydrogen sulfide pathway contributes to ox-LDL-induced
inflammation in macrophage. Cell Signal. 25:2255–2262. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
52
|
Zhang H, Du J, Huang Y, Tang C and Jin H:
Hydrogen sulfide regulates macrophage function in cardiovascular
diseases. Antioxid Redox Signal. 38:45–56. 2023. View Article : Google Scholar : PubMed/NCBI
|
|
53
|
Hu HJ, Qiu J, Zhang C, Tang ZH, Qu SL and
Jiang ZS: Hydrogen sulfide improves ox-LDL-induced expression
levels of Lp-PLA2 in THP-1 monocytes via the p38MAPK
pathway. Mol Med Rep. 23:3582021. View Article : Google Scholar : PubMed/NCBI
|
|
54
|
Hu Q, Zhang R, Zheng J, Song M, Gu C and
Li W: Hydrogen sulfide attenuates uranium-induced kidney cells
pyroptosis via upregulation of PI3K/AKT/mTOR signaling. J Biochem
Mol Toxicol. 37:e232202023. View Article : Google Scholar : PubMed/NCBI
|
|
55
|
Wang L, Meng J, Wang C, Wang Y, Yang C and
Li Y: Hydrogen sulfide attenuates cigarette smoke-induced
pyroptosis through the TLR4/NF-κB signaling pathway. Int J Mol Med.
49:562022. View Article : Google Scholar : PubMed/NCBI
|
|
56
|
Zhu X, Lu R, Zhang G, Fan L, Zhan Y, Chen
G and Zhou L: Diallyl trisulfide attenuates alcohol-induced
hepatocyte pyroptosis via elevation of hydrogen sulfide. Biosci
Biotechnol Biochem. 86:1552–1561. 2022. View Article : Google Scholar : PubMed/NCBI
|
|
57
|
Yu J and Cui X, Zhang X, Cheng M and Cui
X: Advances in the occurrence of pyroptosis: A novel role in
atherosclerosis. Curr Pharm Biotechnol. 22:1548–1558. 2021.
View Article : Google Scholar : PubMed/NCBI
|
|
58
|
Ding Y, Wang H, Geng B and Xu G:
Sulfhydration of perilipin 1 is involved in the inhibitory effects
of cystathionine gamma lyase/hydrogen sulfide on adipocyte
lipolysis. Biochem Biophys Res Commun. 521:786–790. 2020.
View Article : Google Scholar : PubMed/NCBI
|
|
59
|
Xie L, Gu Y, Wen M, Zhao S, Wang W, Ma Y,
Meng G, Han Y, Wang Y, Liu G, et al: Hydrogen sulfide induces Keap1
S-sulfhydration and suppresses diabetes-accelerated atherosclerosis
via Nrf2 activation. Diabetes. 65:3171–3184. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
60
|
Ye X, Li Y, Lv B, Qiu B, Zhang S, Peng H,
Kong W, Tang C, Huang Y, Du J and Jin H: Endogenous hydrogen
sulfide persulfidates Caspase-3 at cysteine 163 to inhibit
doxorubicin-induced cardiomyocyte apoptosis. Oxid Med Cell Longev.
2022:61537722022. View Article : Google Scholar : PubMed/NCBI
|
|
61
|
He TT, Zhang XL, Jia ZL, Wang Y, Ma KT, Li
L and Zhang L: Hydrogen sulfide inhibits oxidized low-density
lipoprotein-induced pyroptosis in vascular endothelial cells by
down-regulating NLRP3/caspase-1 signaling pathway. Chin J
Pathophysiol. 37:1738–1746. 2021.
|
|
62
|
Qian Z, Zhao Y, Wan C, Deng Y, Zhuang Y,
Xu Y, Zhu Y, Lu S and Bao Z: Pyroptosis in the initiation and
progression of atherosclerosis. Front Pharmacol. 12:6529632021.
View Article : Google Scholar : PubMed/NCBI
|
|
63
|
Yang Z, Shi J, Chen L, Fu C, Shi D and Qu
H: Role of pyroptosis and ferroptosis in the progression of
atherosclerotic plaques. Front Cell Dev Biol. 10:8111962022.
