Introduction
Colorectal cancer (CRC) is a common cause of cancer
morbidity and mortality; it is the third most frequently diagnosed
malignancy worldwide and the second most common cause of
cancer-related mortality (1–3).
According to global cancer statistics, >1.85 million new cases
of CRC (9.8% of total cancer cases) and an estimated 850,000
CRC-related deaths (9.2% of total cancer-related deaths) occurred
in 2020 (2,4). Among patients with CRC, 20% have
metastatic CRC and 40% present with recurrence after previous
localized disease treatment. Notably, prognosis of metastatic CRC
is poor, with a 5-year survival rate of <20% (5), and these patients often develop
resistance to systemic therapy. Moreover, 35–40% of patients with
KRAS and NRAS gene mutations do not respond to
targeted therapy (5,6). CRC is a heterogeneous disorder
characterized by a variety of molecular alterations, including the
dysregulation of signaling pathways, leading to tumor initiation,
development and metastasis (7).
Therefore, it is important to identify and develop measures to
effectively prevent the metastasis of CRC cells. Understanding the
pathogenesis of CRC and the expression of related molecules is also
required to develop molecular therapies for the treatment of
metastatic CRC.
The AT-rich interactive domain-containing protein 1A
(ARID1A) gene encodes the BAF250a protein, one of the
accessory subunits of the SWItch/Sucrose Non-Fermentable
chromatin-remodeling multi-subunit complex (8), which specifically modulates promoter
accessibility for transcriptional gene activation or suppression,
and regulates multiple cellular processes, including development,
proliferation, differentiation, DNA repair and tumor suppression
(9,10). Mutations in these subunits can
result in abnormal control of lineage-specific differentiation and
gene expression/suppression, contributing to tumorigenesis in
different types of cancer (8).
According to next-generation sequencing, ARID1A has been
identified as a frequently mutated gene in various types of cancer,
including ovarian clear cell carcinoma (11), breast cancer (12), hepatocellular carcinoma (13), esophageal adenocarcinoma (14) and CRC (15). Furthermore, it has become
increasingly clear that ARID1A variation is associated with the
clinicopathological features of CRC (16,17).
In patients with CRC, the loss of ARID1A expression has been
reported to be more significant as the tumor-node-metastasis (TNM)
stage advances (16), indicating
that loss of ARID1A in CRC could be strongly associated with tumor
progression and metastasis. However, the precise molecular
mechanisms through which ARID1A contributes to the metastasis of
CRC remain poorly elucidated.
Epithelial-mesenchymal transition (EMT) is a highly
dynamic and reversible mechanism that favors malignant cancer
behaviors, such as invasion and metastasis (18). It is a biological process in which
epithelial cells exhibit downregulation of their epithelial
properties and an increase in mesenchymal properties, which is
mainly triggered by cytokines and growth factors secreted by the
tumor microenvironment (19,20).
In CRC, EMT is associated with tumor invasion, metastasis and
resistance to chemotherapy (21).
Previous studies have established a significant association between
ARID1A and EMT. Notably, it has been revealed that the suppression
of ARID1A can enhance migratory capabilities by facilitating EMT in
both breast and gastric cancer cells (22,23);
however, its mechanisms of action in CRC remain to be elucidated.
Hence, the role of ARID1A in the regulation of EMT deserves
consideration. Additionally, the potential involvement of ARID1A in
CRC through its impact on the EMT mechanism has garnered attention.
Therefore, the present study conducted experiments to examine the
function of ARID1A in CRC and to offer preliminary guidance for
targeted therapies in the clinical management of metastatic
CRC.
Materials and methods
Patient samples
A total of 20 samples of formalin-fixed,
paraffin-embedded (FFPE) CRC tissue blocks were provided by Dr
Ratirat Samol (Unit of Pathology, Sawanpracharak Hospital, Nakhon
Sawan, Thailand). The preparation of FFPE blocks was conducted by
the hospital's pathological laboratory following standard
procedures. These samples were diagnosed with different
pathological grades of CRC between January and December 2021, with
samples obtained from 9 men and 11 women (age range, 61–97 years).
The research procedures involving individuals were granted approval
by the Human Ethics Review Board of Sawanpracharak Hospital
(approval no. 16/2560) and the Naresuan University Ethical
Committee for Human Research (Phitsanulok, Thailand; approval no.
P1-0191/2564; certificate of approval no. 178/2021), and adhered to
the principles outlined in The Declaration of Helsinki.
Immunohistochemistry (IHC) and
scoring
IHC was performed on 3-µm FFPE tissue sections from
patients with CRC using a rabbit anti-human ARID1A antibody (1:400
dilution; cat. no. HPA005456; MilliporeSigma). Briefly, the
sections underwent deparaffinization with xylene followed by
rehydration in a decreasing concentration of alcohol solutions. The
sections underwent heat-induced epitope retrieval with citrate
buffer (pH 6) for 15 min at 97°C. Subsequently, endogenous enzyme
activity was quenched with 3%
H2O2/NaN3 for 25 min and
non-specific reactivity was blocked with 0.1% NaN3 for
20 min at room temperature, followed by the primary antibody
incubation at 4°C overnight in a humidified chamber. The bound
antibody was then detected using a goat anti-rabbit IgG (H+L)
antibody [Rabbit specific HRP/DAB Detection IHC Kit; cat. no.
ab64261; Abcam] for 15 min at room temperature. A positive reaction
was developed with ready-to-use DAB substrate (cat. no. ab64238;
Abcam) for 3 min at room temperature. The sections were
subsequently counterstained with hematoxylin, by briefly dipping
the sections in this solution twice. The staining results were
observed under a light microscope, with five distinct areas in both
the cancerous and non-cancerous regions of CRC tissues
systematically captured (Carl Zeiss AG; magnification, ×400).
The immunostained sections were evaluated and
assessed by an expert pathologist, who was blinded to the
clinicopathological information of the patients, according to the
percentage of stained cells and the intensity of staining. The IHC
staining intensity was based on the histochemical scoring (H-score)
assessment and was conducted utilizing a four-point grading system
that assesses the staining intensity of the cells, as follows:
Negative staining (0), weak positive staining (1), moderate positive staining (2), and strong positive staining (3). The H-score was calculated using the
following formula: H-score=[(0× % negative cells) + (1× % weak
positive cells) + (2× % moderate positive cells) + (3× % strong
positive cells)]. The H-score representing IHC staining intensity
was compared between cancerous and adjacent non-cancerous areas on
the same tissue slide.
Cell culture, plasmids, transfection
and transduction
SW48 CRC cells and 293 expressing the large
T-antigen of simian virus 40 (293T) cells were obtained from the
American Type Culture Collection. The cells were maintained in
Dulbecco's modified Eagle's medium supplemented with 10% fetal
bovine serum (FBS), 100 µg/ml streptomycin and 100 U/ml penicillin
(all from Gibco; Thermo Fisher Scientific, Inc.) at 37°C in a
humidified chamber containing 5% CO2.
