Introduction
Chronic myeloid leukemia (CML) is a prevalent form
of leukemia, accounting for ~15% of all leukemia diagnoses and
posing a significant global health burden (1,2). The
discovery of dysregulated tyrosine kinase (TK) activity in CML
pathogenesis facilitated development of TK inhibitors (TKIs) as a
targeted therapy (3). This
approach has improved the prognosis of patients with CML, achieving
long-term remission rates >80% (3). Despite these advances, challenges
remain in optimizing TKI treatment and understanding the long-term
effects. The present study aimed to assess the dose-dependent
impact of TKI on cardiac function to optimize therapy and minimize
adverse effects. A deeper understanding of the mechanisms
underlying TKI action in CML is key for developing patient-specific
treatment regimens that balance anticancer effects with the
prevention or mitigation of cardiotoxic side effects.
TKs are a group of enzymes within the protein kinase
family. They act as molecular switches, regulating cellular
processes such as proliferation, differentiation, metabolism and
apoptosis by attaching phosphate groups from ATP to specific
proteins (4). This phosphorylation
can either activate or inhibit the target protein. Dysregulation of
TKs serves a key role in cancer development and progression
(4). Studies have identified ~58
different TK receptors associated with numerous cancers (5–7).
When activated in cancer cells, TKs promote tumor growth by
stimulating cell proliferation, angiogenesis, resistance to cell
death and metastasis (8).
Therefore, understanding the specific roles of TKs in different
cancer types is essential for developing targeted therapeutic
strategies. By inhibiting these aberrantly active kinases, key
cancer-promoting pathways can be disrupted, offering potentially
more effective treatment options. TKIs have emerged as a drug
against numerous types of cancers, including CML (9,10).
TKIs are classified into generations based on their specificity and
affinity for certain TKs, reflecting the progression in TKI
development aimed at optimizing therapeutic effectiveness while
minimizing side effects for patients. First-generation TKIs like
imatinib, sunitinib, and gefitinib are pioneering drugs with a
broad activity spectrum. They target multiple TKs, including both
cancer-causing targets and non-intended kinases. This wide range of
activity can lead to effective initial tumor reduction but also
increases the likelihood of side effects from inhibiting non-target
essential cellular processes. In contrast, second and
third-generation TKIs, such as dasatinib and ponatinib, show
advancements in design towards greater target specificity. These
newer TKIs aim to inhibit only the cancer-causing tyrosine kinases,
thereby reducing off-target effects and improving patient outcome
(11,12). Despite their similar mode of
action, which involves blocking the activation of receptor TKs
(RTKs) (13,14) TKIs differ in their target
specificity, pharmacokinetics and side effect profiles. While they
all prevent RTKs from triggering downstream signaling pathways, the
precise targeted kinase and drug interactions determine its
effectiveness and potential adverse effects. Understanding these
differences is required for optimizing TKI therapy. Researchers
continuously evaluate tailored approaches that maximize efficacy
while minimizing side effects, leading to better patient outcomes
(15).
While effective in treating various types of
cancers, TKIs pose significant concerns due to their broad-spectrum
targeting, leading to diverse toxicities. Among these,
cardiotoxicity is a key side effect, impacting patient quality of
life and treatment outcomes (16).
For example, imatinib, a first-generation TKI, induces left
ventricular dysfunction in patients and preclinical models
(17,18). Second-generation TKIs) like
dasatinib demonstrate improved selectivity for BCR-ABL proteins.
These BCR-ABL proteins, with their aberrant tyrosine kinase
activity, are critical drivers of chronic myeloid leukemia (CML)
and contribute significantly to treatment resistance. However,
dasatinib, despite its targeted action, can still induce
cardiotoxicities like pulmonary hypertension and heart failure
(19,20). Ponatinib, the only TKI for
T3151-mutant CML, exhibits strong cardiotoxic effects, including
hypertension and heart failure (21,22).
While the cardiotoxic potential of imatinib and ponatinib is
established, understanding the underlying mechanisms is crucial for
developing targeted mitigation strategies. This study addressed
this gap by employing a complementary in vitro and in
vivo approach. H9c2 cardiomyocytes were used for individual
cell analysis to investigate potential apoptotic and hypertrophic
effects, while zebrafish embryos (ZFEs) provided a platform for
whole-organism observations and genetic manipulations. This dual
approach aimed to gain a deeper understanding of TKI-induced
cardiotoxicity mechanisms. We hypothesized that both imatinib and
ponatinib induce cardiomyocyte apoptosis and hypertrophy, with
ponatinib exhibiting a stronger effect. Ultimately, this research
seeks to pave the way for safer and more effective TKI therapies,
improving patient outcomes.
Materials and methods
Culture of H9c2 cardiomyoblasts
This study used H9c2 cardiomyoblasts (European
Collection of Cell Cultures) derived from BDIX embryonic rat
cardiac tissue. Cells were cultured in Dulbecco's Modified Eagle's
Medium Ham's F-12 1:1 (DMEM/F-12; Lonza Group, Ltd.) supplemented
with 10% w/v fetal bovine serum (FBS) (Gibco-Thermo Fisher
Scientific, Inc.) and 1% w/v penicillin-streptomycin. All cultures
were maintained at 37°C in a humidified environment with 5%
CO2 and 95% O2. H9c2 cardiomyoblasts
represent a well-established in vitro model that has been
previously used to investigate the molecular mechanisms of action
of numerous anticancer agents, including TKIs (23–25).
Treatment of H9c2 cardiomyoblasts
H9c2 cardiomyoblasts were treated with imatinib and
ponatinib to investigate their cardiotoxic effects. Stock solutions
of both drugs (50 mM) were prepared by dissolving in Hybri-Max DMSO
(cat. No. D2660; MilliporeSigma). To ensure consistency, the final
concentration of DMSO in the culture media was ≤0.5%. H9c2 cells
were treated with imatinib and ponatinib at 2.5 and 5.0 µM. The
concentrations used in this study were consistent with those used
in previous studies (23,24,26–28).
Control cells received an equivalent volume of vehicle (0.5% v/v
DMSO) only. Following treatment for 24 h at 37°C, cells were
subjected to further experiments.
MTT assay
The viability of H9c2 cardiomyoblasts following
treatment with imatinib or ponatinib was evaluated using MTT assay
(MilliporeSigma). Briefly, cells were seeded in 48-well plates at a
density of 4×104 cells/well and allowed to adhere for 24
h at 37°C. Medium was replaced with fresh Dulbecco's Modified
Eagle's Medium Ham's F-12 1:1 (DMEM/F-12) (Lonza, Basal,
Switzerland) supplemented with 10% FBS) and 1%
penicillin/streptomycin. Cells were then treated with imatinib or
ponatinib at concentrations of 2.5 and 5.0 µM for 24 h at 37°C. The
medium was removed following treatment and 0.5 mg/ml MTT solution
was added to each well. Following incubation for 3 h at 37°C, the
resulting formazan crystals were dissolved in DMSO. Absorbance was
measured at 570 nm using an Epoch 2 optical microplate reader
(Norgen Biotek Corp.). Cell viability was presented as a percentage
of vehicle control. To account for non-specific signal, the mean
absorbance of blank wells containing medium only was subtracted
from all other wells. Background-corrected absorbance values for
each treatment group were normalized to the mean absorbance of the
vehicle control (0.5% DMSO) to enable cross-group comparisons.
Finally, normalized values were multiplied by 100 to present
viability as a percentage of the vehicle control. The experiment
was repeated three times.
Cell surface area measurement of H9c2
cardiomyoblasts
Cell surface area quantification was performed as
previously established (29).
Briefly, H9c2 cardiomyoblasts were seeded at a density of 10,000
cells/35-mm culture dish and allowed to adhere for 24 h at 37°C.
Cells were then treated with imatinib or ponatinib at
concentrations of 2.5 and 5.0 µM for 24 h at 37°C. Following
treatment, cells were washed with 1X phosphate-buffered saline
(PBS), fixed with 4% v/v formaldehyde and stained with 0.5% w/v
crystal violet solution for 20 min at room temperature. Stained
cells were visualized and captured at 20× magnification using an
Axiovert 40 CFL inverted confocal microscope (Carl Zeiss AG). Cell
surface area analysis was performed on 15–30 randomly selected
cells using AxioVision Imaging software 4.8.2 (Carl Zeiss AG). The
experiment was repeated three times. The surface area of the cell
was calculated by normalizing the background-corrected absorbance
values of each treatment group to the mean absorbance of the
vehicle control group (0.5% DMSO) for direct comparison across
groups. Then, the normalized values were multiplied by 100 to
express the viability of each treatment group as a percentage of
the vehicle control.
Flow cytometry analysis of cell death
and size of H9c2 cardiomyoblasts
H9c2 cardiomyoblasts were seeded in 35-mm dishes
containing DMEM-F12 supplemented with 10% w/v FBS and 1% w/v
penicillin-streptomycin. After 24 h 37°C, they were treated with
imatinib or ponatinib at concentrations of 2.5 and 5.0 µM for 24 h
at 37°C. Culture medium was aspirated, and monolayers were rinsed
with sterile PBS to remove serum. Cells were detached using 0.25%
Trypsin [Gibco-Thermo Fisher Scientific (Waltham, MA, USA)] for ≤10
min at 37°C, centrifuged (300–400 g, 5 min, at room temperature),
and washed with PBS/BSA to remove debris. The final pellet was
resuspended in fresh PBS. Cells were stained with annexin-V and
propidium iodide (PI) (BD Biosciences) in 1X annexin binding buffer
for 30 min at room temperature. Flow cytometry was performed using
BD LSRFortessa™ cell analyzer (BD Biosciences) BD FACSDiva software
6.1.3 (BD Biosciences) were used to measure the cell viability.
Apoptotic cells were determined as follows: Viable, PI- and
annexin-FITC-negative; early stage apoptosis, PI-negative and
annexin-FITC-positive; late stage apoptosis PI- and annexin-FITC
positive and necrotic, PI-positive and annexin-FITC-negative as
previously described (30,31). Cell size was measured with forward
light scatter using the flow cytometer and the percentage of cells
in early and late apoptosis was calculated as the total proportion
of apoptotic cells. The experiment was repeated three times.