View Article : Google Scholar : PubMed/NCBI
|
|
64
|
Braunstein I, Engelman R, Yitzhaki O, Ziv
T, Galardon E and Benhar M: Opposing effects of polysulfides and
thioredoxin on apoptosis through caspase persulfidation. J Biol
Chem. 295:3590–3600. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
65
|
Sun F, Luo JH, Yue TT, Wang FX, Yang CL,
Zhang S, Wang XQ and Wang CY: The role of hydrogen sulphide
signalling in macrophage activation. Immunology. 162:3–10. 2021.
View Article : Google Scholar : PubMed/NCBI
|
|
66
|
Chen Y, Jin S, Teng X, Hu Z, Zhang Z, Qiu
X, Tian D and Wu Y: Hydrogen sulfide attenuates LPS-induced acute
kidney injury by inhibiting inflammation and oxidative stress. Oxid
Med Cell Longev. 2018:67172122018. View Article : Google Scholar : PubMed/NCBI
|
|
67
|
Dilek N, Papapetropoulos A, Toliver-Kinsky
T and Szabo C: Hydrogen sulfide: An endogenous regulator of the
immune system. Pharmacol Res. 161:1051192020. View Article : Google Scholar : PubMed/NCBI
|
|
68
|
Zhou T, Qian H, Zheng N, Lu Q and Han Y:
GYY4137 ameliorates sepsis-induced cardiomyopathy via NLRP3
pathway. Biochim Biophys Acta Mol Basis Dis. 1868:1664972022.
View Article : Google Scholar : PubMed/NCBI
|
|
69
|
Wu D, Zhong P, Wang J and Wang H:
Exogenous hydrogen sulfide mitigates LPS + ATP-induced inflammation
by inhibiting NLRP3 inflammasome activation and promoting autophagy
in L02 cells. Mol Cell Biochem. 457:145–156. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
70
|
Ni J, Jiang L, Shen G, Xia Z, Zhang L, Xu
J, Feng Q, Qu H, Xu F and Li X: Hydrogen sulfide reduces pyroptosis
and alleviates ischemia-reperfusion-induced acute kidney injury by
inhibiting NLRP3 inflammasome. Life Sci. 284:1194662021. View Article : Google Scholar : PubMed/NCBI
|
|
71
|
Yue LM, Gao YM and Han BH: Evaluation on
the effect of hydrogen sulfide on the NLRP3 signaling pathway and
its involvement in the pathogenesis of atherosclerosis. J Cell
Biochem. 120:481–492. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
72
|
Zheng Q, Pan L and Ji Y: H2S
protects against diabetes-accelerated atherosclerosis by preventing
the activation of NLRP3 inflammasome. J Biomed Res. 34:94–102.
2019. View Article : Google Scholar : PubMed/NCBI
|
|
73
|
Du J, Huang Y, Yan H, Zhang Q, Zhao M, Zhu
M, Liu J, Chen SX, Bu D, Tang C and Jin H: Hydrogen sulfide
suppresses oxidized low-density lipoprotein (ox-LDL)-stimulated
monocyte chemoattractant protein 1 generation from macrophages via
the nuclear factor κB (NF-κB) pathway. J Biol Chem. 289:9741–9753.
2014. View Article : Google Scholar : PubMed/NCBI
|
|
74
|
Du HP, Li J, You SJ, Wang YL, Wang F, Cao
YJ, Hu LF and Liu CF: DNA methylation in cystathionine-γ-lyase
(CSE) gene promoter induced by ox-LDL in macrophages and in apoE
knockout mice. Biochem Biophys Res Commun. 469:776–782. 2016.
View Article : Google Scholar : PubMed/NCBI
|
|
75
|
Bai L, Dai J, Xia Y, He K, Xue H, Guo Q,
Tian D, Xiao L, Zhang X, Teng X, et al: Hydrogen sulfide
ameliorated high choline-induced cardiac dysfunction by inhibiting
cGAS-STING-NLRP3 inflammasome pathway. Oxid Med Cell Longev.
