Introduction
Skeletal muscles are important for maintaining the
stability of the human body. Myoblast proliferation and
differentiation are complex biological processes during which
myoblasts begin to express myogenic factors such as myogenic factor
4, myoblast determination protein and myogenin. During the late
stage of muscle differentiation, cells upregulate myosin-heavy
chain (MHC) and form a multinuclear structure (1). As they proliferate and differentiate
into myotubes during in vitro culture, the mouse myoblast
cell line C2C12 is widely employed to investigate myoblast
differentiation and associated biochemical pathways (2). Using C2C12 cells, a recent study
found that after differentiation, genes involved in muscle
contraction, autophagy and sarcomeres were more active and that
autophagy increased during muscle cell differentiation, as revealed
by LC3-I to LC3-II conversion (3).
Moreover, chloroquine-mediated inhibition of autophagy can suppress
the expression of muscle-specific genes and muscle fiber formation
(3), indicating that autophagy has
a positive role in muscle cell differentiation and fusion.
Muscle disorders and severe chronic diseases, such
as cancer cachexia, HIV/AIDS, chronic heart failure, chronic
obstructive pulmonary disease and sepsis, can cause muscle atrophy
by decreasing muscle mass and function (4–6). In
the short term, muscle atrophy-induced muscle damage causes
discomfort because of the inability to move freely, whereas in the
long term, it reduces the quality of life and increases mortality.
Most cases of muscle atrophy result from decreased protein
synthesis or increased protein degradation via the
ubiquitin-proteasome system and autophagy (7). The most important markers of muscle
atrophy are the ubiquitin ligases MuRF-1 and atrogin-1 (8). Conversely, excessive muscle
formation, characterized by the over-proliferation and
differentiation of muscle cells, can lead to muscle-related
diseases. For instance, myostatin-related muscle hypertrophy can be
attributed to mutations in myostatin, which cause abnormal muscle
growth and increased muscle mass and strength (9). Rhabdomyosarcoma is a malignant tumor
of the skeletal muscle that disrupts normal muscle formation and
leads to abnormal muscle cell proliferation (10). Therefore, research on inhibiting
muscle cell proliferation and differentiation is crucial for
understanding, preventing and treating these conditions.
Ginkgolic acid (GA), isolated from the leaves and
seed coats of Ginkgo biloba (11), possesses numerous pharmacological
properties, including antitumor, antibacterial, anti-HIV and
anti-inflammatory effects (12–15).
For example, GA can inhibit lipogenic signaling, thereby delaying
pancreatic cancer development (16). GA also activates denosine
monophosphate (AMP)-activated protein kinase (AMPK), inhibits colon
cancer cell invasiveness (17) and
suppresses the migration and metastasis of gastric, breast and lung
cancer cells (18–20). By directly binding to the small
ubiquitin-related modifier (SUMO)-activating enzyme E1, GA was
found to block the formation of the E1-SUMO intermediate, thereby
inhibiting SUMOylation (21).
However, the effects of GA on myoblast
differentiation remain to be elucidated. The present study examined
the effects of GA on the mouse myoblast cell line C2C12 and
investigated the underlying molecular mechanisms. Accordingly, it
aimed to understand how GA influences muscle cell physiology and
provide foundational data for the development of future treatments
for muscle-related diseases.
Materials and methods
Cell line and reagents
C2C12 cells were purchased from American Type
Culture Collection and cultured in growth medium (GM; DMEM
supplemented with 15% fetal bovine serum and 1%
streptomycin/penicillin all from Welgene, Inc.) at 37°C, in a
humidified incubator (with 5% CO2) or differentiation
medium [DM; GM was replaced with DMEM supplemented with 2% horse
serum (Gibco; Thermo Fisher Scientific, Inc.) for 1–3 days]. GA was
purchased from MilliporeSigma. Dimethyl sulfoxide (DMSO) was used
as the control.
MTT assay
Cells (in 100 µl of GM) were seeded in 96-well
plates, cultured for 24 h and then treated with various GA
concentrations for 24 and 48 h. Next, 20 µl of MTT stock solution
(MilliporeSigma) was added to each well, followed by incubation for
2.5 h at 37°C. The medium was then removed and DMSO was added to
each well, followed by cell viability assessment by measuring
absorbance at 570 nm on an INNO microplate reader (LTEK Co.,
Ltd.).
Colony formation assay
C2C12 cells were seeded into six-well plates at a
density of 1,000 cells/well and cultured at 37°C for 24 h. The
medium was then replaced with fresh medium containing various GA
concentrations, followed by culture at 37°C for 4 days until
visible colonies containing at least 50 cells were observed.
Colonies were visualized using crystal violet staining (cat. no.
ab232855; Abcam) according to the manufacturer's protocol. Briefly,
the cells were fixed with 100% methanol at −20°C for 20 min,
stained with 2% crystal violet solution for 20 min at room
temperature, washed and air-dried. Crystal violet-stained cells
were dissolved and absorbance was read at 570 nm on an INNO
microplate reader (LTEK Co., Ltd.).
Apoptosis analysis
Apoptosis was evaluated using the Muse Annexin V and
Dead Cell Kit (cat. no. MCH100105; Luminex Corporation). This assay
kit detects PS on apoptotic cell surfaces using fluorescently
labeled (PE) Annexin V, along with the dead cell marker 7-AAD,
which stains cells with compromised membranes. C2C12 cells were
cultured in six-well plates at a density of 5×104
cells/well and treated with various GA concentrations for 24 or 48
h. The cells were detached using trypsin and resuspended in a fresh
medium. Next, 100 µl of Muse Annexin V and Dead Cell reagent was
added to the cell suspension, followed by incubation for 20 min at
room temperature in the dark. Apoptosis was evaluated using a Guava
Muse Cell Analyzer (Luminex Corporation) according to the
manufacturer's instructions.
Flow cytometric Ki67 analysis
The Muse Ki67 Proliferation Kit (cat. no. MCH100114;
Luminex Corporation) was used to determine the proportion of
proliferating cells based on Ki67 expression. Briefly, the cells
were harvested and fixed with 1X fixation solution for 15 min at
room temperature. Subsequently, the cells were washed with 1X assay
buffer, resuspended and treated with the permeabilization solution
for 15 min. The cells were then washed and incubated with Muse Hu
IgG1-PE (1:20; cat. no. 4700-1669; Luminex Corporation) or Muse Hu
Ki67-PE (1:20; cat. no. 4700-1667; Luminex Corporation) antibodies
for 30 min, followed by flow cytometry using a Guava Muse Cell
Analyzer (Luminex Corporation).