ZFEs
Adult zebrafish (AB strain) were maintained in
recirculating systems (AQUA NEERING ZD560) at the Qatar University
Biomedical Research Center under a 14 h light/10 h dark cycle with
controlled temperatures (room: 26°C, water: 28°C). All procedures
adhered to approved protocols (QU-IACUC, QU-IBC-2022/014) and
national/international zebrafish guidelines (27). Standard practices (32) guided breeding. Fish were maintained
in reconstituted saltwater tanks and fed twice daily with fresh
brine shrimp. For embryo collection, male and female zebrafish were
separated overnight using a mesh barrier in a mating tank. The next
morning, the barrier was removed, allowing mating for 20 min.
Fertilized embryos were collected in freshly prepared E3 medium
(32) and staged/fixed according
to established protocols (33).
Treatment of ZFE
To assess the cardiac effects of TKIs in a
developing organism, fertilized ZFEs were collected and maintained
at 28°C in N-phenylthiourea (PTU) water at a standard concentration
of 0.003% (200 µM) to suppress pigmentation and aid observation and
time-lapse video acquisition. Experiments used 12-well Falcon
Tissue Culture Plate, flat bottom with low evaporation lid (Corning
Life Sciences, Netherlands), housing 20 embryos each. For optimal
detection of potential toxicity, treatment commenced occurred at 6
h post-fertilization (hpf), corresponding to the ‘high stage’ as
described by Kimmel et al (33). This stage is characterized by rapid
cell division and organogenesis, rendering the developing embryo
highly susceptible to disruptions caused by toxic substances due to
the presence of a largely undifferentiated cell population.
Following PTU removal, 2 ml drug solution was added. TKI
concentrations (2.5, 5.0 and 10.0 µM) were selected based on
preliminary tissue culture experiments, ensuring relevance to
clinically relevant ranges. The following controls were used:
Control, maintained in PTU; negative control, treated with the DMSO
vehicle (0.1% v/v) and the positive control (PC), treated with
known cardiotoxic agent aristolochic acid I (AA; 1 µM). All drug
solutions were prepared in DMSO at a final concentration of 0.1%.
Treatment was conducted at 28°C for 24 h for SR, 48 h for both SR
and HR, and 72 h for SR, cardiac function assessment, and cardiac
gene assay.
Observation and analysis of ZFEs
embryos
At 24–72 hpf, ZFEs were examined every 24 h under a
SteREO Discovery V8 light microscope (Carl Zeiss AG) with a
Hamamatsu Orca Flash camera (Hamamatsu Photonics UK Limited) and
HCImage software 2.0.4 (Hamamatsu Photonics UK Limited) to monitor
developmental stage, mortality, hatching, spontaneous movement,
response to touch, presence of deformities and heart rate. The
phenotypical aberrations were recorded at each point and compared
with the controls. Images of the embryos were captured and the
number of similar phenotypes in the experimental group noted.
Survival and morphological abnormalities were assessed; opaque,
coagulated embryos lacking a heartbeat were considered non-viable
and removed. The mortality percentage was determined by counting
the number of dead ZFEs/group at 24, 48 and 72 hpf divided by the
total number of injected embryos ×100. Dead embryos were removed at
each time point. Potential neurological or muscular defects at 24
hpf were assessed by tail-flicking (burst/min) using Danio Scope
software, EthoVision XT9.0 (Noldus Information Technology). The
hatching percentage was determined by counting the number of
hatched ZFEs/group at 48 and 72 hpf, divided by the total number of
injected embryos ×100.
Experiments exceeding 20% mortality in the negative
control group were excluded. Observed abnormalities were
documented. At 72 hpf, six embryos/group were immobilized with 3%
methylcellulose at room temperature for 30 sec to conduct
cardiovascular analysis. Each embryo underwent 10 sec bright field
video recording at 100 frames/sec (fps) of the beating heart and
the body. MicroZebralab 3.6 software (Viewpoint) was used to
determine blood flow velocity, arterial pulse and vessel diameter
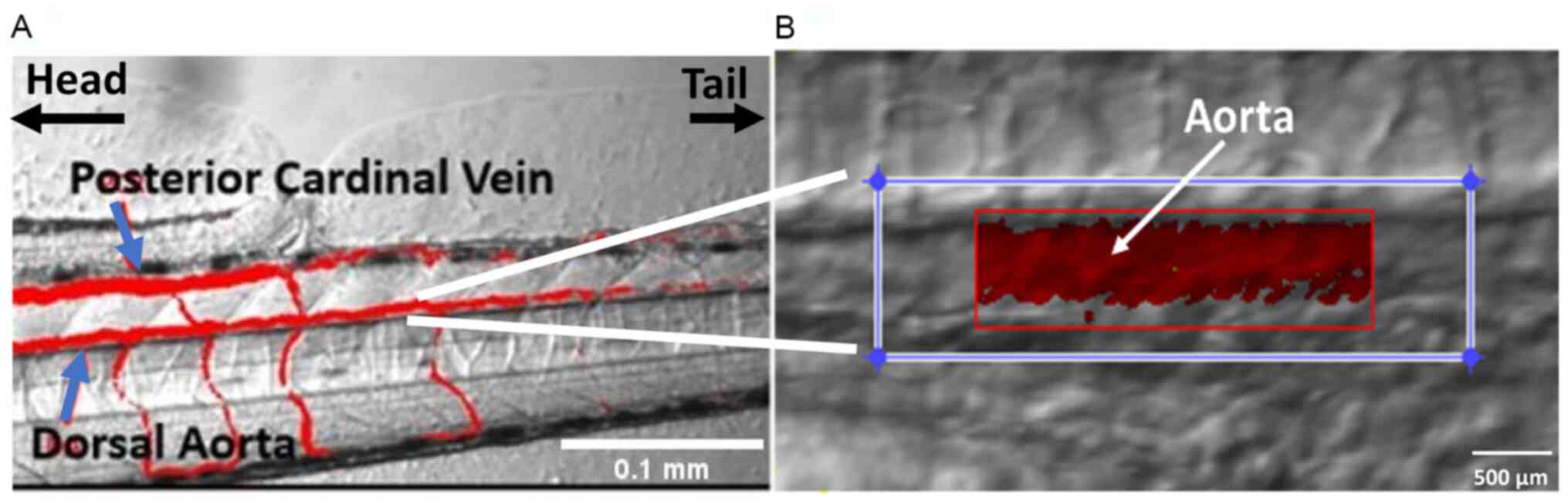
in the dorsal aorta (DA) and posterior cardinal vein, ensuring
consistency across samples (34,35)
(Fig. 1).
Cardiac function assessment
The cardiac function of treated and negative control
embryos was evaluated at 72 hpf. Time-lapse video capturing beating
ventricles and red blood cell (RBC) movement in major vessels was
analyzed as described previously (35). An in-house algorithm implemented in
Viewpoint ZebraLab software version 3.4.4 (Viewpoint) tracked RBCs,
enabling the measurement of aorta blood velocity, heartbeat and
aorta diameter (Fig. 1). The mean
of these measurements was used to estimate frictional shear stress
levels in the cardiovascular system using the formula: Shear stress
(τ; dynes/cm2)=[4 × blood viscosity
(dynes/cm2) × average blood velocity (µm/sec)]/vessel
diameter (µm). flow rate (nl/min), was calculated as follows:
Average blood velocity (µm/s) × vessel diameter (µm) (34–36).
Gene expression analysis in ZFE
Gene expression changes were detected by reverse
transcription-quantitative PCR which involved a 2 min Uracil DNA
glycosylase incubation at 50°C, a 2-min polymerase activation at
95°C, a 1-sec denaturation at 95°C, and a final 30-sec annealing at
60°C. Total RNA was isolated from TKI-treated and negative control
embryos using the IBI DNA/RNA/Protein Extraction kit (cat. no.
IB47702; IBI Scientific) following the manufacturer's instructions.
First-strand cDNA was synthesized from the extracted RNA using the
SuperScript™ IV VILO™ Master Mix kit (cat. no. 11756050; Thermo
Fisher Scientific, Inc.). Quantitative analysis of specific mRNA
expression was performed using TaqMan Fast Advanced Master Mix
(Applied Biosystems; Thermo Fisher Scientific, Inc.) and specific
primers (Table I) and probes
constructed against the genes of interest. These include atrial
natriuretic peptide (ANP; Applied Biosystems; Thermo Fisher
Scientific, Inc.) and brain natriuretic peptide (BNP; Applied
Biosystems, Thermo Fisher Scientific, Inc.). The signal was
detected using the ABI 7500 Real-Time PCR System (Applied
Biosystems; Thermo Fisher Scientific, Inc.). mRNA levels were
quantified using the 2−ΔΔCq method (37) and normalized to the internal
reference gene B2M. This approach ensured accurate and consistent
gene expression analysis across all samples.
 | Table I.Primers used in reverse-transcription
quantitative PCR. |
Table I.
Primers used in reverse-transcription
quantitative PCR.
| Gene | RefSeq | Cat. no. | Assay ID |
|---|
| ANP-nppa
zebrafish | NM_198800 | 4331348 | TaqMan Gene
Expression Assay ID APGZVJD |
| BNP-nppa
zebrafish | NM_001327776 | 4331348 | TaqMan Gene
Expression Assay ID APGZVJD, |
| B2M | NM_131163.2 | 4351372 | TaqMan Gene
Expression Assay Dr03432699_m1 |
|
| NM_001159768.1 |
|
|
Statistical analysis
Statistical analysis was performed using GraphPad
Prism software, version 8 (GraphPad LLC; Dotmatics,).
D'Agostino-Pearson normality test confirmed the distribution of
parametric data, which were analyzed by one-way ANOVA with Sidak's
or Dunnett's post hoc test and two-way mixed ANOVA with either
Sidak's or Tukey's post hoc test. P<0.05 was considered to
indicate a statistically significant difference. All data points
are presented as mean ± SEM. Each experiment was performed three
times.