2022:13928962022. View Article : Google Scholar : PubMed/NCBI
|
|
76
|
Gong W, Zhang S, Chen Y, Shen J, Zheng Y,
Liu X, Zhu M and Meng G: Protective role of hydrogen sulfide
against diabetic cardiomyopathy via alleviating necroptosis. Free
Radic Biol Med. 181:29–42. 2022. View Article : Google Scholar : PubMed/NCBI
|
|
77
|
Qin M, Long F, Wu W, Yang D, Huang M, Xiao
C, Chen X, Liu X and Zhu YZ: Hydrogen sulfide protects against
DSS-induced colitis by inhibiting NLRP3 inflammasome. Free Radic
Biol Med. 137:99–109. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
78
|
Gao Y, Zhang H, Wang Y, Han T, Jin J, Li
J, Tang Y and Liu C: L-cysteine alleviates myenteric neuron injury
induced by intestinal ischemia/reperfusion via inhibitin the
macrophage NLRP3-IL-1β pathway. Front Pharmacol. 13:8991692022.
View Article : Google Scholar : PubMed/NCBI
|
|
79
|
Zhang N, Zhou Z, Huang Y, Wang G, Tang Z,
Lu J, Wang C and Ni X: Reduced hydrogen sulfide production
contributes to adrenal insufficiency induced by hypoxia via
modulation of NLRP3 inflammasome activation. Redox Rep.
28:21633542023. View Article : Google Scholar : PubMed/NCBI
|
|
80
|
Fan J, Zheng F, Li S, Cui C, Jiang S,
Zhang J, Cai J, Cui Q, Yang J, Tang X, et al: Hydrogen sulfide
lowers hyperhomocysteinemia dependent on cystathionine γ lyase
S-sulfhydration in ApoE-knockout atherosclerotic mice. Br J
Pharmacol. 176:3180–3192. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
81
|
Yang K, Coburger I, Langner JM, Peter N,
Hoshi T, Schönherr R and Heinemann SH: Modulation of K+
channel N-type inactivation by sulfhydration through hydrogen
sulfide and polysulfides. Pflugers Arch. 471:557–571. 2019.
View Article : Google Scholar : PubMed/NCBI
|
|
82
|
Luo S, Kong C, Ye D, Liu X, Wang Y, Meng
G, Han Y, Xie L and Ji Y: Protein persulfidation: Recent progress
and future directions. Antioxid Redox Signal. 39:829–852. 2023.
View Article : Google Scholar : PubMed/NCBI
|
|
83
|
Wu Q, Zhao B, Weng Y, Shan Y, Li X, Hu Y,
Liang Z, Yuan H, Zhang L and Zhang Y: Site site-specific
quantification of persulfidome by combining an isotope-coded
affinity tag with strong cation-exchange-based fractionation. Anal
Chem. 91:14860–14864. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
84
|
Bibli SI, Hu J, Sigala F, Wittig I,
Heidler J, Zukunft S, Tsilimigras DI, Randriamboavonjy V, Wittig J,
Kojonazarov B, et al: Cystathionine γ lyase sulfhydrates the RNA
binding protein human antigen r to preserve endothelial cell
function and delay atherogenesis. Circulation. 139:101–114. 2019.
View Article : Google Scholar : PubMed/NCBI
|
|
85
|
Luo S, Kong C, Zhao S, Tang X, Wang Y,
Zhou X, Li R, Liu X, Tang X, Sun S, et al: Endothelial
HDAC1-ZEB2-NuRD complex drives aortic aneurysm and dissection
through regulation of protein S-sulfhydration. Circulation.
147:1382–1403. 2023. View Article : Google Scholar : PubMed/NCBI
|
|
86
|
Wu D, Tan B, Sun Y and Hu Q: Cystathionine
γ lyase S-sulfhydrates Drp1 to ameliorate heart dysfunction. Redox
Biol. 58:1025192022. View Article : Google Scholar : PubMed/NCBI
|
|
87
|
Altaany Z, Ju Y, Yang G and Wang R: The
coordination of S-sulfhydration, S-nitrosylation, and
phosphorylation of endothelial nitric oxide synthase by hydrogen
sulfide. Sci Signal. 7:ra872014. View Article : Google Scholar : PubMed/NCBI
|