The lentivirus-mediated gene overexpression
(pLenti-puro-ARID1A; cat. no. 39478) and empty vector (pLenti-puro;
cat. no. 39481) systems were purchased from Addgene, Inc., with
lentiviral vectors carrying puromycin tags. For the 2nd-generation
lentiviral system, the lentiviral vectors were co-transfected into
293T cells with pCMV–VSV-G and psPAX2 plasmids (Addgene, Inc.) in a
1.5:1:1 µg ratio, using Lipofectamine® 3000 (Invitrogen;
Thermo Fisher Scientific, Inc.). After 16 h at 37°C, the cell
culture media was refreshed. A total of 48 h after transfection,
the harvested viral supernatants were centrifuged at 2,000 × g for
5 min at 4°C, filtered through 0.45-µm membrane filters, and
transduced into SW48 cells at a multiplicity of infection of 2.5,
utilizing 8 mg/ml polybrene reagent (MilliporeSigma). A total of 24
h post-transduction, the lentivirus-infected cells were selected in
selection medium containing 500 ng/ml puromycin (Invitrogen; Thermo
Fisher Scientific, Inc.) for 3–5 days. The remaining living cells
were collected and used in the subsequent experiments.
Cell staining and
immunofluorescence
Lentivirus-infected SW48 cells were fixed in 4%
paraformaldehyde for 10 min and stained with 0.4% crystal violet
for 30 min at room temperature, facilitating the observation of
morphological changes through a light microscope (Olympus IX70;
Olympus Corporation). The expression of EMT protein markers was
observed in ARID1A-overexpressing SW48 cells by
immunofluorescence staining. Cells were grown in a 96-well plate
until they reached 50% confluence within 48 h, after which, they
were rinsed with 1X PBS and then fixed with 4% paraformaldehyde for
30 min at room temperature. After fixation, the cells underwent two
washes with 1X PBS. Subsequently, they were permeabilized with 0.2%
Triton X-100 solution for 5 min and nonspecific immunoreactivity
was blocked with 2% bovine serum albumin (MilliporeSigma) for 1 h
at room temperature. The cells were then exposed individually to
primary antibodies against E-cadherin (1:1,000 dilution; cat. no.
ab40772; Abcam), zonula occludens 1 (ZO-1; 1:1,000 dilution; cat.
no. ab216880; Abcam), vimentin (1:500 dilution; cat. no. ab92547;
Abcam) and zinc finger E-box binding homeobox 1 (ZEB1; 1:500
dilution; cat. no. ab203829; Abcam) overnight at 4°C. After washing
with PBS three times, the bound antibodies were visualized using
Alexa Fluor® 488-conjugated goat anti-rabbit IgG
antibody (1:1,000 dilution; cat. no. A11008; Invitrogen; Thermo
Fisher Scientific, Inc.) for 1 h at room temperature in the absence
of light. Subsequent counterstaining was performed with 1 µg/ml
DAPI for 30 min in the dark. Finally, the fluorescent images were
visualized using a laser scanning confocal microscope (Carl Zeiss™
AXio Vert.A1; Carl Zeiss AG).
Reverse transcription-quantitative PCR
(RT-qPCR)
Total RNA was extracted from sub-confluent cell
cultures with TRIzol® reagent (Invitrogen; Thermo Fisher
Scientific, Inc.) according to the manufacturer's guidelines. The
isolated RNA was quantified using a NanoDrop ND-1000
spectrophotometer (NanoDrop Technologies) and cDNA was then
synthesized using a SuperScript Vilo cDNA synthesis kit
(Invitrogen; Thermo Fisher Scientific, Inc.), according to the
manufacturer's protocol. The Luna Universal qPCR Master Mix (New
England BioLabs, Inc.) was utilized to perform qPCR using the
CFX96™ Optics Module (Bio-Rad Laboratories, Inc.). Gene
amplification was carried out according to the standard
instructions: An initial DNA denaturation step at 95°C for 3 min;
40 cycles of denaturation at 95°C for 15 sec, annealing at 60°C for
30 sec and extension at 72°C for 30 sec; followed by a melt curve
(58–95°C). All RT-qPCR assays were performed in duplicate and were
repeated in at least three independent experiments. The relative
fold gene expression levels of the samples were calculated using
the 2−∆∆Cq method (24)
and were normalized to the control, human GAPDH expression.
The specific oligonucleotide primer sequences are presented in
Table I.
 | Table I.Sequences of primers used in reverse
transcription-quantitative PCR analysis of gene expression. |
Table I.
Sequences of primers used in reverse
transcription-quantitative PCR analysis of gene expression.
| Gene | NCBI accession
no. | Forward, 5′-3′ | Reverse, 5′-3′ | Product size,
bp |
|---|
| ARID1A | NM_006015.6 |
TCCCACCTGGCTCTGTTGAA |
CATCATTACCCGCCATGCC | 101 |
| CDH1 | NM_004360.5 |
ATTTTTCCCTCGACACCCGAT |
TCCCAGGCGTAGACCAAGA | 109 |
| TJP1 | NM_003257.5 |
CCCTCAAGGAGCCATTC |
CAGTTTGCTCCAACGAGA | 264 |
| VIM | NM_003380.5 |
CCTTGAACGCAAAGTGGAATC |
AGGTCGGGCTTGGAAACATC | 138 |
| ZEB1 | NM_001128128.3 |
ACCCTTGAAAGTGATCCAGC |
TTGGGCGGTGTAGAATCAGA | 111 |
| GAPDH | NM_002046.7 |
CGACCACTTTGTCAAGCTCA |
AGGGGTCTACATGGCAACTG | 228 |
Wound healing assay
Cells were placed in a 24-well plate and incubated
for 24 h at a final cell density of 80%. The cell surface was
gently scratched across the center of the well with a 200-µl
sterile pipette tip to create an equally wide single scratch. The
plate then underwent three washes with PBS in order to eliminate
any detached cells and debris. To facilitate the survival of the
cells, they were subsequently cultured with fresh culture medium
supplemented with 10% FBS (25)
and were incubated in a humidified incubator containing 5%
CO2 at 37°C. Areas of wound healing were examined under
an inverted light microscope (Olympus IX70; Olympus Corporation)
and images were captured immediately after the scratch was
generated (T0), as well as at 24 h (T24), 48 h (T48) and 72 h (T72)
after incubation at 37°C using imaging software (magnification,
×100; cellScens Standard.Ink, version 2.3; Olympus Corporation).
The images of the same areas were acquired. The change in cell-free
area and wound closure rate were calculated by referring to the
image at T0 using ImageJ (version no. 1.54 g; National Institutes
of Health) (26).
Transwell cell migration assay
Cell migration was measured in SPLInsert™ Hanging
8.0-µm pore polycarbonate membrane inserts (SPL Life Sciences).