Flow cytometric cell cycle
analysis
The Muse Cell Cycle Kit (cat. no. MCH100106; Luminex
Corporation) was used for cell cycle analysis. Briefly, cells were
centrifuged at 300 × g for 5 min at room temperature and fixed
using 70% ethanol at −20°C for 3 h. Next, the cells were stained
with the Muse Cell Cycle Reagent (Luminex Corporation) and
incubated in the dark for 30 min at room temperature, followed by
flow cytometry on a Guava Muse Cell Analyzer (Luminex
Corporation).
Western blotting
Western blotting was performed as described
previously (22) using the
following primary antibodies: anti-MHC (1:200; cat. no. MF20;
Developmental Studies Hybridoma Bank), anti-myogenin (1:200; cat.
no. F5D; Developmental Studies Hybridoma Bank), anti-myoblast
determination protein 1 (MyoD; 1:500; cat. no. 554130; BD
Biosciences), anti-poly (ADP-ribose) polymerase (PARP; 1:1,000;
cat. no. 9542S; Cell Signaling Technology, Inc.), anti-Caspase-3
(1:1,000; cat. no. 9665S; Cell Signaling Technology, Inc.),
anti-cleaved Caspase-3 (1:1,000; cat. no. 9664S; Cell Signaling
Technology, Inc.), anti-phosphorylated (p)-MEK1/2 (1:1,000; cat.
no. 9121S; Cell Signaling Technology, Inc.), anti-phospho-p44/42
MAPK (Erk1/2; 1:1,000; cat. no. 9106s; Cell Signaling Technology,
Inc.), anti-phospho-p44/42 MAPK (1:1,000; cat. no. 4695S; Cell
Signaling Technology, Inc.), anti-Lamin B1 (1:1,000; cat. no.
13435s; Cell Signaling Technology, Inc.), anti-LC3B (1:1,000; cat.
no. 2775s; Cell Signaling Technology, Inc.), anti-β-actin (1:1,000;
cat. no. 4967S, Cell Signaling Technology), anti-MEK-1 (1:500;
sc-219), anti-HSP90 (1:500; cat. no. sc-13119) and anti-GAPDH
(1:500; cat. no. sc-166574; Santacruz Biotechnology). After
washing, the membranes were incubated with the diluted horseradish
peroxidase (HRP)-conjugated secondary antibodies (1:5,000; cat.
nos. 7074S or 7076S; Cell Signaling Technology, Inc.) for 1 h at
room temperature. Protein signals were developed and quantified
using an Azure imaging system (c280; cat. no. AC2801; Azure
Biosystems, Inc.). After initial detection, the membranes were
stripped of antibodies using a stripping buffer (cat. no. S2039;
Biosesang), followed by reblotting to detect other target proteins.
The strips were washed three times with TBS containing 0.05% Tween
before immunoblotting.
Immunofluorescence
MHC immunostaining was performed as described
previously (22). Briefly, C2C12
cells were seeded at a density of 1×104 cells per well
using circular glass coverslips (18 mm) placed in 12-well plates.
Cells were fixed in 4% paraformaldehyde for 10 min at room
temperature and permeabilized with 0.2% Triton X-100 in
phosphate-buffered saline (PBS) for 5 min at room temperature.
Subsequently, cells were blocked with 1% bovine serum albumin
(MilliporeSigma) in PBS for 30 min, followed by three washes with
0.2% Triton X-100 in PBS for 5 min each. The cells were then
incubated with an anti-MHC (1:100; cat. no. MF20; Developmental
Studies Hybridoma Bank) antibody at 4°C overnight. Afterward, the
cells were washed three times with 0.2% Triton X-100 in PBS for 5
min each and then probed with a secondary antibody (Fab2-Alexa
Fluor 488; 1:500; cat. no. 4408S; Cell Signaling Technology, Inc.)
for 1 h at room temperature. The nuclei were counterstained using
ProLong Gold Antifade Mountant with DAPI (Invitrogen; Thermo Fisher
Scientific, Inc.) for 10 min at room temperature, followed by cell
examination under a fluorescence microscope (EVOS FL Cell Imaging
System; Thermo Fisher Scientific, Inc.). Images were captured using
a 20X objective lens.
Fusion index
Nuclei were counted using the ImageJ 1.53a software
(National Institutes of Health) and the fusion index was calculated
as the ratio between the number of nuclei within each myotube and
the total number of nuclei.
Separation of nuclear and cytoplasmic
proteins
To examine extracellular signal-regulated kinase
(ERK) signaling, NE-PER Nuclear and Cytoplasmic Extraction Reagent
(cat. no. 78833; Thermo Fisher Scientific, Inc.) was used to
isolate cytoplasmic and nuclear fractions according to the
manufacturer's guidelines. Briefly, cells were washed with ice-cold
PBS, followed by the addition of cytoplasmic extraction reagent
(CER) I to the cell pellet, vortexing and incubation on ice. CER II
was then added and the cytoplasmic fraction was isolated by
centrifugation at 10,000 × g for 10 min at 4°C. Nuclei-containing
pellets were washed with ice-cold PBS, then mixed with nuclear
extraction reagent (Thermo Fisher Scientific, Inc.) by vortexing
and incubated and centrifuged at 10,000 × g for 10 min at 4°C to
obtain the nuclear fraction.
Flow cytometric autophagy
assessment
The Muse Cell Analyzer and Muse Autophagy
LC3-Antibody Based Kit (MCH200109; Luminex Corporation) were used
to assess GA-mediated autophagy, according to the manufacturer's
instructions. Briefly, cultured untreated or treated cells were
detached and incubated with an anti-LC3-Alexa Fluor 555 antibody
and 1X Autophagy Reagent for 30 min in the dark at room
temperature. The cells were then resuspended in 1X assay buffer,
followed by flow cytometry using a Muse Cell Analyzer (Luminex
Corporation). Autophagy induction is presented as the signal ratio
between the test and control sample fluorescence.
Statistical analyses
GraphPad Prism version 8.0 (Dotmatics) was used for
the statistical analysis. Data are presented as mean ± standard
deviation. The statistical significance of the differences between
the GA-treated and control (untreated) groups was determined using
Student's unpaired t-test. Statistical differences between the
means of multiple groups were compared using one-way analysis of
variance, followed by Tukey's multiple comparison test or Dunnett's
multiple comparison test if variances were equal. P<0.05 was
considered to indicate a statistically significant difference.