Results
Ponatinib induces dose-dependent
cytotoxicity in H9c2 cells
To assess the cardiotoxic potential of imatinib and
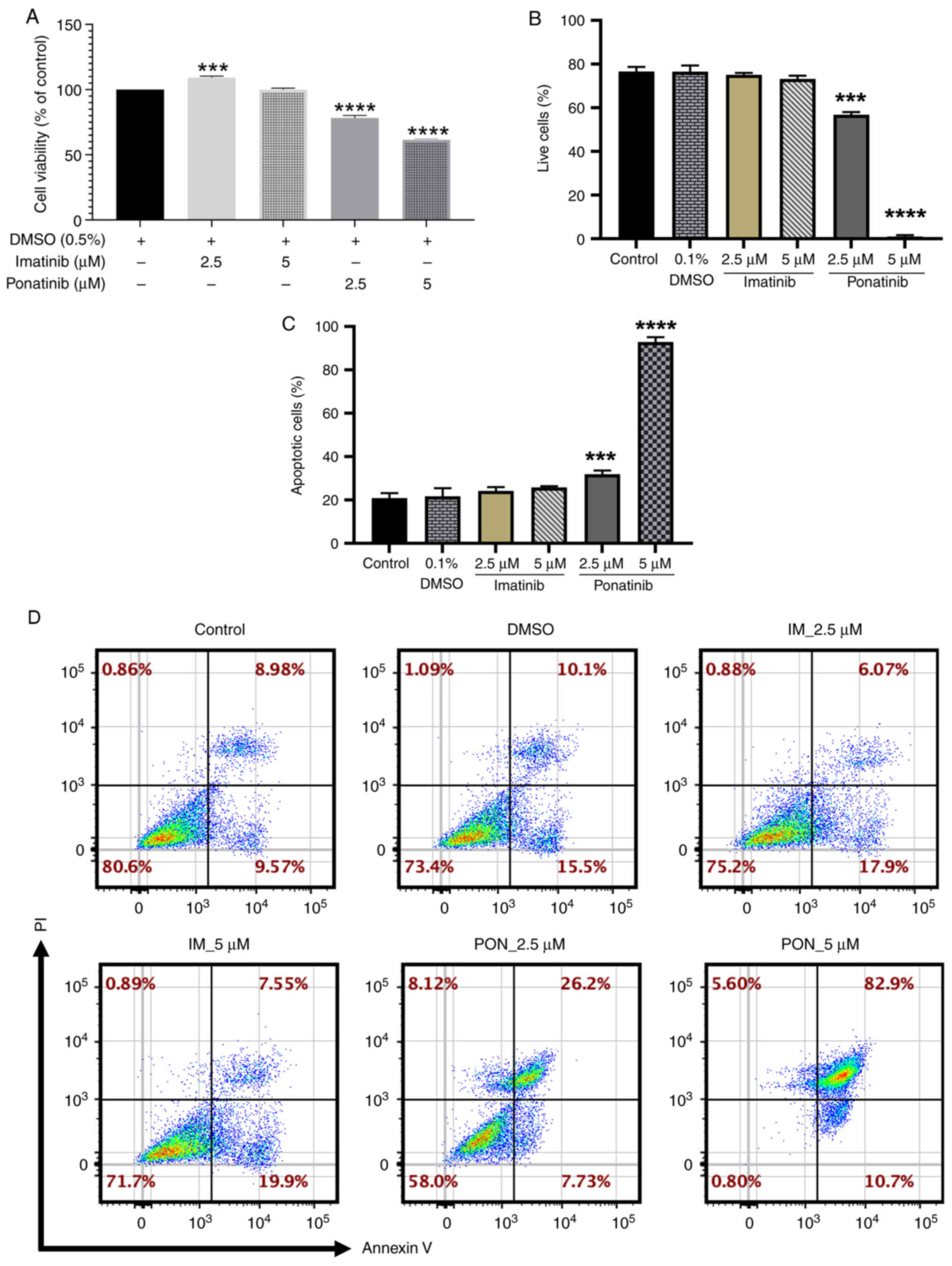
ponatinib, H9c2 cardiomyoblast viability was evaluated using an MTT
assay following 24 h treatment. A significant increase in cell
viability (109.19±2.35%) compared with the negative control was
demonstrated with 2.5 µM imatinib treatment, however, no
significant changes were demonstrated at 5 µM. Ponatinib induced
significant dose-dependent reductions in cell viability, with a 22%
decrease at 2.5 µM (viability, 78.24±3.80%) and a 40% decrease at 5
µM (viability, 61.46±1.49%) compared with the negative control
(Fig. 2A).
Flow cytometry confirmed these contrasting effects.
While both 2.5 and 5.0 µM ponatinib significantly reduced the
number of live cells in the sample population for each group
(59.10±2.14 and 6.83±0.07%, respectively) compared with the
negative control. Imatinib treatment at each concentration showed
no significant change in the number of live cells compared with
negative control (Fig. 2B).
To assess the underlying mechanism of cell death,
the percentage of apoptotic cells was assessed using flow cytometry
and annexin-V/PI staining. Ponatinib induced significant
dose-dependent increases in the percentage of apoptotic cells
compared with negative control at 2.5 (30.62±2.47%) and 5.0 µM
(92.4±2.96%). Imatinib did not induce a significant change in the
percentage of apoptotic cells at either concentration (Fig. 2C). Representative flow cytometry
plots are shown in (Fig. 2D).
Ponatinib induces morphological
changes and decreases number of H9c2 cardiomyoblasts
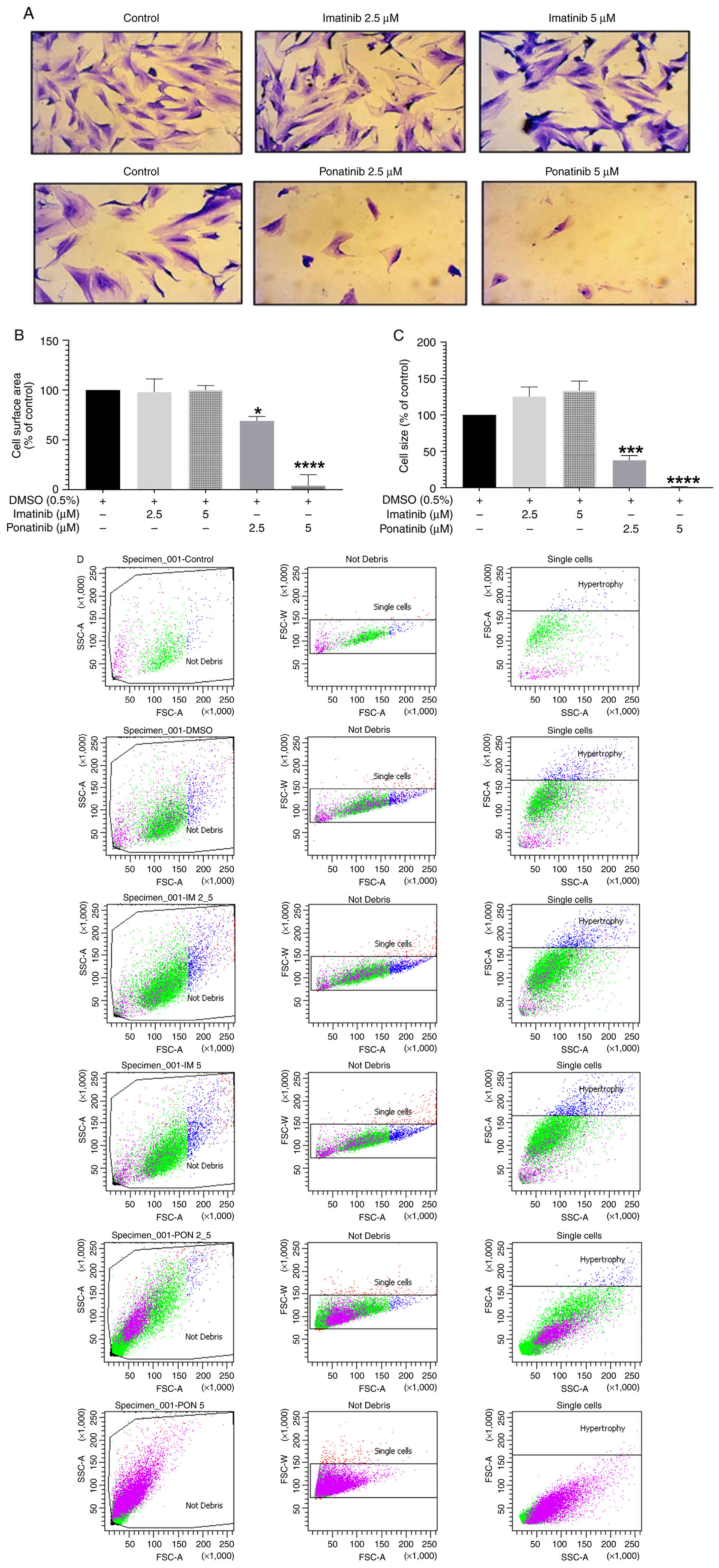
The effect of imatinib and ponatinib on cell
morphology and cardiomyocyte hypertrophic markers, including cell
surface area and size were assessed. Treatment with 2.5 or 5.0 µM
ponatinib reduced cellular density and increased cellular
detachment and cellular shrinkage compared with the Negative
control (DMSO 0.1% v/v) (Fig. 3A).
Imatinib treatment increased cell size, indicative of hypertrophy.
Ponatinib, but not imatinib, impacted H9c2 cardiomyoblast
morphology. At 2.5 µM, ponatinib cause a significant reduction in
cell surface area (69.23±9.86%) compared with the negative control,
indicating cell shrinkage. Furthermore, 5 µM ponatinib resulted in
severe cell loss, making surface area measurement impossible.
(Fig. 3A and B).
To confirm these results, flow cytometry was used to
measure cell size. As expected, 2.5 and 5.0 µM ponatinib
significantly reduced cell size by 37.59±14.35 and 1.26±0.22%,
respectively, compared with negative control. Imatinib treatment
did not induce any significant change in cell size (Fig. 3C). Representative flow cytometry
plots for the cell size are shown in Fig. 3D.