Vector control or ARID1A-overexpressing cells were seeded
into the insert at a density of 2×104 cells/well in
serum-free medium (upper chamber). The insert was then transferred
into a 24-well plate containing culture medium supplemented with
10% FBS (lower chamber). After incubation in a humidified
atmosphere at 37°C for 24 h, cells remaining on the surface of the
upper chamber that had not penetrated the filter were removed with
a cotton swab. The cells that had migrated to the lower surface of
the insert were fixed with 4% paraformaldehyde for 2 min at room
temperature, followed by a 20-min permeabilization with 100%
methanol at room temperature. Subsequently, the samples were washed
with PBS and stained with 0.4% crystal violet dye for 15 min at
room temperature. The migrated cells were observed and images were
captured under a bright-field microscope. The number of migrating
cells in each chamber was quantified by counting from at least four
fields and three experiments, using ImageJ software analysis
(26).
Statistical analysis
Three independent experiments were performed.
Results are presented as the mean ± standard deviation or mean ±
standard error of the mean. Statistical analysis was conducted
using GraphPad Prism version 9.5.1 (Dotmatics). To test for
differences in ARID1A protein expression, a paired t-test was used
to compare variance in cancerous and adjacent non-cancerous areas
from the same CRC tissues. Differences between two independent
groups were analyzed using Student's t-test, whereas multiple
comparisons between >2 groups were assessed using one-way
analysis of variance and Tukey's post hoc test. P<0.05 was
considered to indicate a statistically significant difference.
Results
ARID1A exhibits a low expression in
human CRC tissues
Since mutations in ARID1A have been
demonstrated to result in the absence of protein expression in
tumors (11–15), the expression of ARID1A in CRC
tissues was initially investigated. IHC was employed to analyze the
expression levels of ARID1A in the cancerous regions (n=20), as
well as in the corresponding adjacent non-cancerous regions (n=20)
of tissues. All CRC tissues exhibited ARID1A expression in adjacent
normal mucosal epithelial cells, with strong, moderate and weak
positive staining observed in 11 cases (55%), 8 cases (40%) and 1
case (5%), respectively. By contrast, ARID1A expression was
detected in cancerous regions, with strong, moderate and weak
positive staining observed in 1 case (5%), 14 cases (70%) and 5
cases (25%), respectively. These data revealed that ARID1A
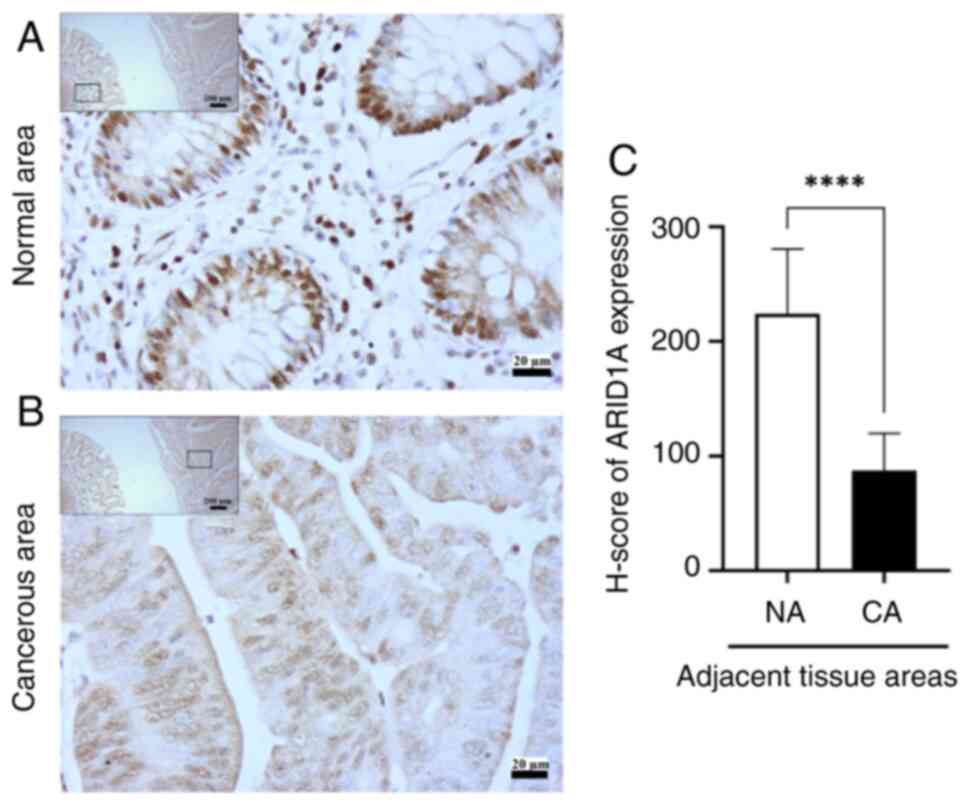
exhibited a lower expression in the cancerous regions (Fig. 1B) than in the adjacent normal areas
(Fig. 1A). Similarly, the average
H-score of ARID1A, as determined by IHC, was significantly lower in
cancerous areas than that in the adjacent non-cancerous regions
(P<0.0001; Fig. 1C). ARID1A
expression was reduced in CRC tissues, probably due to the fact
that ARID1A is frequently mutated in CRC (15). Moreover, the loss of ARID1A has
been shown to be closely linked to CRC metastasis (16). The subsequent experiments focused
on the process of EMT and its association with CRC metastasis,
specifically focusing on the involvement of ARID1A.
Overexpression of ARID1A is associated
with morphological alterations in SW48 cells
To distinguish the effect of ARID1A on an EMT-like
phenotype in CRC, SW48 cells were transduced with a lentivirus
expressing human full-length ARID1A, and its overexpression
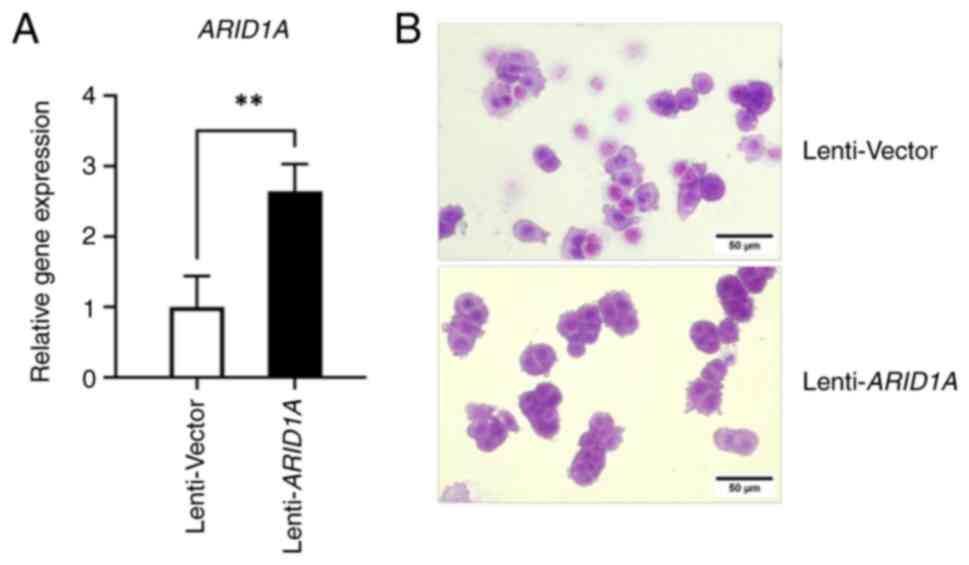
was confirmed by RT-qPCR. The mRNA expression levels of
ARID1A were increased by ~2-fold in SW48 cells that
overexpressed ARID1A, confirming the successful
establishment of ARID1A-stabilized cells (Fig. 2A). Cell morphology was observed
under a phase-contrast microscope following 0.4% crystal violet
staining. Control lentivirus-infected SW48 cells originally
exhibited an epithelial phenotype with concentric nuclei and the
cytoplasm was spread out in a uniform direction; in addition, the
shape of the cells was round, cuboid or columnar (Fig. 2B). However, SW48 cells with
ARID1A overexpression exhibited a more pronounced epithelial
cell shape and appeared to exhibit slightly enhanced cell adhesion,
resulting in a more rounded cellular structure when compared with
the control cells (Fig. 2B). These
results suggested that the changes in the physical structure of
cells overexpressing ARID1A, which resembled an epithelial
phenotype, may indicate that ARID1A has a role in inhibiting the
spread of metastatic CRC by regulating biological mechanisms that
contribute to EMT.