Results
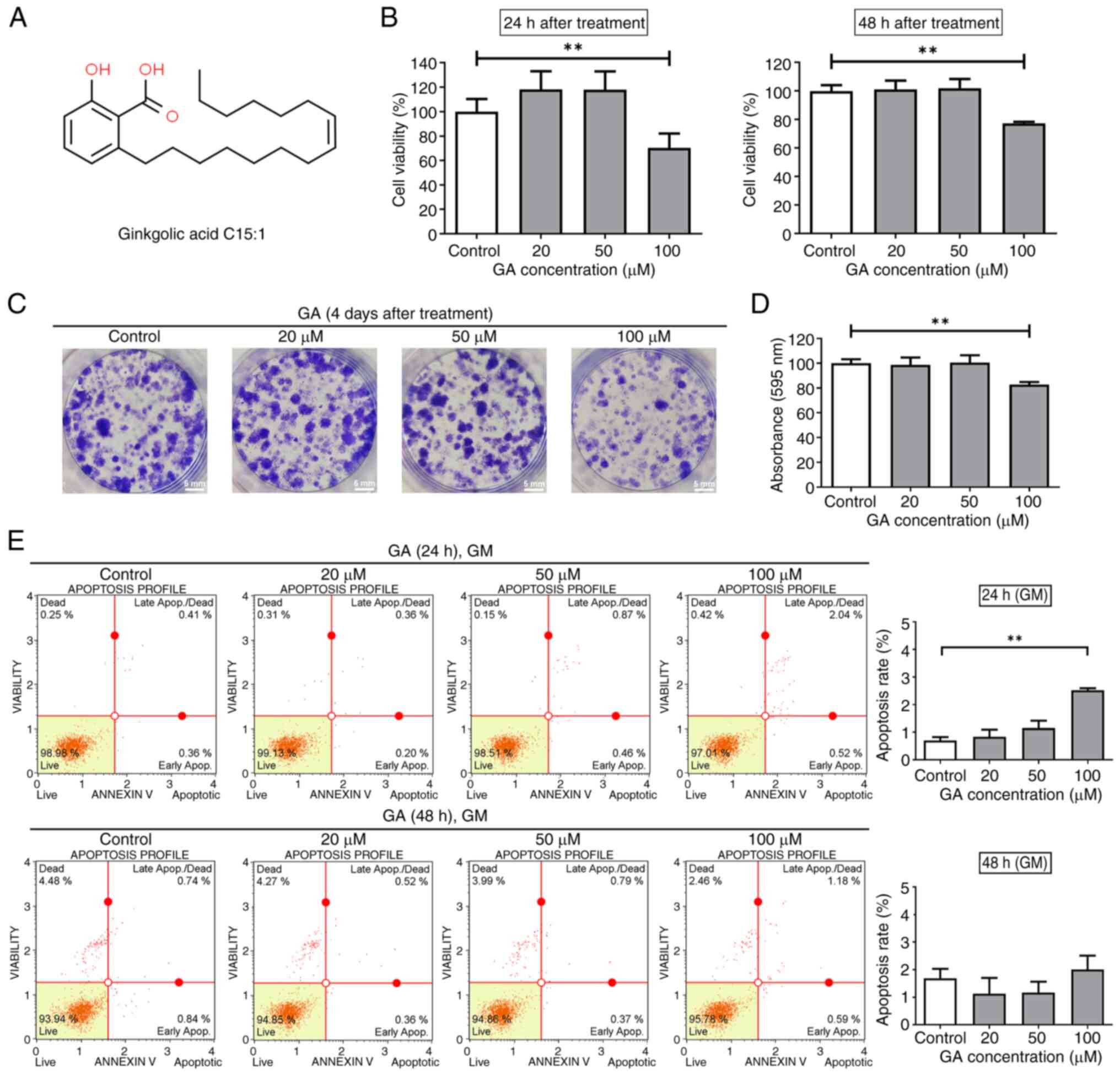
GA inhibits C2C12 cell viability and
colony formation
The present study first performed the MTT assay to
evaluate the cytotoxicity of GA on C2C12 cells treated with various
GA concentrations (0–100 µM) for 24 or 48 h (Fig. 1A). Based on the MTT assay results,
cell viability decreased significantly to 70 and 77% in cells
exposed to 100 µM GA for 24 and 48 h, respectively, as shown in
Fig. 1B (left and right graphs). A
colony formation assay was performed to examine the effect of GA on
C2C12 cell survival. Crystal violet staining revealed that colonies
treated with GA at 20, 50 and 100 µM for four days were smaller
when compared with those in the control group (Fig. 1C). Treatment with GA at 100 µM
reduced absorbance at 595 nm to 82% (Fig. 1D). These findings indicated that
treatment with GA significantly affected the viability and colony
formation capacity of C2C12.
GA does not influence C2C12
apoptosis
To investigate the mechanism of action of GA in
C2C12 cells, its effects on apoptosis were examined. C2C12 cells
were treated with increasing concentrations of GA for 24 or 48 h,
followed by apoptosis analysis using Annexin V/7-AAD staining. When
C2C12 cells were treated with 100 µM GA for 24 h, the apoptosis
rate significantly increased compared to the control, reaching
~2.52%, although more than 95% of the cells remained viable
(Fig. 1E, upper panel).
Furthermore, we performed western blotting to detect
apoptosis-related proteins, including PARP, cleaved PARP, Caspase-3
and cleaved Caspase-3, after 24 h treatment with GA (Fig. S1). The results showed no
differences in the expression levels of these proteins between
GA-treated and control cells, indicating that this small increase
in apoptosis is likely not biologically significant and represents
background levels. For the 48 h treatment period, there were no
significant changes in apoptosis at all tested concentrations of GA
(Fig. 1E, right panel), suggesting
that GA does not effectively induce apoptosis in C2C12 cells.