Imatinib and ponatinib induce
malformations in ZFEs
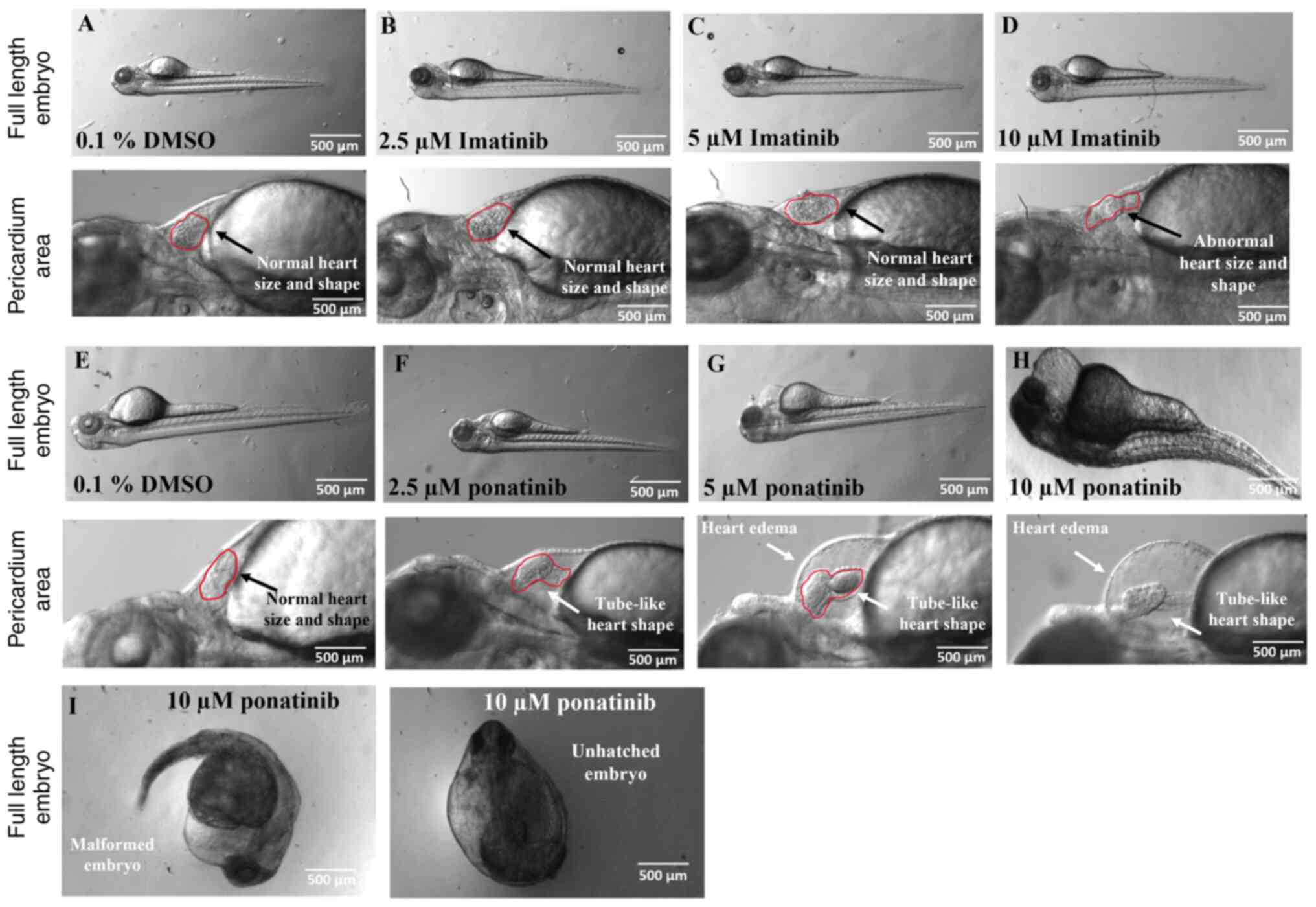
To confirm cardiotoxic effects of imatinib and
ponatinib, ZFEs were used as an in vivo model. Embryos were
treated with TKI and toxicity analyses were conducted by monitoring
the survival and morphology for up to 72 hpf. For the 2.5 µM
imatinib and ponatinib treatment, several phenotypes were observed,
including edema and/or lordosis. Edema was the most common
disfigurement in all ponatinib concentrations. Edema typically
included cardiac malformations that disrupted the sinus rhythm of
the heart. These malformations usually did not result in death of
the animals. As these animals aged, they maintained their bent
stature but could otherwise function normally, for example swimming
and feeding. The decreased diameter of DA and posterior cardinal
vein (PCV) was consistently observed in 2.5 µM imatinib and
ponatinib treated embryos compared with the negative control
embryos. The animals that displayed this malformation exhibited
notable difficulties swimming at later time points. Dorsalization
was observed in rare cases in the 10 µM ponatinib-treated embryos.
Embryos exhibiting this phenotype seldom survived and failed to
hatch because of the extreme nature of the deformities. Exposure to
TKIs significantly increased malformation rates in zebrafish
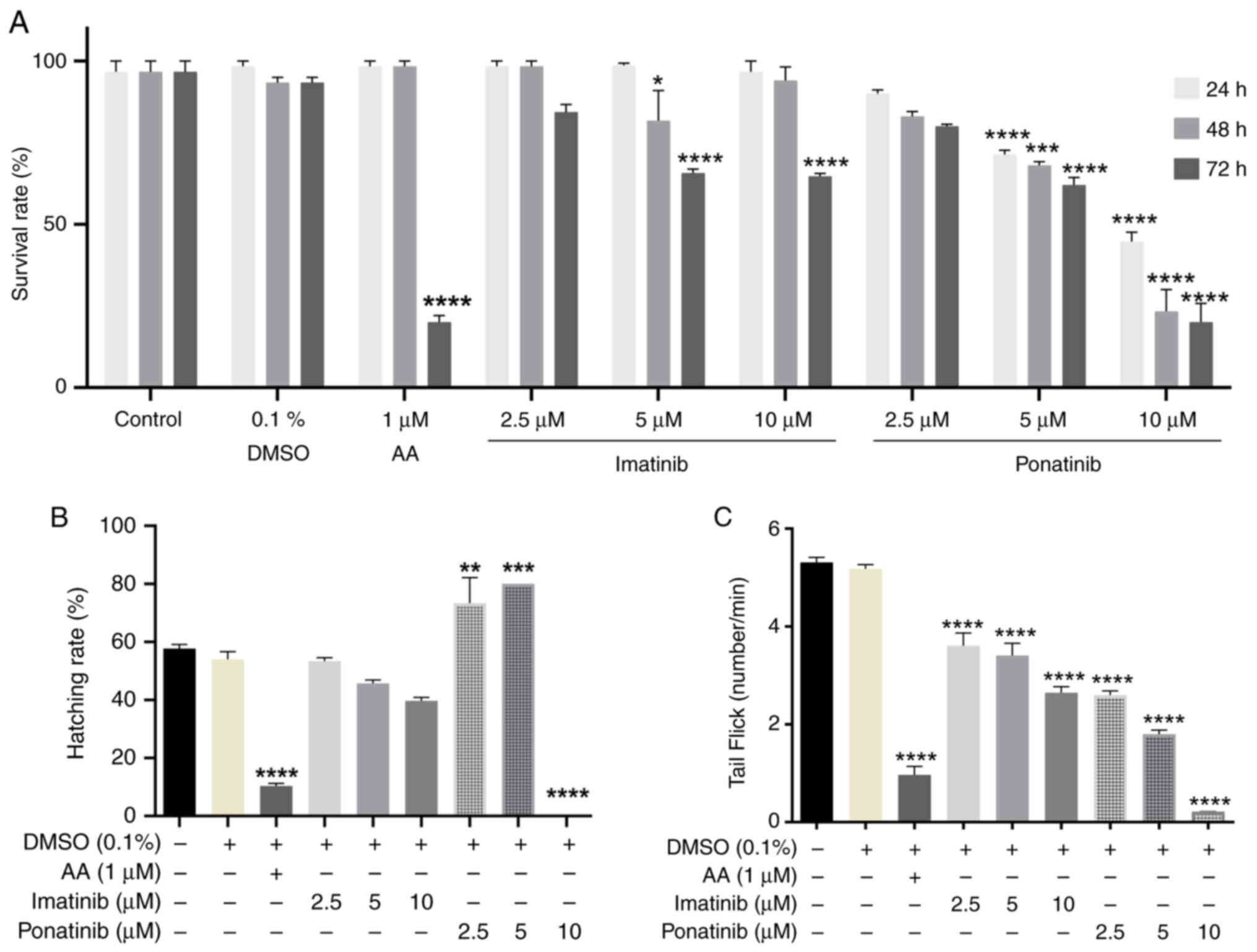
embryos compared to controls treated with 1% DMSO (Fig. 4; Table II). Control embryos showed a low
baseline incidence of malformations, with only 5% exhibiting edema.
Imatinib impact was dose-dependent; at lower concentrations (2.5
and 5 µM), it did not significantly increase malformation rates.
However, at the highest concentration (10 µM), it caused a moderate
rise in edema (10%) and a substantial increase in cardiac
disruptions (40%). Ponatinib demonstrated a stronger teratogenic
effect than Imatinib. Even at its lowest dose (2.5 µM), Ponatinib
significantly increased the prevalence of edema (20%) and cardiac
disruptions (25%). The malformation rates increased as the
concentration of Ponatinib rose, with 65% of embryos showing edema
and 60% having cardiac issues at 5 µM. At the highest dose (10 µM),
nearly all embryos displayed malformations, with 85% showing edema,
90% exhibiting lordosis, and 95% experiencing cardiac
disruptions.
| Table II.Dorsal aorta blood flow analysis for
zebrafish embryos. |
Table II.
Dorsal aorta blood flow analysis for
zebrafish embryos.