Overexpression of ARID1A induces
alterations in cell junction molecules and the cytoskeleton in SW48
cells
Following the induction of ARID1A
overexpression, there were notable alterations in the morphology of
SW48 cells. Since the EMT process has been associated with a
profound reorganization of the cytoskeleton, in order to weaken
cell-cell attachment and strengthen cell-matrix adhesions (27), the present study observed the
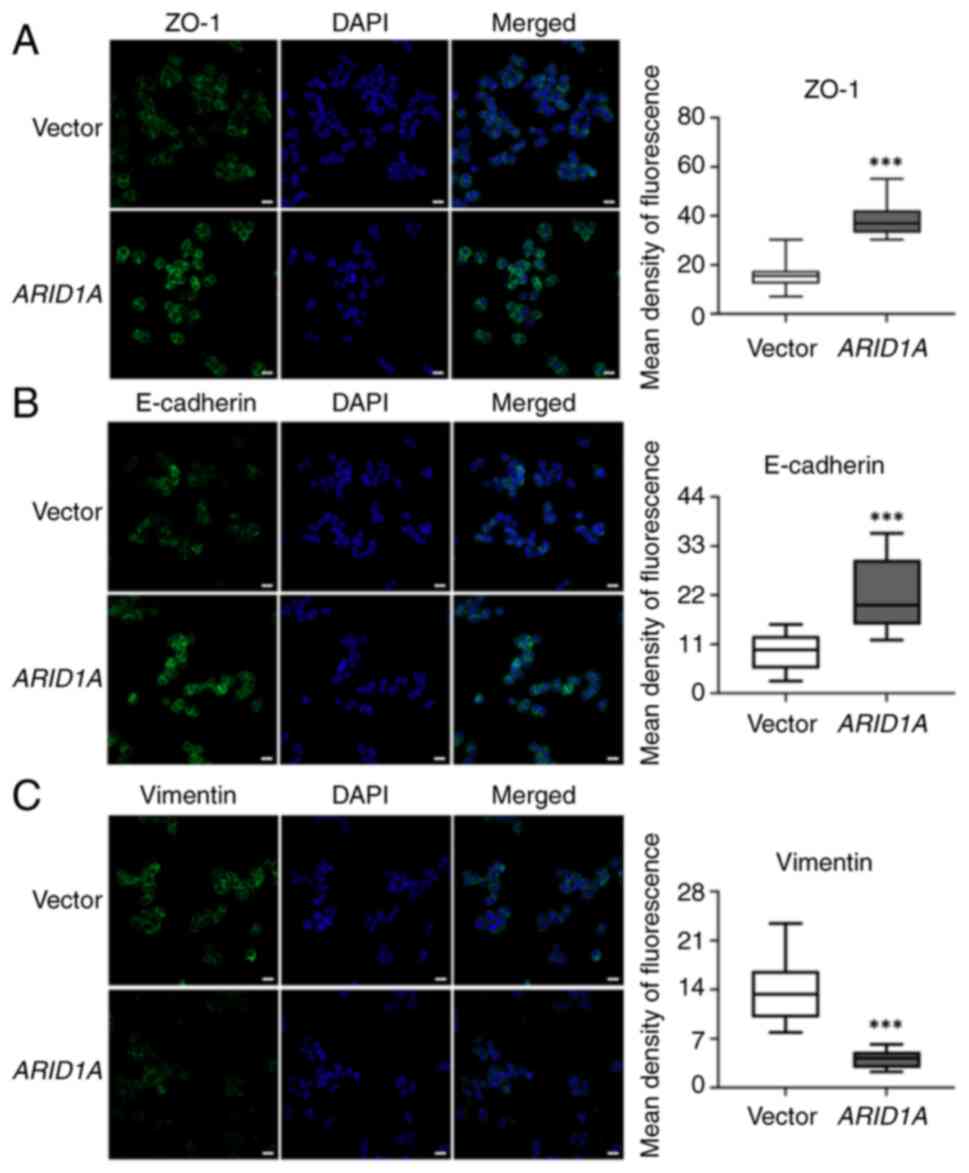
levels of ZO-1, E-cadherin and vimentin in SW48 cells
overexpressing ARID1A. Immunofluorescence staining was used
to determine the expression of ZO-1, E-cadherin and vimentin, and
to evaluate their location in SW48 cells; quantitative analysis of
immunofluorescence intensity is displayed in box-and-whisker plots.
The results showed that ZO-1 and E-cadherin were present in the
cell membrane, and were distinctly increased in
ARID1A-overexpressing cells (Fig. 3A and B). By contrast, vimentin was
detected in the cytoplasm and exhibited a decrease in cells with
ARID1A overexpression (Fig.
3C). The expression levels of these proteins indicated that
overexpression of ARID1A may affect cell shape remodeling,
potentially resulting in suppressed EMT characteristics.