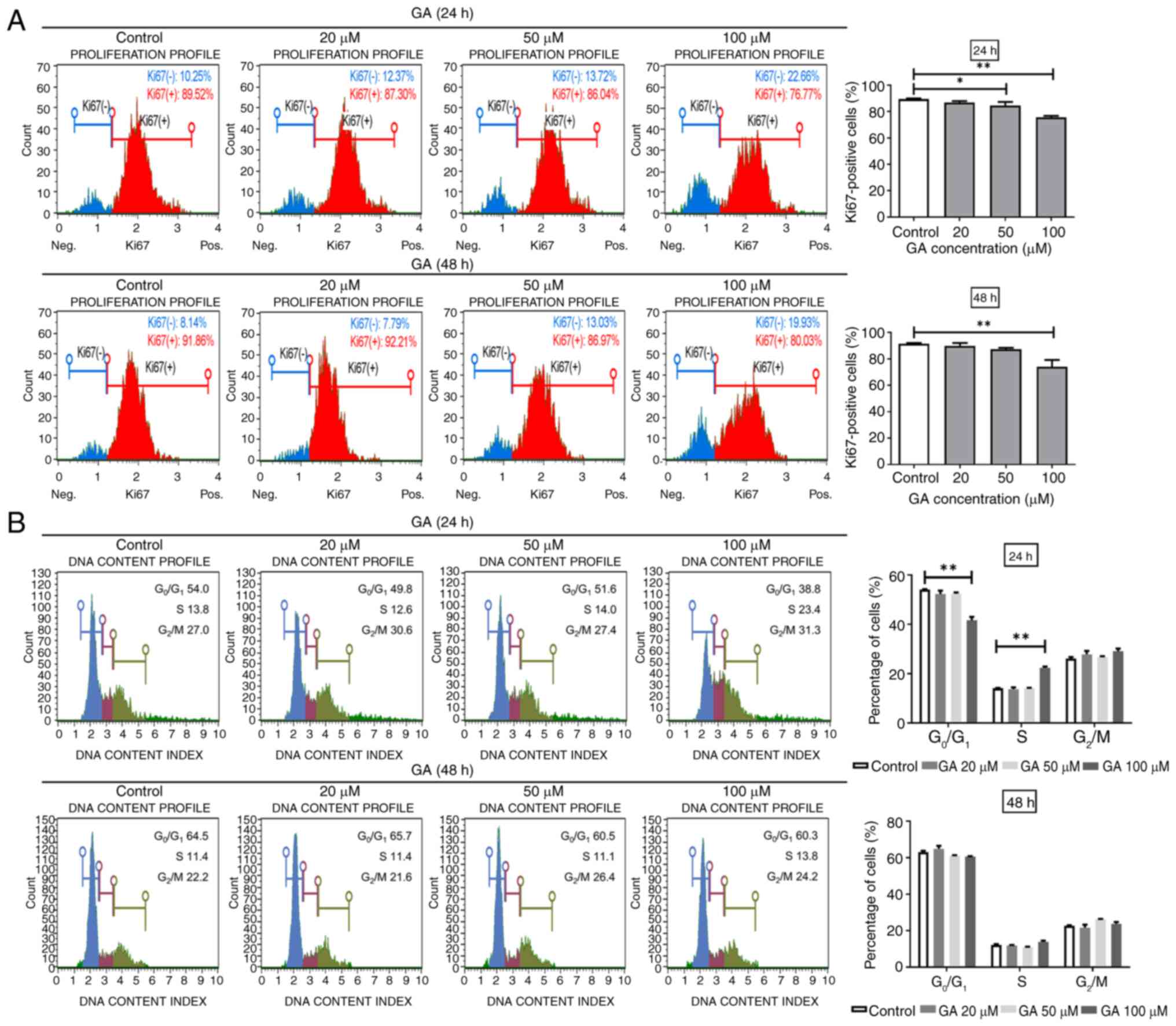
GA suppresses C2C12 cell
proliferation
C2C12 cell proliferation was assessed by staining
for Ki67, a proliferation marker (Fig.
2A) and quantified using flow cytometry. Following treatment
with GA at 100 µM, the proportion of Ki67-positive cells decreased
from 89.44–75.72% after 24 h and from 91.5–74.04% after 48 h
(Fig. 2A, upper and lower panels,
respectively). These results indicated that treatment with GA could
exert anti-proliferative effects on C2C12 cells.
GA-mediated cell cycle arrest
contributes to C2C12 cell growth inhibition
The cell cycle is crucial for cell growth. To
investigate whether the GA-mediated inhibition of C2C12 cell
proliferation was associated with cell cycle arrest, the cells were
treated with various GA concentrations for 24 and 48 h and the cell
cycle profiles assessed using the Guava Muse Cell Analyzer.
Treatment with GA at 100 µM significantly increased the proportion
of cells in the S phase after 24 h but not after 48 h (Fig. 2B, upper and lower panels,
respectively). Cells in the S phase are mainly engaged in DNA
replication and preparation for cell division. These findings
indicated that GA-mediated cell cycle arrest partially contributed
to the inhibition of C2C12 cell growth.
GA inhibits C2C12 myoblast
differentiation
The effects of GA on myogenic differentiation were
further investigated by treating C2C12 cells with GA at 20 and 50
µM for three days, followed by western blotting. Replacing GM with
DM for three days significantly enhanced the expression of myogenic
markers such as MHC and myogenin (23). Although treatment with GA at 20 µM
did not notably affect the expression of MyoD or myogenin (Fig. 3A), the high GA concentration (50
µM) markedly reduced MHC, MyoD and myogenin expression compared
with the untreated control (Fig.
3B). These results indicated that GA could suppress C2C12 cell
differentiation.