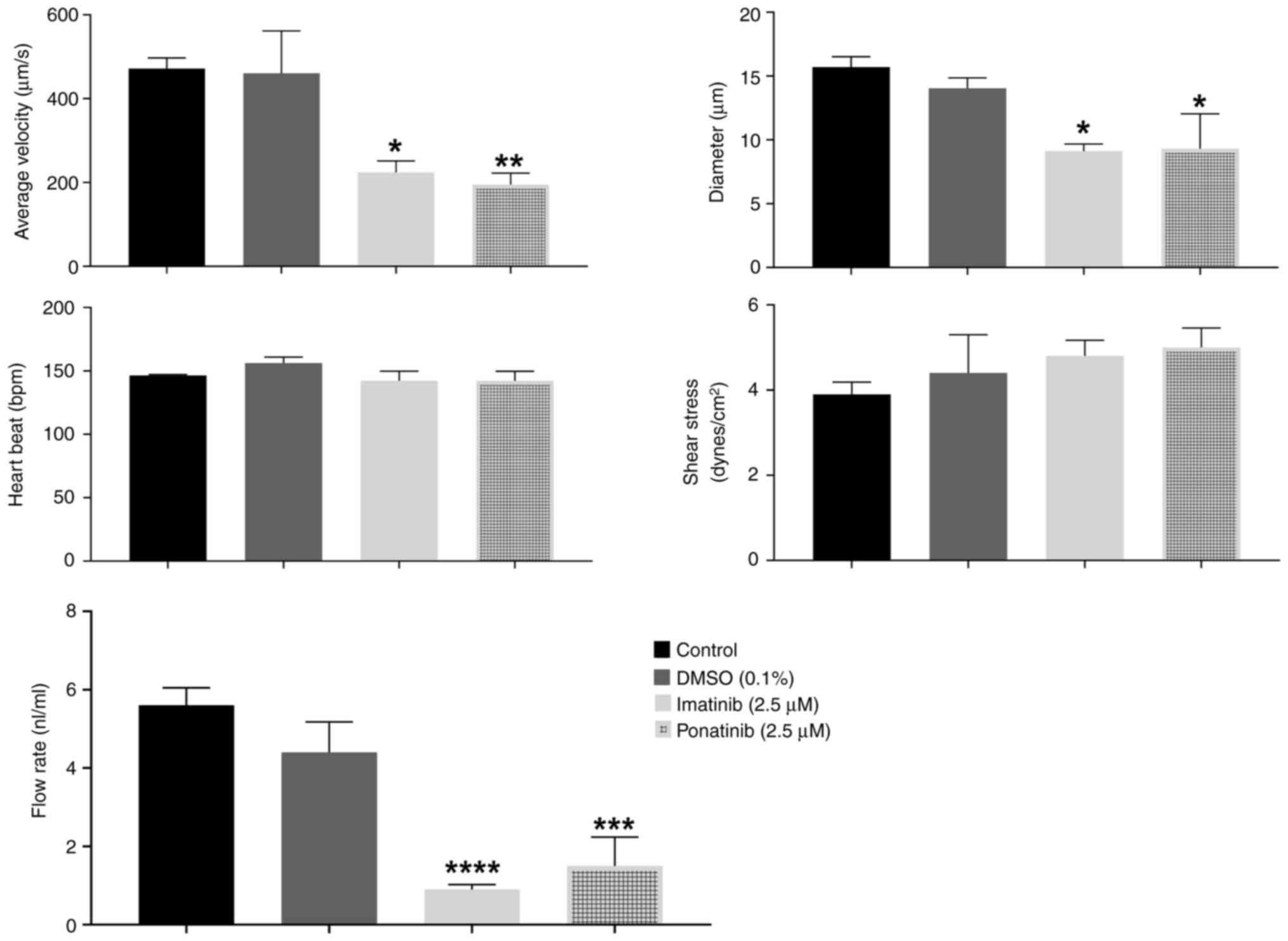
| Property | Control | 0.1% DMSO | 2.5 µM
imatinib | 2.5 µM
ponatinib |
|---|
| Blood flow
velocity, µm/sec | 472.00±25.30 | 460.40±101.30 |
223.80±27.40a |
194.90±27.40b |
| Diameter, µm | 15.70±2.00 | 14.03±2.020 | 9.10±1.40 | 9.30±6.70 |
| Heartbeat, bpm | 146.30±1.90 | 156.20±11.50 | 142.00±318.30 | 142.20±18.30 |
| Sheer stress,
dynes/cm2 | 4.80±0.90 | 5.20±1.120 | 3.90±0.70 | 4.40±2.20 |
| Cardiac output,
nl/ml | 5.60±1.10 | 4.40±1.90 | 0.90±0.03 | 1.50±1.80 |
Imatinib and ponatinib induce
developmental toxicity in ZFE
To assess the potential teratogenic effects of
imatinib and ponatinib, treated embryos were visually inspected
compared with the negative controls at 24, 48 and 72 hpf. As a PC
for heart failure induction, 1 µM AA was used, consistent with
previous studies (38,39). Concentration-dependent decreases in
survival rate and tail-flicking activity were observed in embryos
treated with imatinib and ponatinib compared with the negative
control (Fig. 5A and C). Ponatinib
significantly affected survival rate at 5 and 10 µM concentrations.
This effect began at the first day (24 h post fertilization, hpf)
and persisted until 48 and 72 hpf. Both Imatinib and Ponatinib
significantly affected tail flicking at all concentrations tested.
However, Ponatinib exhibited a stronger inhibitory effect on both
survival rate and tail flocking compared to Imatinib.
Furthermore, changes in hatching rates were observed
at 48 hpf. While 2.5 and 5.0 µM ponatinib significantly increased
the hatching rate compared with the control, 10 µM ponatinib
significantly decreased hatching rate compared with the control.
Imatinib treatment did not significantly affect hatching at any
concentration. As expected, 1 µM AA significantly reduced the
hatching rate compared with the negative control (0.1% v/v DMSO)
(Fig. 5B).
Imatinib and ponatinib disrupt cardiac
function and structure in ZFE
DA blood flow analysis demonstrated the detrimental
effects of both TKIs on cardiac function. Notably, 5 and 10 µM
ponatinib nearly arrested blood flow (Fig. 4I), precluding further analysis at
higher concentrations. At 2.5 µM, ponatinib and imatinib
significantly impaired cardiac function. Compared with negative
control embryos, 2.5 µM, ponatinib and imatinib caused a
significant 48% decrease in DA blood flow velocity, indicating
decreased flow rate. Furthermore, 2.5 µM ponatinib and imatinib
induced a significant 27% narrowing of the aorta diameter,
suggesting structural abnormalities. Both TKIs caused a 73%
reduction in overall flow rate compared with negative controls
(Fig. 6). These findings suggest
the cardiotoxic potential of both ponatinib and imatinib in
developing ZFEs.
Imatinib and ponatinib upregulate
cardiac failure markers in ZFE
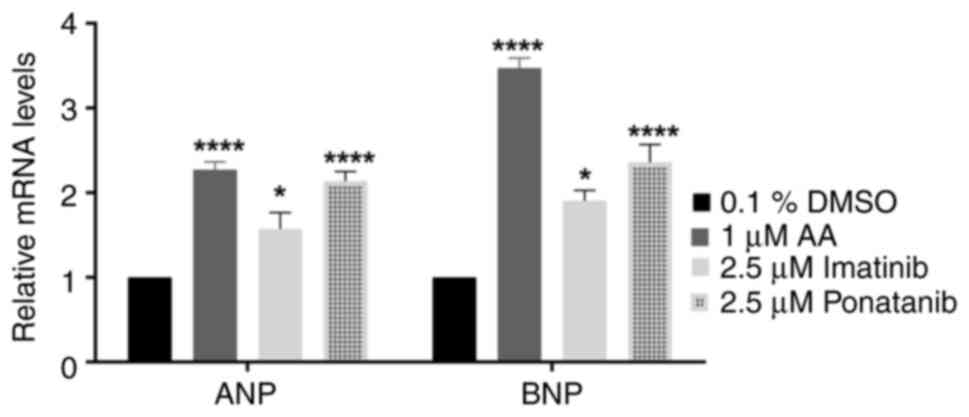
Imatinib and ponatinib significantly increased mRNA
expression of ANP and BNP compared with the controls (Fig. 7). ANP and BNP are established
markers of cardiac failure (40).
Ponatinib treatment significantly increased ANP and
BNP mRNA expression by approximately twofold compared to the
negative control. In contrast, Imatinib only caused a onefold
increase in both ANP and BNP mRNA expression compared to the
negative control.
These changes in gene expression mirrored those
observed in embryos treated with 1 µM Aristolochic acid I (AA), a
positive control for ZFE cardiotoxicity. This suggests that both
TKIs (Ponatinib and Imatinib) significantly affect zebrafish
embryonic development. Notably, Ponatinib's effect was comparable
to the positive control (Fig.
7).
Discussion
Cardio-oncology emphasizes the importance of early
detection and management of cardiotoxic effects during cancer
treatment (41). These adverse
effects can occur acutely, subacutely or chronically, depending on
the drug and patient context (42–44).
While numerous anticancer medications affect the cardiovascular
system (4), multi-targeted TKIs
such as imatinib and ponatinib raise concerns due to their
well-established cardiotoxic potential (45–47).
Novel strategies are required to mitigate TKI-induced
cardiotoxicity.
Among small-molecule kinase inhibitors used for CML
treatment, ponatinib carries the highest risk of cardiotoxicity,
manifesting as congestive heart failure (CHF), cardiac arrhythmia
and hypertension (22,48). Clinical trials report that 7% of
ponatinib-treated patients experience CHF or left ventricular
dysfunction, with potentially life-threatening consequences
(49–51). This heightened risk is further
emphasized by the black box warning issued by the U.S. Food and
Drug Administration (FDA). Black box warnings are the FDA's most
stringent warnings, highlighting medications with potentially
serious, long-lasting, or even fatal risks. The present study
evaluated the effects of TKIs using H9c2 cells and ZF as models.
Initial observations with H9c2 cells identified ponatinib more
cardiotoxic than imatinib, which was confirmed in the ZF model.
H9c2 cells were chosen for their established use in exploring
cellular mechanisms of TKI-induced cardiotoxicity (23,25,52–56).
Moreover, the ZF model offers an efficient and cost-effective means
for in vivo toxicity assessment. Despite differences between
ZF and human biology, notable genetic overlap and ability to mimic
cardiotoxic effects observed in patients with cancer make them
valuable tools for assessing cardiovascular toxicity associated
with cancer treatment (34,56–59).
Notably, zebrafish embryos offer a high-throughput screening
approach but are limited to early developmental stages, it offers
advantages over other in vitro and in vivo models,
their rapid development, ease of use and large numbers of embryos
enable quick testing of many compounds. Alternative models such as
adult ZF, chick embryos and mice should be used for comprehensive
analyses, including extended assessments of acute or chronic
effects.
A previous study assessed the involvement of p90
ribosomal S6 kinase and autophagy in TKI-induced cardiotoxicity
(60). Based on previous analyses,
where cardiotoxic effects of clinically approved TKIs (Dasatinib,
Nilotinib, Ponatinib) were compared and the distinct effects of
Ponatinib on H9c2 cells were identified (61–64).
Imatinib has well-established clinical use and relatively low
cardiotoxic profile compared with other TKIs studied; therefore,
ponatinib and imatinib were assessed in the present study (60,62–64).
Cytotoxic drugs such as imatinib and ponatinib
affect cellular processes, including morphology, proliferation,
attachment and viability (65). A
particular concern is in the context of the heart, where
cardiomyocyte loss serves a key role in the development of heart
failure (66,67). Numerous mechanisms including
autophagy, apoptosis and necrosis contribute to this loss (66,67).
Moreover, pathological cardiac hypertrophy, often triggered by
factors including elevated blood pressure, can lead to heart
failure (68). This condition is
characterized by enlarged cardiomyocytes and increased expression
of natriuretic peptides such as ANP and BNP (68).
Given the link between healthy cardiomyocytes and
heart failure, the potential cardiotoxic effects of imatinib and
ponatinib on H9c2 cardiomyoblasts were assessed by two key
indicators: Cell viability and hypertrophic response. ponatinib
significantly decreased viability in H9c2 cells, demonstrating
potent effect. The MTT assay indicated ~20 and ~40% reductions for
2.5 and 5 µM ponatinib, respectively, after 24 h. These findings
align with other in vitro studies (23,24,65).