ARID1A overexpression influences the
mRNA expression levels of EMT-related genes in SW48 cells
Subsequently, the present study explored the
potential association between these changes and modulation of the
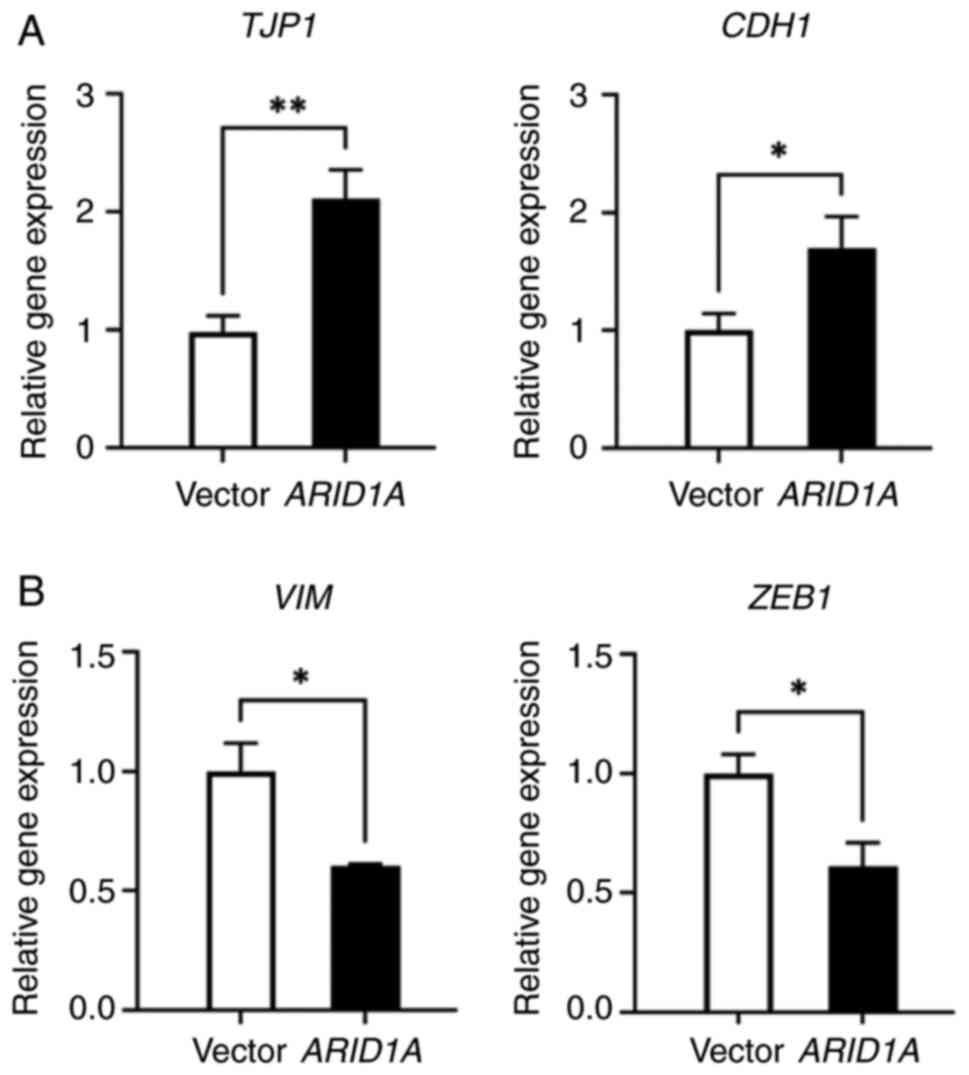
EMT pathway in CRC cells mediated by ARID1A. First, the expression
levels of the epithelial cell markers, TJP1/ZO-1 and
CDH1/E-cadherin, and the mesenchymal markers,
VIM/vimentin and ZEB1, were detected in the cells
with ARID1A overexpression. RT-qPCR was carried out to
detect the expression levels of these EMT-related genes in
ARID1A-overexpressing SW48 cells (Fig. 4). The mRNA expression levels of the
epithelial marker genes, TJP1/ZO-1 and
CDH1/E-cadherin, were obviously upregulated (Fig. 4A). By contrast, there was a notable
decrease in the expression levels of VIM/vimentin and
ZEB1, both of which are considered mesenchymal marker genes
(Fig. 4B). These RT-qPCR results
demonstrated that ARID1A may negatively regulate the EMT process by
influencing the expression of EMT-related genes in CRC.
ARID1A inhibits the motility and
migration of SW48 cells
The progression of cancer is driven by cancer cell
migration, which is a functional hallmark of EMT. To observe the
effects of ARID1A overexpression on EMT-associated
phenotypes, wound healing and cell migration assays were performed.
The bright-field images from the wound healing experiment indicated
that the motility of ARID1A-overexpressing cells was much
slower when compared with control cells at each time point
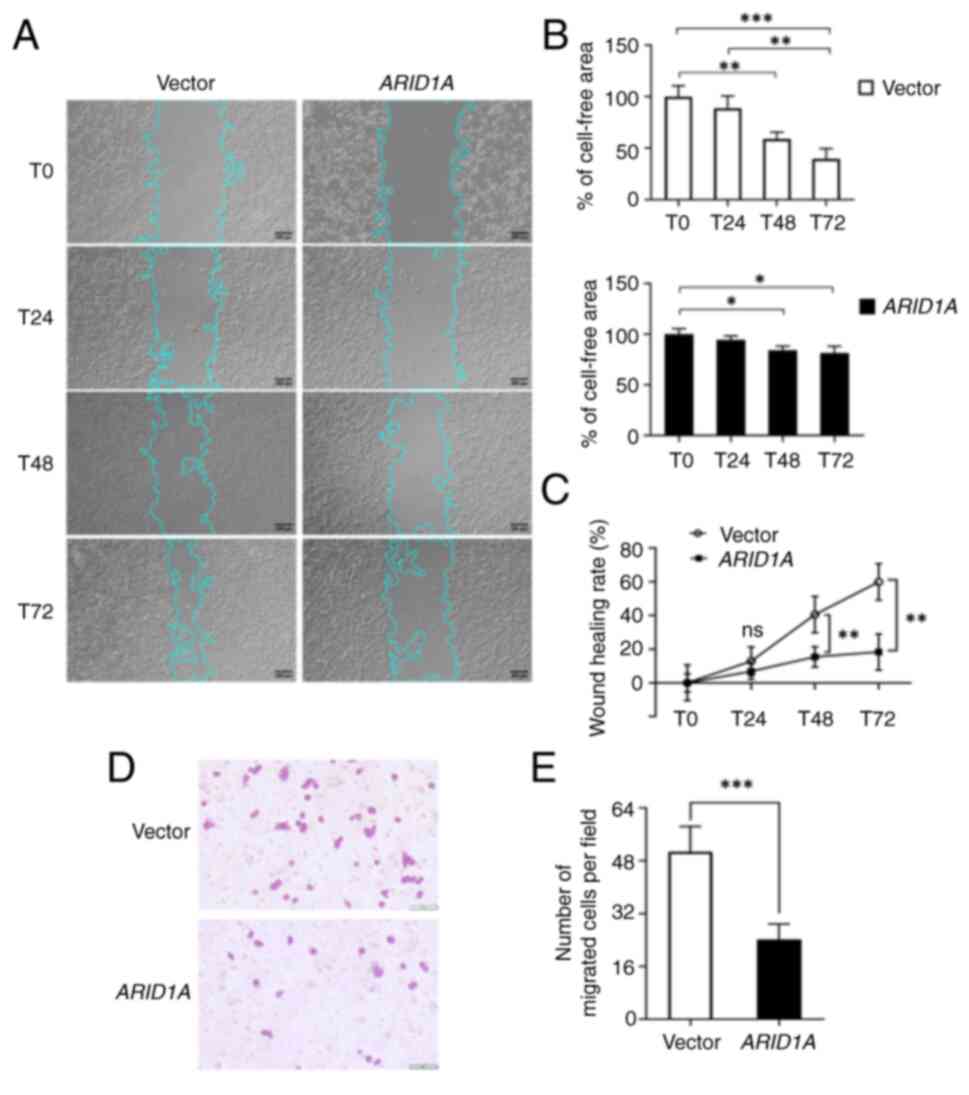
(Fig. 5A). The cell-free area in
control cells remained at 58.83±6.68% at T48 and 39.74±9.78% at T72
(Fig. 5B). In cells overexpressing
ARID1A, the cell-free area was 84.44±3.77 and 81.38±6.71% at
T48 and T72, respectively (Fig.