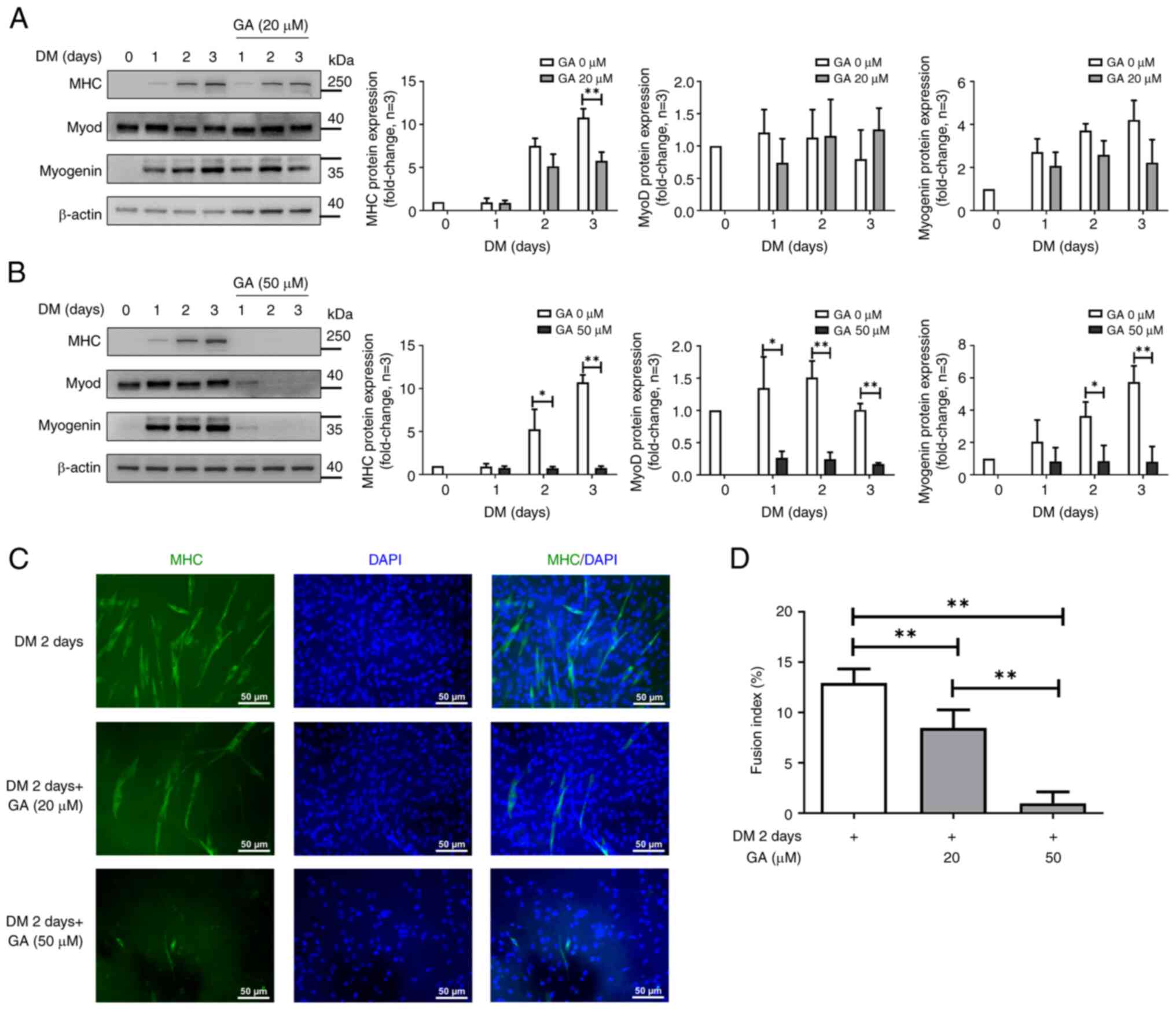 | Figure 3.GA regulates C2C12 cell myogenic
differentiation. (A) C2C12 cells were treated with GA (20 and 50
µM) or with DMSO, followed by differentiation induction for three
days and western blotting using antibodies against MHC, MyoD,
myogenin and β-actin (loading control). (B) Quantification of MHC,
MyoD and myogenin protein levels. The bar graphs show relative MHC,
MyoD and myogenin protein levels. Relative protein levels were
determined based on protein band density using ImageJ. *P<0.05
and **P<0.01 vs. the untreated group (Student's unpaired
t-test). (C) Fluorescence images of C2C12 myotube cells
differentiated for two days following treatment with GA (20 and 50
µM) and staining with an anti-MHC antibody (green, myotube
morphology) and DAPI (blue, nuclei). Scale bar, 50 µm. (D) Fusion
index quantification in the control and GA treatment groups. The GA
group shows a significant reduction in the fusion index when
compared with the control group. *P<0.05 and **P<0.01 vs.
each group (one-way ANOVA with Tukey's post hoc test). Experiments
were repeated thrice. GA, ginkgolic acid; DMSO, dimethyl sulfoxide;
MHC, myosin heavy chain; MyoD, myoblast determination protein 1;
DAPI, 4′,6-diamidino-2-phenylindole; DM, differentiation
medium. |
GA impairs myoblast fusion
To determine whether GA treatment inhibits myotube
formation, we induced the differentiation of GA-treated C2C12 cells
for two days, followed by immunostaining with an anti-MHC antibody
and DAPI counterstaining. At 20 or 50 µM, GA induced cell
morphological changes but did not promote cell differentiation
(Fig. S2). Compared with control
cells, GA-treated C2C12 cells did not form myotubes after two days
(Fig. 3C). Quantitative data
(fusion index) also revealed that GA reduced the percentage of
MHC-positive cells two days after C2C12 differentiation (Fig. 3D). These results indicated that GA
significantly inhibited myogenic differentiation in C2C12 cells, as
indicated by the reduced levels of MHC-positive myotubes compared
to those in control cells.