While MTT assay offers a convenient and widely used method for
assessing cell viability, its limitations must be acknowledged,
particularly when evaluating drugs such as ponatinib that target
the mitochondria (69,70). MTT assay measures formazan
production, a byproduct of mitochondrial activity, as an indirect
measure of cell viability (71).
This can be misleading as drugs such as ponatinib primarily affect
mitochondrial function without inducing substantial cell death
(72). Moreover, factors beyond
cell viability, such as cellular morphology and metabolic activity,
influence formazan production, potentially leading to inaccuracy
(71,73). Ponatinib is known to inhibit
BCR-ABL TK, a protein located in the mitochondria (74). This can directly impair
mitochondrial function, leading to decreased formazan production in
viable cells (75). Therefore,
relying solely on the MTT assay for assessing cell viability in
response to ponatinib treatment may not accurately reflect the true
impact on cell survival. It is key to consider using complementary
assays that directly assess cell viability, such as trypan blue
exclusion or flow cytometry based on viability dyes. Combining MTT
assay with these alternative methods can provide a more
comprehensive and reliable picture of cell viability (76), particularly when studying drugs
with potential mitochondrial effects, such as Ponatinib.
Compared with MTT assay, flow cytometry showed more
significant reductions in cell viability (>90% for imatinib and
35–80% for ponatinib) due to the increased sensitivity of flow
cytometry (65,77). Imatinib and ponatinib induced
different morphological alterations. Ponatinib caused significant
shrinkage and detachment of cardiomyoblasts, while significant
increase in cell viability compared with the negative control was
demonstrated with 2.5 µM imatinib treatment, however, no
significant changes were demonstrated at 5 µM. Cell shrinkage
suggests apoptosis, while decreased surface area may indicate
impaired hypertrophic response (68,78,79).
These findings align with previous observations of the effect of
ponatinib on Neonatal rat ventricular myocytes (80). While the adverse effects of
ponatinib on cardiomyocyte morphology are concerning, further
studies are needed to confirm its hypertrophic potential.
To evaluate the developmental and cardiac impacts of
imatinib and ponatinib, ZFEs were treated 2.5, 5.0 and 10.0 µM
imatinib or ponatinib for 72 h. To ensure minimal interference from
the solvent, 0.1% DMSO was used to adhere to established minimal
toxicity recommendations and The Organization for Economic
Cooperation and Development guidelines (81,82).
Early embryo lethality at <12 hpf was attributed to unfertilized
embryos mimicking normal appearance but lacking viability. Prompt
removal of these and inclusion of an untreated control group
ensured that the observed lethality accurately reflected TKI
exposure.
Exposure to both TKIs resulted in dose-dependent
malformation, with ponatinib causing stronger effects. Notably, the
consistent presence of edema across all ponatinib concentrations
directly contributed to cardiac disruption and ultimately impaired
long-term swimming ability, Edema is the accumulation of fluid in
tissues, leading to swelling. When edema occurs in zebrafish
embryos around the heart, it can physically compress the organ,
impairing its ability to effectively pump blood. Additionally, the
fluid from edema may carry molecules that disrupt normal signaling
pathways within the heart muscle, potentially resulting in weakened
contractions or abnormal heart rhythms. Decreased blood vessel
diameter further emphasized the detrimental impact on
cardiovascular function. The rare but severe dorsalization
phenotype at 10 µM ponatinib concentrations highlighted potential
teratogenic effects. Visual inspection confirmed the teratogenic
potential of both TKIs, evidenced by decreased survival and
tail-flicking activity and altered hatching rates. Ponatinib
exhibited a stronger inhibitory effect on hatching compared to
imatinib. Ponatinib displayed a biphasic response, with low doses
(2.5, 5 µM) stimulating hatching while higher concentrations (10
µM) significantly suppressed it. This suggests a dose-dependent
mechanism for ponatinib's effect on hatching, distinct from that of
imatinib. Notably, the AA PC further validated these results and
emphasized the teratogenicity of the tested TKIs. These findings
demonstrate the cardiotoxic potential of imatinib and ponatinib,
extending beyond in vitro models and underlining potential
clinical risks.
The cardiac structure and function measurement
revealed that while heart rate remained consistent across
TKI-treated animals, significant cardiovascular disruptions
occurred even at the lowest tested concentration of 2.5 µM. Both
drugs significantly decreased aortic blood flow velocity,
suggesting impaired flow rate and circulation. This effect was
further confirmed by the complete absence of blood flow at 10 µM
ponatinib concentrations. Furthermore, the decrease in blood vessel
diameter caused by ponatinib further supported its detrimental
impact on the cardiovascular system. The abnormalities in heart
shape and size aligned with the known cardiotoxic effects of TKIs
and potentially explain these functional impairments (59). However, no significant differences
in shear stress were demonstrated, suggesting that the mechanical
hemodynamic effects are unlikely to be the primary driver of
deformities and dysfunctions. These findings reinforce the need for
further investigation into specific molecular pathways underlying
TKI-induced cardiotoxicity.
Both TKIs significantly affected cardiac marker gene
expression of ANP and BNP levels, Notably, Ponatinib effect was
comparable to the positive control, indicating severe
cardiotoxicity and potential hypertrophy. This aligns with previous
studies demonstrating ponatinib-induced p90RSK phosphorylation
in vitro, which promotes cardiomyocyte hypertrophy (60,83,84).
The present study serves as a primary screening of
the cardiotoxic potential of TKIs in a ZF model. While ANP and BNP
results suggest a molecular mechanism, further molecular assessment
is recommended to achieve more comprehensive understanding of
potential cardiotoxic effects. Future work should evaluate
additional cardiomyocyte injury markers and conduct signaling
pathway analysis for other markers such as Reactive Oxygen Species
related genes and Vascular endothelial growth factor receptor 2
(VEGFR2). This may provide deeper knowledge of underlying
mechanisms and long-term consequences associated with these
drugs.
Despite their effectiveness as tyrosine kinase
inhibitors (TKIs) for targeting ABL in cancers like CML, both
imatinib and ponatinib present a risk of cardiotoxicity. This
off-target effect likely stems from distinct pathways. Suppression
of ABL by imatinib may trigger endoplasmic reticulum stress and ROS
(reactive oxygen species) accumulation. This, in turn, could
cripple mitochondrial function and lead to cardiotoxicity, as
supported by clinical observations and damage-mimicking experiments
(65,70,71).
While it also promotes ROS generation and mitochondrial
dysfunction, ponatinib raises additional clinical concerns like
vascular complications and hypertension (16,72).
Its ability to hinder blood vessel formation suggests potential
off-target inhibition of VEGFR2, a receptor involved in
angiogenesis (blood vessel growth) (73,74).
Despite imatinib and ponatinib sharing the mechanism
of blocking RTK activity, their targeted kinases, pharmacokinetics
and side effects differ. The specific pathways affected by each
drug require further elucidation. Understanding the molecular
mechanism is required for developing targeted interventions.
Advanced tools such as transcriptome analysis and specific
inhibitors should be used to identify these unique mechanisms.
In summary, the present study demonstrated that both
imatinib and ponatinib exhibited distinct cardiotoxic effects,
highlighting the need for further investigations of TKIs and their
developmental impacts. Early detection and a comprehensive
understanding of these adverse effects are crucial for improving
patient outcomes and quality of life. Looking ahead, targeted
delivery systems using nanoparticles offer a promising solution to
minimize systemic side effects while maintaining the therapeutic
efficacy of TKIs.
Building upon the established CML zebrafish model,
in vivo assessments of these nanoparticle-based drug
delivery systems can evaluate their effectiveness and safety
(50). This approach holds
significant promise for reducing off-target toxicity and improving
the overall patient experience.
Acknowledgements
The authors would like to thank Ms Jensa Mariam
Joseph, College of Pharmacy, Qatar University, Doha 2713, Qatar)
for technical support.
Funding
The present study was supported by Qatar University internal
funds (grant nos. QUST-BRC-SPR2017-1 and QUUG-BRC-2017-3).
Availability of data and materials
The data generated in the present study may be
requested from the corresponding author.
Authors' contributions
FM, HY and HK conceived the study. ZZ, MA and MS
designed the methodology. FA, FM, FB, SS, SU and HK analyzed data.
ZZ performed experiments. FA, FM, HY and HK collected data. ZZ, MS
and FB wrote the manuscript. FM, HY and ZZ revised the manuscript.
FM and HY supervised the study. ZZ, MA and MS performed study
administration. FM, HY confirm the authenticity of the raw data.
All authors have read and approved the final manuscript.
Ethics approval and consent to
participate
All experiments were approved by under Qatar
University Institutional Animal Care and Use Committee (approval
no. QU-IACUC 13-11/2018-REN1) and Institutional Biohazard Committee
(approval no. QU-IBC-2018/057-REN2).
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Heron MP and Anderson RN: National Center
for Health Statistics: Changes in the leading cause of death:
recent patterns in heart disease and cancer mortality. US
Department of Health and Human Services, Centers for Disease
Control and Prevention. National Center for Health Statistics;
Hyattsville, MD: 2016
|
|
2
|
Hochhaus A and Kantarjian H: The
development of dasatinib as a treatment for chronic myeloid
leukemia (CML): From initial studies to application in newly
diagnosed patients. J Cancer Res Clin Oncol. 139:1971–1984. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
3
|
López-Otín C and Hunter T: The regulatory
crosstalk between kinases and proteases in cancer. Nat Rev Cancer.
10:278–292. 2010. View
Article : Google Scholar : PubMed/NCBI
|
|
4
|
Ségaliny AI, Tellez-Gabriel M, Heymann MF
and Heymann D: Receptor tyrosine kinases: Characterisation,
mechanism of action and therapeutic interests for bone cancers. J
Bone Oncol. 4:1–12. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Zhang N and Li Y: Receptor tyrosine
kinases: Biological functions and anticancer targeted therapy.
MedComm (2020). 4:e4462023.PubMed/NCBI
|
|
6
|
Chen Y, McAndrews KM and Kalluri R:
Clinical and therapeutic relevance of cancer-associated
fibroblasts. Nat Rev Clin Oncol. 18:792–804. 2021. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
K Bhanumathy K, Balagopal A, Vizeacoumar
FS, Vizeacoumar FJ, Freywald A and Giambra V: Protein tyrosine
kinases: Their roles and their targeting in leukemia. Cancers
(Basel). 13:1842021. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Paul MK and Mukhopadhyay AK: Tyrosine
kinase-role and significance in cancer. Int J Med Sci. 1:101–115.