5B). In addition, the cell wound closure rate was calculated as
a reduction of cell-free area. As shown in Fig. 5C, the wound closure rate from T0 to
T72 in the vector control group was obviously increased, whereas
SW48 cells overexpressing ARID1A did not show a different
wound closure rate over time (Fig.
5C). The results of the wound healing assay indicated that the
motility of SW48 cells was markedly inhibited by ARID1A
overexpression. Additionally, the Transwell cell migration assay
revealed that ARID1A overexpression attenuated the migratory
abilities of SW48 cells (Fig. 5D)
and caused a significant reduction in the number of migrated cells
(Fig. 5E). The wound healing and
cell migration assays demonstrated that the strength of cell
adhesion in SW48 cells overexpressing ARID1A may be due to
ARID1A obstructing cell motility and migration related to EMT,
which is a hallmark of malignancy.
Discussion
Recent studies have reported on the discovery of
novel biomarkers and molecular targets in various types of cancer,
with a particular emphasis on innovative diagnostic methods
(28), the underlying molecular
processes of cancer (29–33) and genetic changes (5,6,13).
Dysregulation in gene expression serves a crucial role in cell
proliferation and is a significant contributor to tumor
development. Consequently, focusing on particular genes that are
associated with CRC could offer a promising therapeutic strategy
for its treatment (15). The
ARID1A gene has been categorized as a novel tumor suppressor
due to the association observed between ARID1A/BAF250a
expression and various cancer types, including CRC (11–15).
ARID1A has been identified as a highly mutated gene in CRC,
with loss of its expression becoming increasingly significant as
the TNM stage progresses, suggesting a strong association between
ARID1A loss in CRC and the advancement of tumor progression and
metastasis (16). Metastasis is a
leading factor in the mortality of patients with CRC, and EMT is
recognized as a pivotal process in cancer metastasis. The silencing
of ARID1A has been reported to enhance migratory
capabilities by promoting EMT in breast and gastric cancer cells
(22,23). Although a previous study has
reported that ARID1A knockdown results in upregulation of
VIM and downregulation of CDH1 in CRC cells (34), the EMT mechanisms that underlie the
involvement of ARID1A in CRC metastasis have not been well
characterized. The present study confirmed the low expression of
ARID1A in CRC tissues, which was not assessed in previous research.
Additionally, this study demonstrated the function of ARID1A in the
processes of EMT and its association with ZEB1, a key
transcriptional regulator of EMT in CRC. The findings showed that
ARID1A overexpression can obstruct the EMT-like phenotype by
increasing the expression of epithelial markers and decreasing
those of mesenchymal markers, thereby inhibiting the EMT process of
CRC cells and preventing CRC cell migration.
The initial findings of the current study indicated
that the expression of ARID1A in human CRC tissue samples was lower
compared with that in adjacent normal tissues. This confirms that
ARID1A expression is commonly lost in human CRC, as reported in
previous studies (16,35,36).
In addition, the reduction of ARID1A has been associated with
clinicopathological significance in patients with CRC. The loss of
ARID1A has been reported to be significantly correlated with age,
sex, location and tumor size (36), as well as distant metastasis and
advanced TNM stage (16). Numerous
studies have attempted to explore the fundamental molecular process
behind the reduction of ARID1A expression in CRC. For example, loss
of ARID1A expression has been shown to be associated with mismatch
repair deficiency and somatic hypermethylation, as a key driver
event in CRC (36). In addition,
the deletion of Arid1a in mice has led to the formation of
invasive adenocarcinoma in the colon (37). However, the molecular mechanisms
underlying the role of ARID1A in CRC metastasis remain to be
elucidated. EMT is a crucial element in the process of tumor
metastasis and invasion, playing a pivotal role in the progression
of cancer, notably in CRC (21).
Hence, the present study focused on assessing the role of ARID1A in
the EMT pathway, which presents an intriguing area of research in
the context of cell migration, a metastatic feature of cancer.
A recent study revealed diverse mRNA expression
levels of ARID1A in different CRC cell lines, ranging from
high levels in HCT116 and HT-29/219 cells to nearly undetectable
levels in SW48 cells (35).
Therefore, this facilitated the design of the ARID1A
overexpression experimental model in SW48 cells. Transduction of a
lentivirus containing the human full-length ARID1A sequence
had a significant impact on cellular morphological alterations,
leading to a greater prevalence of cells with an epithelial
phenotype and enhanced cell-to-cell contact. Since epithelial cells
exhibit polarity from their apical to basal surfaces, establishing
adhesion and communication among themselves via specialized
intercellular junctions (38), the
present study revealed that the tight junction molecule, ZO-1, and
the adherens junction molecule, E-cadherin (epithelial cadherin),
were significantly increased in cells overexpressing ARID1A.
By contrast, the intermediate filament protein, vimentin, was
decreased. Key events in EMT include the dissolution of epithelial
cell-cell junctions, loss of apical-basal polarity, reorganization
of the cytoskeletal architecture and alterations in cellular
morphology (27,30,39).
Therefore, the present results indicated that the overexpression of
ARID1A could potentially inhibit EMT-like characteristics.
These results were confirmed by the detection of the mRNA
expression levels of EMT-related genes.
ARID1A-overexpressing cells exhibited upregulation of the
epithelial markers TJP1/ZO-1 and CDH1/E-cadherin,
whereas the mesenchymal markers VIM/vimentin and ZEB1
were downregulated. The EMT process is distinguished by the loss of
epithelial markers, such as E-cadherin, and the gain of mesenchymal
markers, such as vimentin (40,41).
Several studies have reported that as most types of cancer
progresses, there is an observed increase in vimentin levels
(31,41), while E-cadherin levels tend to
decrease (41,42). Regarding the present data,
ARID1A overexpression enhanced E-cadherin levels and reduced
vimentin levels, indicating the functional role of ARID1A in
negatively regulating the EMT process. Consequently, the migratory
capabilities of cells overexpressing ARID1A were
suppressed.
The present findings are consistent with the
findings of previous reports. In a previous study, ARID1A
silencing was shown to induce E-cadherin downregulation, enhancing
gastric cancer cell migration and invasion (23). Baldi et al (34) reported that ARID1A
deficiency could promote cell proliferation and migration via
VIM activation and CDH1 suppression in colon cancer.
In addition to E-cadherin and vimentin, other EMT-related markers,
TJP1/ZO-1 and ZEB1 were also revealed to be
associated with ARID1A-regulated EMT. ZO-1 is essential for tight
junction formation. The tight junctions of cells disintegrate
during EMT, accompanied by reduced levels of ZO-1 expression
(38). Zhang et al
(43) detected a decline in ZO-1
expression in hepatocellular carcinoma. Accordingly, overexpression
of ZO-1 suppressed HepG2 cell proliferation. ZEB1 has been
recognized as a key transcriptional regulator of EMT, and
inhibiting ZEB1 has been reported to reduce the migration and
invasion of prostate cancer (29).