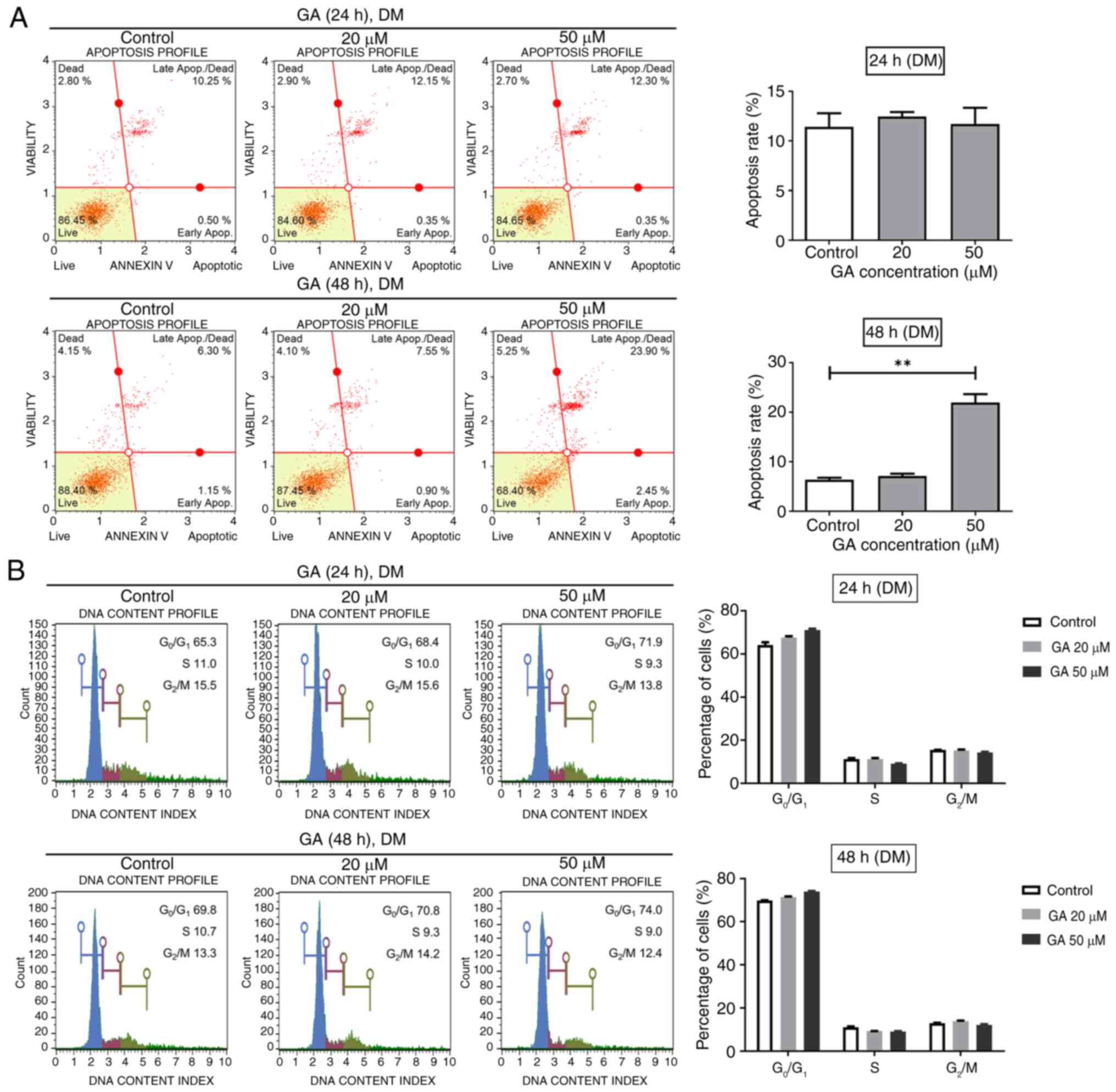
GA promotes apoptosis but does not
affect the cell cycle during C2C12 cell differentiation
Upon establishing that GA exerts toxic effects
against differentiated C2C12 cells, flow cytometry was used to
investigate the effects of GA on apoptosis. Cells were cultured in
DM with GA at concentrations of 0, 20 and 50 µM for 24 or 48 h.
Treatment of DM with GA for 24 h did not affect the total apoptotic
rate of the cells (Fig. 4A, upper
panel). However, after 48 h of differentiation, GA significantly
increased the total apoptotic rate of cells (Fig. 4A, lower panel). The role of GA in
differentiated C2C12 cells was further investigated by flow
cytometric cell cycle and Ki67 analyses, revealing no significant
differences between GA-treated cells in DM and untreated cells at
24 and 48 h (Figs. 4B and S3). These results indicated that GA
could promote apoptosis but does not affect the cell cycle in
differentiating C2C12 myogenic cells.
GA affects cell proliferation through
Erk phosphorylation
Mitogen-activated protein kinase kinase (MEK) and
ERK signaling have been found to promote the proliferation of
various cell types, including myoblasts (24). To assess the upstream signaling
pathways involved in GA-mediated inhibition of C2C12 cell
proliferation, the effects of GA treatment on MEK and ERK
phosphorylation, which is crucial for enzyme activation of MEK and
ERK, were detected. Compared with untreated C2C12 cells, which
exhibited increased levels of ERK phosphorylation after 48 h,
treatment with GA strongly decreased ERK phosphorylation in a
dose-dependent manner after 48 h without affecting MEK
phosphorylation (Fig. 5A). To
determine the cellular location of GA-mediated suppression of ERK
phosphorylation, nuclear fractions of C2C12 cells treated with GA
(0, 20, 50 and 100 µM) for 48 h were subjected to western blotting.
It was found that ERK phosphorylation primarily occurred in the
nucleus and that GA suppressed nuclear ERK phosphorylation
(Fig. 5B). These results indicated
that treatment with GA decreases ERK phosphorylation in a
dose-dependent manner without affecting MEK phosphorylation and
that in C2C12 cells, it primarily suppressed nuclear ERK
phosphorylation.
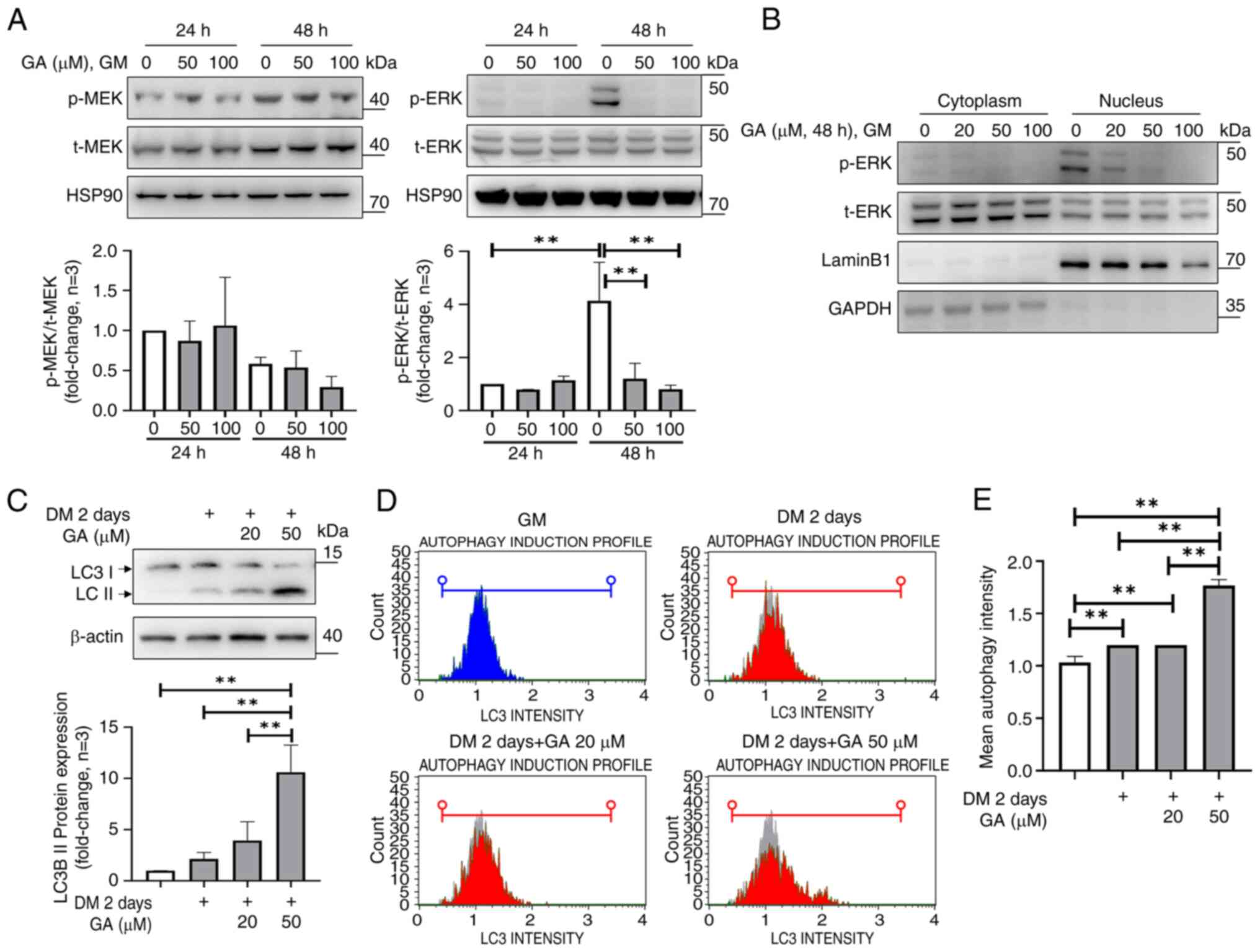 | Figure 5.GA inhibits ERK phosphorylation in
proliferating C2C12 cells and induces autophagy in differentiated
C2C12 cells. (A) Cells were treated with (or without) GA at 50 and
100 µM for 24 and 48 h in proliferating conditions, followed by
immunoblotting for p-MEK, t-MEK, p-ERK and ERK and band intensity
quantification using ImageJ (lower panel). HSP90 was used as a
loading control. (B) Western blotting of the cytoplasmic and