2004. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Petrelli A and Giordano S: From single- to
multi-target drugs in cancer therapy: When aspecificity becomes an
advantage. Curr Med Chem. 15:422–432. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Jabbour E and Kantarjian H: Chronic
myeloid leukemia: 2018 Update on diagnosis, therapy and monitoring.
Am J Hematol. 93:442–459. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Bukowski RM: Third generation tyrosine
kinase inhibitors and their development in advanced renal cell
carcinoma. Front Oncol. 2:132012. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Stasi I and Cappuzzo F: Second generation
tyrosine kinase inhibitors for the treatment of metastatic
non-small-cell lung cancer. Transl Respir Med. 2:22014. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Yewale C, Baradia D, Vhora I, Patil S and
Misra A: Epidermal growth factor receptor targeting in cancer: A
review of trends and strategies. Biomaterials. 34:8690–8707. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Segaliny A, Tellez-Gabriel M, Heymann MF
and Heymann D: Receptor tyrosine kinases: Characterisation,
mechanism of action and therapeutic interests for bone cancers. J
Bone Oncol. 4:1–12. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Yang Y, Li S, Wang Y, Zhao Y and Li Q:
Protein tyrosine kinase inhibitor resistance in malignant tumors:
Molecular mechanisms and future perspective. Signal Transduct
Target Ther. 7:3292022. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Moslehi JJ: Cardiovascular toxic effects
of targeted cancer therapies. N Engl J Med. 375:1457–1467. 2016.
View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Kerkelä R, Grazette L, Yacobi R, Iliescu
C, Patten R, Beahm C, Walters B, Shevtsov S, Pesant S, Clubb FJ, et
al: Cardiotoxicity of the cancer therapeutic agent imatinib
mesylate. Nat Med. 12:908–916. 2006. View
Article : Google Scholar : PubMed/NCBI
|
|
18
|
Sayegh N, Yirerong J, Agarwal N, Addison
D, Fradley M, Cortes J, Weintraub NL, Sayed N, Raval G and Guha A:
Cardiovascular toxicities associated with tyrosine kinase
inhibitors. Curr Cardiol Rep. 25:269–280. 2023. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Kantarjian H, Shah NP, Hochhaus A, Cortes
J, Shah S, Ayala M, Moiraghi B, Shen Z, Mayer J, Pasquini R, et al:
Dasatinib versus imatinib in newly diagnosed chronic-phase chronic
myeloid leukemia. N Engl J Med. 362:2260–2270. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Montani D, Bergot E, Günther S, Savale L,
Bergeron A, Bourdin A, Bouvaist H, Canuet M, Pison C, Macro M, et
al: Pulmonary arterial hypertension in patients treated by
dasatinib. Circulation. 125:2128–2137. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Cortes JE, Kim DW, Pinilla-Ibarz J, le
Coutre P, Paquette R, Chuah C, Nicolini FE, Apperley JF, Khoury HJ,
Talpaz M, et al: A phase 2 trial of ponatinib in Philadelphia
chromosome-positive leukemias. N Engl J Med. 369:1783–1796. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Dorer DJ, Knickerbocker RK, Baccarani M,
Cortes JE, Hochhaus A, Talpaz M and Haluska FG: Impact of dose
intensity of ponatinib on selected adverse events: Multivariate
analyses from a pooled population of clinical trial patients. Leuk
Res. 48:84–91. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Korashy HM, Al-Suwayeh HA, Maayah ZH,
Ansari MA, Ahmad SF and Bakheet SA: Mitogen-activated protein
kinases pathways mediate the sunitinib-induced hypertrophy in rat
cardiomyocyte H9c2 cells. Cardiovasc Toxicol. 15:41–51. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Zhao Y, Xue T, Yang X, Zhu H, Ding X, Lou
L, Lu W, Yang B and He Q: Autophagy plays an important role in
sunitinib- mediated cell death in H9c2 cardiac muscle cells.
Toxicol Appl Pharmacol. 248:20–27. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Will Y, Dykens JA, Nadanaciva S, Hirakawa
B, Jamieson J, Marroquin LD, Hynes J, Patyna S and Jessen BA:
Effect of the multitargeted tyrosine kinase inhibitors imatinib,
dasatinib, sunitinib, and sorafenib on mitochondrial function in
isolated rat heart mitochondria and H9c2 cells. Toxicol Sci.
106:153–161. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Talbert DR, Doherty KR, Trusk PB, Moran
DM, Shell SA and Bacus S: A multi-parameter in vitro screen in
human stem cell-derived cardiomyocytes identifies ponatinib-induced
structural and functional cardiac toxicity. Toxicol Sci.
143:147–155. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Doherty KR, Wappel RL, Talbert DR, Trusk
PB, Moran DM, Kramer JW, Brown AM, Shell SA and Bacus S:
Multi-parameter in vitro toxicity testing of crizotinib, sunitinib,
erlotinib, and nilotinib in human cardiomyocytes. Toxicol Appl
Pharmacol. 272:245–255. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
French KJ, Coatney RW, Renninger JP, Hu
CX, Gales TL, Zhao S, Storck LM, Davis CB, McSurdy-Freed J, Chen E
and Frazier KS: Differences in effects on myocardium and
mitochondria by angiogenic inhibitors suggest separate mechanisms
of cardiotoxicity. Toxicol Pathol. 38:691–702. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Pembrey RS, Marshall KC and Schneider RP:
Cell surface analysis techniques: What do cell preparation
protocols do to cell surface properties? Appl Environ Microbiol.
65:2877–2894. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Prabhu KS, Siveen KS, Kuttikrishnan S,
Iskandarani A, Tsakou M, Achkar IW, Therachiyil L, Krishnankutty R,
Parray A, Kulinski M, et al: Targeting of X-linked inhibitor of
apoptosis protein and PI3-kinase/AKT signaling by embelin
suppresses growth of leukemic cells. PLoS One. 12:e01808952017.
View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Khan AQ, Siveen KS, Prabhu KS,
Kuttikrishnan S, Akhtar S, Shaar A, Raza A, Mraiche F, Dermime S
and Uddin S: Curcumin-mediated degradation of S-phase kinase
protein 2 induces cytotoxic effects in human
papillomavirus-positive and negative squamous carcinoma cells.
Front Oncol. 8:3992018. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Westerfield M: The zebrafish book: A guide
for the laboratory use of zebrafish (Danio rerio). 4th edition.
University of Oregon Press; Eugene: 2000, http://zfin. org/zf_info/zfbook/zfbk.html
|
|
33
|
Kimmel CB, Ballard WW, Kimmel SR, Ullmann
B and Schilling TF: Stages of embryonic development of the
zebrafish. Dev Dyn. 203:253–310. 1995. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Benslimane FM, Zakaria ZZ, Shurbaji S,
Abdelrasool MKA, Al-Badr MAHI, Al Absi ESK and Yalcin HC: Cardiac
function and blood flow hemodynamics assessment of zebrafish (Danio
rerio) using high-speed video microscopy. Micron. 136:1028762020.
View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Yalcin HC, Amindari A, Butcher JT, Althani
A and Yacoub M: Heart function and hemodynamics analysis for
zebrafish embryos. Dev Dyn. 246:868–880. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Benslimane FM, Alser M, Zakaria ZZ, Sharma
A, Abdelrahman HA and Yalcin HC: Adaptation of a mice doppler
echocardiography platform to measure cardiac flow velocities for
embryonic chicken and adult zebrafish. Front Bioeng Biotechnol.
7:962019. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Rao X, Huang X, Zhou Z and Lin X: An
improvement of the 2ˆ(−delta delta CT) method for quantitative
real-time polymerase chain reaction data analysis. Biostat
Bioinforma Biomath. 3:71–85. 2013.PubMed/NCBI
|
|
38
|
Huang CC, Chen PC, Huang CW and Yu J:
Aristolochic acid induces heart failure in zebrafish embryos that
is mediated by inflammation. Toxicol Sci. 100:486–494. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Narumanchi S, Wang H, Perttunen S,
Tikkanen I, Lakkisto P and Paavola J: Zebrafish heart failure
models. Front Cell Dev Biol. 9:6625832021. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Januzzi JL Jr: Natriuretic peptides as
biomarkers in heart failure. J Investig Med. 61:950–955. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Wickramasinghe CD, Nguyen KL, Watson KE,
Vorobiof G and Yang EH: Concepts in cardio-oncology: Definitions,
mechanisms, diagnosis and treatment strategies of cancer
therapy-induced cardiotoxicity. Future Oncol. 12:855–870. 2016.
View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Dolci A, Dominici R, Cardinale D, Sandri
MT and Panteghini M: Biochemical markers for prediction of
chemotherapy-induced cardiotoxicity: Systematic review of the
literature and recommendations for use. Am J Clin Pathol.
130:688–695. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Pai VB and Nahata MC: Cardiotoxicity of
chemotherapeutic agents: Incidence, treatment and prevention. Drug
Saf. 22:263–302. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Albini A, Pennesi G, Donatelli F,
Cammarota R, De Flora S and Noonan DM: Cardiotoxicity of anticancer
drugs: The need for cardio-oncology and cardio-oncological
prevention. J Natl Cancer Inst. 102:14–25. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Sheng CC, Amiri-Kordestani L, Palmby T,
Force T, Hong CC, Wu JC, Croce K, Kim G and Moslehi J: 21st Century
cardio-oncology: Identifying cardiac safety signals in the era of
personalized medicine. JACC Basic Transl Sci. 1:386–398. 2016.
View Article : Google Scholar : PubMed/NCBI
|
|
46
|
Cortes JE, Kim DW, Pinilla-Ibarz J, Le
Coutre P, Paquette R, Chuah C, Nicolini FE, Apperley JF, Khoury HJ,
Talpaz M, et al: Long-term follow-up of ponatinib efficacy and
safety in the phase 2 PACE trial. Blood. 124:31352014. View Article : Google Scholar
|
|
47
|
Sayed-Ahmed MM, Alrufaiq BI, Alrikabi A,
Abdullah ML, Hafez MM and Al-Shabanah OA: Carnitine supplementation
attenuates sunitinib-induced inhibition of AMP-activated protein
kinase downstream signals in cardiac tissues. Cardiovasc Toxicol.
19:344–356. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
48
|
Jin Y, Xu Z, Yan H, He Q, Yang X and Luo
P: A comprehensive review of clinical cardiotoxicity incidence of
FDA-approved small-molecule kinase inhibitors. Front Pharmacol.