Despite these findings, to the best of our knowledge, the present
study is the first to report that ARID1A overexpression may
inhibit CRC cell migration through the suppression of the EMT
process by enhancing ZO-1 expression while decreasing ZEB1
expression levels. Recently, our proteomic analysis investigation
demonstrated that the overexpression of ARIDA had a notable
impact on the modification of multiple proteins associated with the
Wnt signaling pathway in CRC cells (44). The activation of Wnt signaling in
colon cancer is responsible for driving tumorigenesis and
facilitating advanced metastasis through the promotion of the EMT
program (32). However, the
present study did not investigate the involvement of the Wnt
signaling pathway in the regulation of EMT in CRC. Future research
involving gene knockdown experiments, the use of specific
inhibitors against the Wnt signaling pathway, and
immunohistochemistry for Wnt-related proteins is required to
enhance the understanding of the mechanisms observed in the present
study.
In conclusion, the findings of the present study
indicated that expression of the tumor suppressor ARID1A is
frequently lost in CRC tissues. The in vitro experiments
demonstrated that ARID1A may have the potential to counteract
EMT-like characteristics and cell migration by modifying the
expression of EMT-related genes. In simpler terms, ARID1A might
impede the EMT process in CRC, leading to the inhibition of
malignant progression. Consequently, additional investigations are
necessary to identify which EMT pathways are regulated by ARID1A,
and to establish a stronger connection between the clinical
treatment of CRC and the utilization of ARID1A as a biomarker and
therapeutic target for CRC metastasis.
Acknowledgements
The authors would like to thank Mr. Olalekan Isreal
Aiikulola (Faculty of Medical Science, Naresuan University,
Phitsanulok, Thailand) for providing an English editing service.
Additionally, the authors would like to thank Dr Ratirat Samol
(Unit of Pathology, Sawanpracharak Hospital, Nakhon Sawan,
Thailand), for providing the FFPE tissue blocks.
Funding
This research received funding support from the National
Science, Research, and Innovation Fund via the Program Management
Unit for Human Resources & Institutional Development, Research
and Innovation (grant no. B05F640168).
Availability of data and materials
The data generated in the present study may be
requested from the corresponding author.
Authors' contributions
SW contributed to the conception and design of the
study, conducted experiments, performed data analysis and
visualization, and was a major contributor to manuscript writing.
SA, KS and WM participated in experiments and data analysis. NS
contributed to the conception and design of the study, data
analysis, supervision, funding acquisition and manuscript editing.
SW and NS confirm the authenticity of all the raw data. All authors
read and approved the final version of the manuscript.
Ethics approval and consent to
participate
All procedures performed involving human
participants adhered to the ethical guidelines set by the
institutional and/or national research committee, as well as The
1964 Declaration of Helsinki and its subsequent amendments, or
equivalent ethical standards. The study received authorization from
the Human Ethics Review Board of Sawanpracharak Hospital (approval
no. 16/2560) and the Naresuan University Ethical Committee for
Human Research (approval no. P1-0191/2564; certificate of approval
no. 178/2021). Written informed consent was obtained from all
subjects involved in the study.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Siegel RL, Miller KD, Wagle NS and Jemal
A: Cancer statistics, 2023. CA Cancer J Clin. 73:17–48. 2023.
View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Siegel RL, Wagle NS, Cercek A, Smith RA
and Jemal A: Colorectal cancer statistics, 2023. CA Cancer J Clin.
73:233–254. 2023. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Brenner H, Heisser T, Cardoso R and
Hoffmeister M: Reduction in colorectal cancer incidence by
screening endoscopy. Nat Rev Gastroenterol Hepatol. 21:125–133.
2024. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Bhupender S and Chhikara KP: Global Cancer
Statistics 2022: The trends projection analysis. Chem Biol Lett.
10:4512023.
|
|
5
|
Biller LH and Schrag D: Diagnosis and
treatment of metastatic colorectal cancer: A review. JAMA.
325:669–685. 2021. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Malki A, ElRuz RA, Gupta I, Allouch A,
Vranic S and Al Moustafa AE: Molecular mechanisms of colon cancer
progression and metastasis: Recent insights and advancements. Int J
Mol Sci. 22:1302020. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Vogelstein B, Fearon ER, Hamilton SR, Kern
SE, Preisinger AC, Leppert M, Smits AM and Bos JL: Genetic
alterations during colorectal-tumor development. N Engl J Med.
319:525–532. 1988. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Wilson BG and Roberts CWM: SWI/SNF
nucleosome remodellers and cancer. Nat Rev Cancer. 11:481–492.
2011. View
Article : Google Scholar : PubMed/NCBI
|
|
9
|
Reisman D, Glaros S and Thompson E: The
SWI/SNF complex and cancer. Oncogene. 28:1653–1668. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Shain AH and Pollack JR: The spectrum of
SWI/SNF mutations, ubiquitous in human cancers. PLoS One.
8:e551192013. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Wiegand KC, Shah SP, Al-Agha OM, Zhao Y,
Tse K, Zeng T, Senz J, McConechy MK, Anglesio MS, Kalloger SE, et
al: ARID1A mutations in endometriosis-associated ovarian
carcinomas. N Engl J Med. 363:1532–1543. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Mamo A, Cavallone L, Tuzmen S, Chabot C,
Ferrario C, Hassan S, Edgren H, Kallioniemi O, Aleynikova O,
Przybytkowski E, et al: An integrated genomic approach identifies
ARID1A as a candidate tumor-suppressor gene in breast cancer.
Oncogene. 31:2090–2100. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Guichard C, Amaddeo G, Imbeaud S, Ladeiro
Y, Pelletier L, Maad IB, Calderaro J, Bioulac-Sage P, Letexier M,
Degos F, et al: Integrated analysis of somatic mutations and focal
copy-number changes identifies key genes and pathways in
hepatocellular carcinoma. Nat Genet. 44:694–698. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Streppel MM, Lata S, DelaBastide M,
Montgomery EA, Wang JS, Canto MI, Macgregor-Das AM, Pai S, Morsink
FH, Offerhaus GJ, et al: Next-generation sequencing of endoscopic
biopsies identifies ARID1A as a tumor-suppressor gene in Barrett's
esophagus. Oncogene. 33:347–357. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Muzny DM, Bainbridge MN, Chang K, Dinh HH,
Drummond JA, Fowler G, Kovar CL, Lewis LR, Morgan MB, Newsham IF,
et al: Comprehensive molecular characterization of human colon and
rectal cancer. Nature. 487:330–337. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Wei XL, Wang DS, Xi SY, Wu WJ, Chen DL,
Zeng ZL, Wang RY, Huang YX, Jin Y, Wang F, et al: Clinicopathologic
and prognostic relevance of ARID1A protein loss in colorectal
cancer. World J Gastroenterol. 20:18404–18412. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Ye J, Zhou Y, Weiser MR, Gönen M, Zhang L,
Samdani T, Bacares R, DeLair D, Ivelja S, Vakiani E, et al:
Immunohistochemical detection of ARID1A in colorectal carcinoma:
Loss of staining is associated with sporadic microsatellite
unstable tumors with medullary histology and high TNM stage. Hum
Pathol. 45:2430–2436. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Toiyama Y, Yasuda H, Saigusa S, Tanaka K,
Inoue Y, Goel A and Kusunoki M: Increased expression of Slug and
Vimentin as novel predictive biomarkers for lymph node metastasis
and poor prognosis in colorectal cancer. Carcinogenesis.