nuclear fractions of C2C12 cells treated with GA at 20 and 50 µM
for 48 h under proliferating conditions. Lamin B1 and GAPDH were
used as loading controls for nuclear and cytoplasmic fractions,
respectively. (C) Western blotting of the levels of the
autophagy-related protein, LC3 I/II, in differentiated C2C12 cells
treated with (or without) GA for two days. Band intensities were
quantified using ImageJ (lower panel). (D and E) Cells were treated
with GA at 20 and 50 µM in differentiation conditions, followed by
autophagy quantification using a Muse Autophagy LC3-antibody-based
kit on a MUSE cell analyzer. Data are shown as mean ± standard
deviation of three independent experiments. **P<0.01 vs. each
group (one-way ANOVA with Tukey's post hoc test). GA, ginkgolic
acid; GM, growth medium; DM, differentiation medium; p-,
phosphorylated; t-, total; ERK, extracellular signal-regulated
kinase; MEK, mitogen-activated protein kinase kinase. |
GA induces autophagy through LC3
activation in differentiated C2C12 cells
Autophagy is a fundamental process that maintains
homeostasis under normal and cellular stress conditions (25). To assess autophagy induction,
western blotting and flow cytometry were used to determine LC3
protein levels, a marker of autophagosome formation. LC3 II
degradation occurs via lysosome fusion (25). Treatment of differentiated C2C12
cells with GA at 20 and 50 µM for two days increased LC3 II levels,
indicating LC3 I to LC3 II conversion (Fig. 5C). Flow cytometry was used to
quantify LC3 levels in differentiated C2C12 cells treated with GA.
This analysis revealed an autophagy induction ratio of 1.7 in
differentiated C2C12 cells treated with GA at 50 µM (Fig. 5D and E). However, in C2C12 cells
cultured in GM, treatment with GA at 50 and 100 µM for 24 or 48 h
did not induce LC3 expression differences (Fig. S4A and B). These results indicated
that GA induces autophagy and programmed cell death during C2C12
cell differentiation.
Discussion
The present study demonstrated that the treatment
with GA reduced the viability and colony formation of C2C12 cells.
Although GA suppressed C2C12 cell proliferation, it did not affect
apoptosis. GA-mediated cell cycle arrest partly contributed to the
inhibition of C2C12 cell growth. Moreover, continued treatment with
GA at 50 µM downregulated MHC, MyoD and myogenin expression under
differentiation conditions, thereby substantially delaying myotube
formation (fusion index). Additionally, GA suppressed ERK
phosphorylation, particularly in the nucleus, highlighting its role
in regulating cell proliferation. Furthermore, in differentiated
C2C12 cells, GA triggered autophagy, as evidenced by elevated LC3
II levels, indicating the conversion of LC3 I to LC3 II (Fig. 6).