11:8912020. View Article : Google Scholar : PubMed/NCBI
|
|
49
|
Moslehi JJ and Deininger M: Tyrosine
kinase inhibitor-associated cardiovascular toxicity in chronic
myeloid leukemia. J Clin Oncol. 33:4210–4218. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
50
|
Shah RR and Morganroth J: Update on
cardiovascular safety of tyrosine kinase inhibitors: With a special
focus on QT interval, left ventricular dysfunction and overall
risk/benefit. Drug Saf. 38:693–710. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
51
|
Cortes JE, Kim DW, Pinilla-Ibarz J, le
Coutre P, Paquette R, Chuah C, Nicolini FE, Apperley JF, Khoury HJ,
Talpaz M, et al: Long-term follow-up of ponatinib efficacy and
safety in the phase 2 PACE trial. Blood. 124:31352014. View Article : Google Scholar
|
|
52
|
Zordoky BN and El-Kadi AOS: H9c2 cell line
is a valuable in vitro model to study the drug metabolizing enzymes
in the heart. J Pharmacol Toxicol Methods. 56:317–322. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
53
|
Watkins SJ, Borthwick GM and Arthur HM:
The H9C2 cell line and primary neonatal cardiomyocyte cells show
similar hypertrophic responses in vitro. In Vitro Cell Dev Biol
Anim. 47:125–131. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
54
|
Witek P, Korga A, Burdan F, Ostrowska M,
Nosowska B, Iwan M and Dudka J: The effect of a number of H9C2 rat
cardiomyocytes passage on repeatability of cytotoxicity study
results. Cytotechnology. 68:2407–2415. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
55
|
Kobuszewska A, Tomecka E, Zukowski K,
Jastrzebska E, Chudy M, Dybko A, Renaud P and Brzozka Z:
Heart-on-a-Chip: An investigation of the influence of static and
perfusion conditions on cardiac (H9C2) cell proliferation,
morphology, and alignment. SLAS Technol. 22:536–546. 2017.
View Article : Google Scholar : PubMed/NCBI
|
|
56
|
Bouleftour W, Mery B, Rowinski E, Rivier
C, Daguenet E and Magne N: Cardio-oncology preclinical models: A
comprehensive review. Anticancer Res. 41:5355–5364. 2021.
View Article : Google Scholar : PubMed/NCBI
|
|
57
|
Khan FR and Alhewairini SS: Zebrafish
(Danio rerio) as a model organism. Curr Trends Cancer manage.
27:3–18. 2018.
|
|
58
|
Lane S, More LA and Asnani A: Zebrafish
models of cancer therapy-induced cardiovascular toxicity. J
Cardiovasc Dev Dis. 8:82021.PubMed/NCBI
|
|
59
|
Al-Thani HF, Shurbaji S, Zakaria ZZ, Hasan
MH, Goracinova K, Korashy HM and Yalcin HC: Reduced cardiotoxicity
of ponatinib-loaded PLGA-PEG-PLGA nanoparticles in zebrafish
xenograft model. Materials (Basel). 15:39602022. View Article : Google Scholar : PubMed/NCBI
|
|
60
|
Suleiman M: The role of P90 ribosomal S6
kinase and autophagy in sunitinib and ponatinib-induced
cardiotoxicity. 2019.
|
|
61
|
Lekes D, Szadvari I, Krizanova O, Lopusna
K, Rezuchova I, Novakova M, Novakova Z, Parak T and Babula P:
Nilotinib induces ER stress and cell death in H9c2 cells. Physiol
Res. 65 (Suppl 4):S505–S514. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
62
|
Wang H, Wang Y, Li J, He Z, Boswell SA,
Chung M, You F and Han S: Three tyrosine kinase inhibitors cause
cardiotoxicity by inducing endoplasmic reticulum stress and
inflammation in cardiomyocytes. BMC Med. 21:1472023. View Article : Google Scholar : PubMed/NCBI
|
|
63
|
Lamore SD, Kohnken RA, Peters MF and
Kolaja KL: Cardiovascular toxicity induced by kinase inhibitors:
Mechanisms and preclinical approaches. Chem Res Toxicol.
33:125–136. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
64
|
Sun S, Qin J, Liao W, Gao X, Shang Z, Luo
D and Xiong S: Mitochondrial dysfunction in cardiotoxicity induced
by BCR-ABL1 tyrosine kinase inhibitors-underlying mechanisms,
detection, potential therapies. Cardiovasc Toxicol. 23:233–254.
2023. View Article : Google Scholar : PubMed/NCBI
|
|
65
|
Méry B, Guy JB, Vallard A, Espenel S,
Ardail D, Rodriguez-Lafrasse C, Rancoule C and Magné N: In vitro
cell death determination for drug discovery: A landscape review of
real issues. J Cell Death. 10:11796707176912512017. View Article : Google Scholar : PubMed/NCBI
|
|
66
|
Yussman MG, Toyokawa T, Odley A, Lynch RA,
Wu G, Colbert MC, Aronow BJ, Lorenz JN and Dorn GW II:
Mitochondrial death protein Nix is induced in cardiac hypertrophy
and triggers apoptotic cardiomyopathy. Nat Med. 8:725–730. 2002.
View Article : Google Scholar : PubMed/NCBI
|
|
67
|
Kostin S, Pool L, Elsässer A, Hein S,
Drexler HC, Arnon E, Hayakawa Y, Zimmermann R, Bauer E, Klövekorn
WP and Schaper J: Myocytes die by multiple mechanisms in failing
human hearts. Circ Res. 92:715–724. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
68
|
Tham YK, Bernardo BC, Ooi JY, Weeks KL and
McMullen JR: Pathophysiology of cardiac hypertrophy and heart
failure: Signaling pathways and novel therapeutic targets. Arch
Toxicol. 89:1401–1438. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
69
|
Ghasemi M, Turnbull T, Sebastian S and
Kempson I: The MTT assay: Utility, limitations, pitfalls, and
interpretation in bulk and single-cell analysis. Int J Mol Sci.
22:128272021. View Article : Google Scholar : PubMed/NCBI
|
|
70
|
Rai Y, Pathak R, Kumari N, Sah DK, Pandey
S, Kalra N, Soni R, Dwarakanath BS and Bhatt AN: Mitochondrial
biogenesis and metabolic hyperactivation limits the application of
MTT assay in the estimation of radiation induced growth inhibition.
Sci Rep. 8:15312018. View Article : Google Scholar : PubMed/NCBI
|
|
71
|
van Meerloo J, Kaspers GJ and Cloos J:
Cell sensitivity assays: The MTT assay. Methods Mol Biol.
731:237–245. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
72
|
Yu T, Cao J, Alaa Eddine M, Moustafa M,
Mock A, Erkut C, Abdollahi A, Warta R, Unterberg A, Herold-Mende C
and Jungwirth G: Receptor-tyrosine kinase inhibitor ponatinib
inhibits meningioma growth in vitro and in vivo. Cancers (Basel).
13:58982021. View Article : Google Scholar : PubMed/NCBI
|
|
73
|
Ghasemi M, Liang S, Luu QM and Kempson I:
The MTT assay: A method for error minimization and interpretation
in measuring cytotoxicity and estimating cell viability. Methods
Mol Biol. 2644:15–33. 2023. View Article : Google Scholar : PubMed/NCBI
|
|
74
|
Gustafson D, Fish JE, Lipton JH and Aghel
N: Mechanisms of cardiovascular toxicity of BCR-ABL1 tyrosine
kinase inhibitors in chronic myelogenous leukemia. Curr Hematol
Malig Rep. 15:20–30. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
75
|
Loren CP, Aslan JE, Rigg RA, Nowak MS,
Healy LD, Gruber A, Druker BJ and McCarty OJ: The BCR-ABL inhibitor
ponatinib inhibits platelet immunoreceptor tyrosine-based
activation motif (ITAM) signaling, platelet activation and
aggregate formation under shear. Thromb Res. 135:155–160. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
76
|
Menyhárt O, Harami-Papp H, Sukumar S,
Schäfer R, Magnani L, de Barrios O and Győrffy B: Guidelines for
the selection of functional assays to evaluate the hallmarks of
cancer. Biochim Biophys Acta. 1866:300–319. 2016.PubMed/NCBI
|
|
77
|
Wang X, Xia Y, Liu L, Liu M, Gu N, Guang H
and Zhang F: Comparison of MTT assay, flow cytometry, and RT-PCR in
the evaluation of cytotoxicity of five prosthodontic materials. J
Biomed Mater Res B Appl Biomater. 95:357–364. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
78
|
Yurinskaya V, Aksenov N, Moshkov A, Model
M, Goryachaya T and Vereninov A: A comparative study of U937 cell
size changes during apoptosis initiation by flow cytometry, light
scattering, water assay and electronic sizing. Apoptosis.
22:1287–1295. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
79
|
Chen L, Zhao L, Samanta A, Mahmoudi SM,
Buehler T, Cantilena A, Vincent RJ, Girgis M, Breeden J, Asante S,
et al: STAT3 balances myocyte hypertrophy vis-à-vis autophagy in
response to Angiotensin II by modulating the AMPKα/mTOR axis. PLoS
One. 12:e01798352017. View Article : Google Scholar : PubMed/NCBI
|
|
80
|
Hasinoff BB, Patel D and Wu X: The
myocyte-damaging effects of the BCR-ABL1-targeted tyrosine kinase
inhibitors increase with potency and decrease with specificity.
Cardiovasc Toxicol. 17:297–306. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
81
|
Hoyberghs J, Bars C, Ayuso M, Van Ginneken
C, Foubert K and Van Cruchten S: DMSO concentrations up to 1% are
safe to be used in the zebrafish embryo developmental toxicity
assay. Front Toxicol. 3:8040332021. View Article : Google Scholar : PubMed/NCBI
|
|
82
|
OECD, . OECD guidelines for the testing of
chemicals. Oecd. 1994.
|
|
83
|
Jaballah M, Mohamed IA, Alemrayat B,
Al-Sulaiti F, Mlih M and Mraiche F: Na+/H+ exchanger isoform 1
induced cardiomyocyte hypertrophy involves activation of p90
ribosomal s6 kinase. PLoS One. 10:e01222302015. View Article : Google Scholar : PubMed/NCBI
|
|
84
|
Yamaguchi N, Chakraborty A, Pasek DA,
Molkentin JD and Meissner G: Dysfunctional ryanodine receptor and
cardiac hypertrophy: Role of signaling molecules. Am J Physiol
Heart Circ Physiol. 300:H2187–H2195. 2011. View Article : Google Scholar : PubMed/NCBI
|