34:2548–2557. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Ye X and Weinberg RA:
Epithelial-Mesenchymal plasticity: A central regulator of cancer
progression. Trends Cell Biol. 25:675–686. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Brabletz T, Kalluri R, Nieto MA and
Weinberg RA: EMT in cancer. Nat Rev Cancer. 18:128–134. 2018.
View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Gurzu S, Silveanu C, Fetyko A, Butiurca V,
Kovacs Z and Jung I: Systematic review of the old and new concepts
in the epithelial-mesenchymal transition of colorectal cancer.
World J Gastroenterol. 22:6764–6775. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Wang T, Gao X, Zhou K, Jiang T, Gao S, Liu
P, Zuo X and Shi X: Role of ARID1A in epithelial-mesenchymal
transition in breast cancer and its effect on cell sensitivity to
5-FU. Int J Mol Med. 46:1683–1694. 2020.PubMed/NCBI
|
|
23
|
Yan HB, Wang XF, Zhang Q, Tang ZQ, Jiang
YH, Fan HZ, Sun YH, Yang PY and Liu F: Reduced expression of the
chromatin remodeling gene ARID1A enhances gastric cancer cell
migration and invasion via downregulation of E-cadherin
transcription. Carcinogenesis. 35:867–876. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Livak KJ and Schmittgen TD: Analysis of
relative gene expression data using real-time quantitative PCR and
the 2(−Delta Delta C(T)) method. Methods. 25:402–408. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Tian X, Wei Z, Wang J, Liu P, Qin Y and
Zhong M: MicroRNA-429 inhibits the migration and invasion of colon
cancer cells by targeting PAK6/cofilin signaling. Oncol Rep.
34:707–714. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Schneider CA, Rasband WS and Eliceiri KW:
NIH Image to ImageJ: 25 years of image analysis. Nat Methods.
9:671–675. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Yilmaz M and Christofori G: EMT, the
cytoskeleton, and cancer cell invasion. Cancer Metastasis Rev.
28:15–33. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Wang R, Yang S, Wang M, Zhou Y, Li X, Chen
W, Liu W, Huang Y, Wu J and Cao J: A sustainable approach to
universal metabolic cancer diagnosis. Nat Sustainability.
7:602–615. 2024. View Article : Google Scholar
|
|
29
|
Graham TR, Zhau HE, Odero-Marah VA,
Osunkoya AO, Kimbro KS, Tighiouart M, Liu T, Simons JW and O'Regan
RM: Insulin-like growth factor-I-dependent up-regulation of ZEB1
drives epithelial-to-mesenchymal transition in human prostate
cancer cells. Cancer Res. 68:2479–2488. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Leggett SE, Hruska AM, Guo M and Wong IY:
The epithelial-mesenchymal transition and the cytoskeleton in
bioengineered systems. Cell Commun Signal. 19:322021. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Niknami Z, Eslamifar A, Emamirazavi A,
Ebrahimi A and Shirkoohi R: The association of vimentin and
fibronectin gene expression with epithelial-mesenchymal transition
and tumor malignancy in colorectal carcinoma. EXCLI J.
16:1009–1017. 2017.PubMed/NCBI
|
|
32
|
Zhao H, Ming T, Tang S, Ren S, Yang H, Liu
M, Tao Q and Xu H: Wnt signaling in colorectal cancer: Pathogenic
role and therapeutic target. Mol Cancer. 21:1442022. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Yang Q, Huang W, Hsu JC, Song L, Sun X, Li
C, Cai W and Kang L: CD146-targeted nuclear medicine imaging in
cancer: State of the art. View (Beijing). 4:202200852023.PubMed/NCBI
|
|
34
|
Baldi S, Zhang Q, Zhang Z, Safi M, Khamgan
H, Wu H, Zhang M, Qian Y, Gao Y, Shopit A, et al: ARID1A
downregulation promotes cell proliferation and migration of colon
cancer via VIM activation and CDH1 suppression. Cell Mol Med.
26:5984–5997. 2022. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Erfani M, Hosseini SV, Mokhtari M, Zamani
M, Tahmasebi K, Alizadeh Naini M, Taghavi A, Carethers JM, Koi M,
Brim H, et al: Altered ARID1A expression in colorectal cancer. BMC
Cancer. 20:3502020. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Chou A, Toon CW, Clarkson A, Sioson L,
Houang M, Watson N, DeSilva K and Gill AJ: Loss of ARID1A
expression in colorectal carcinoma is strongly associated with
mismatch repair deficiency. Hum Pathol. 45:1697–1703. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Mathur R, Alver BH, San Roman AK, Wilson
BG, Wang X, Agoston AT, Park PJ, Shivdasani RA and Roberts CW:
ARID1A loss impairs enhancer-mediated gene regulation and drives
colon cancer in mice. Nat Genet. 49:296–302. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Huang RYJ, Guilford P and Thiery JP: Early
events in cell adhesion and polarity during epithelial-mesenchymal
transition. J Cell Sci. 125:4417–4422. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Lamouille S, Xu J and Derynck R: Molecular
mechanisms of epithelial-mesenchymal transition. Nat Rev Mol Cell
Biol. 15:178–196. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Gout S and Huot J: Role of cancer
microenvironment in metastasis: Focus on colon cancer. Cancer
Microenviron. 1:69–83. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Xiao S, Liu L, Lu X, Long J, Zhou X and
Fang M: The prognostic significance of bromodomain PHD-finger
transcription factor in colorectal carcinoma and association with
vimentin and E-cadherin. J Cancer Res Clin Oncol. 141:1465–1474.
2015. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Kolijn K, Verhoef EI and van Leenders GJ:
Morphological and immunohistochemical identification of
epithelial-to-mesenchymal transition in clinical prostate cancer.
Oncotarget. 6:24488–24498. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Zhang X, Wang L, Zhang H, Tu F, Qiang Y
and Nie C: Decreased expression of ZO-1 is associated with tumor
metastases in liver cancer. Oncol Lett. 17:1859–1864.
2019.PubMed/NCBI
|
|
44
|
Aluksanasuwan S, Somsuan K, Wanna-Udom S,
Roytrakul S, Morchang A, Rongjumnong A and Sakulsak N: Proteomic
insights into the regulatory function of ARID1A in colon cancer
cells. Oncology Letters. 28:3922024. View Article : Google Scholar : PubMed/NCBI
|