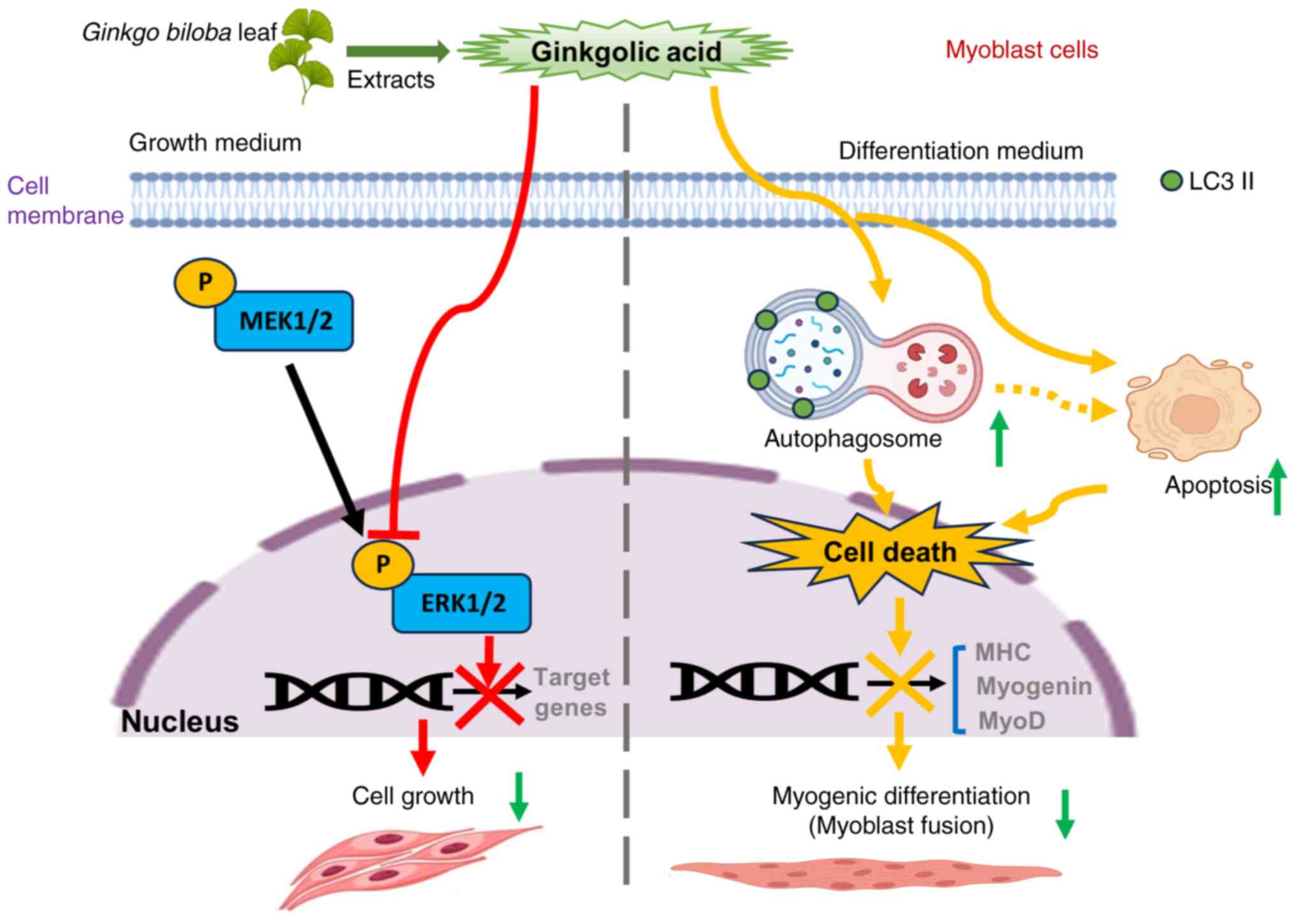 | Figure 6.Schematic diagram illustrating the
effects of GA on C2C12 cells under two conditions. GA inhibits ERK
phosphorylation, particularly in the nucleus, highlighting its role
in regulating cell proliferation pathways. Moreover, GA triggers
autophagy in differentiated C2C12 cells, as indicated by elevated
LC3 II levels, indicating LC3 I to LC3 II conversion. Consequently,
treatment with GA can impair myoblast differentiation, resulting in
a reduced expression of the myogenesis markers. MHC, MyoD and
myogenin and also inhibit myotube formation. GA, ginkgolic acid;
ERK, extracellular signal-regulated kinase; MEK, mitogen-activated
protein kinase kinase; MHC, myosin heavy chain; MyoD, myoblast
determination protein 1. |
Myoblast proliferation and differentiation are
essential for muscle development, growth and repair. These
processes involve myoblast migration, proliferation,
differentiation and fusion with mature myofibers. After birth,
muscle growth mainly involves an increase in myofiber size, whereas
activated satellite cells repair muscle damage (26). The precise regulation of cell
proliferation and differentiation is crucial for embryonic and
post-embryonic skeletal muscle development. The present study
showed that GA inhibited the proliferation and delayed the
differentiation of C2C12 cells. In multicellular organisms, cell
proliferation is regulated by external growth factors and involves
complex processes, including the MAPK pathway. ERK plays a critical
role in MEK-ERK signaling, a well-characterized MAPK signaling
pathway (27). The present study
showed that GA can inhibit C2C12 cell proliferation by suppressing
nuclear ERK phosphorylation. Although serum deprivation typically
triggers differentiation, this process was enhanced by GA-induced
apoptosis and autophagy. Furthermore, as indicated by the reduced
MyoD and myogenin expression in differentiated C2C12 cells, GA
inhibited myogenesis. MyoD is a key myogenic transcription factor
that binds to hundreds of muscle gene promoters and promotes
myoblast proliferation (28). By
inducing myoblast myogenic differentiation, myogenin induces cell
cycle exit and initiates fusion with multinucleated myofibers
(29). The present study detected
an increased fusion index even after two days of differentiation,
which was completely suppressed by GA. Collectively, the data
indicated that GA potently regulates gene expression during C2C12
myogenesis.
The present study explored the role of GA in
myogenic development, an area not extensively studied hitherto.
Previous research has examined the involvement of GA in various
biological processes, but the present study is the first, to the
best of the authors' knowledge, to specifically investigate its
effects on myogenesis, showing that GA critically affects both
myogenesis and general cellular processes. This enhances our
understanding of muscle biology and the broader physiological roles
of GA. Additionally, the present study is the first to demonstrate
that GA can induce muscle loss in an in vitro model,
typically involving decreased protein synthesis or increased
protein degradation via the ubiquitin-proteasome and autophagy
pathways (7). Elevated expression
levels of the atrophic markers MuRF-1 and atrogin-1 have been
associated with activation of the ubiquitin-proteasome
pathway-induced atrophy (8).
Substances that inhibit muscle differentiation, such as GA, can be
used to treat specific conditions to suppress excessive muscle
formation or the growth of certain cancer cells (9,10).
Additionally, research exploring GA-mediated inhibition of muscle
differentiation can provide valuable insights into the mechanisms
and development of treatments for muscle-related diseases, such as
sarcopenia and muscle atrophy. For example, the synthetic
glucocorticoid analog dexamethasone upregulates muscle-specific E3
ubiquitin ligase genes (8) and has
been used to model muscle atrophy (30). Additionally, dexamethasone is
widely used to treat various diseases, including cancer and
autoimmune disorders (31,32).
Studies indicate that apoptosis regulates the number
of muscle cells and mediates myogenesis. In humans and rodents,
muscle cell apoptosis leads to skeletal muscle atrophy and
sarcopenia (33). Numerous
apoptotic factors activate complex and multistep processes of
myoblast differentiation (34).
Therefore, elucidating the mechanisms underlying muscle cell
apoptosis is crucial to comprehensively clarify skeletal muscle
development.
Autophagy is critical for the maintenance of
cellular balance in skeletal muscles, particularly during metabolic
stress. Insufficient or excessive autophagy can trigger
pathological processes leading to muscle weakness and atrophy
(35). In C2C12 cells, autophagy
is induced during muscle differentiation despite mTOR activation
(36). The inhibition of autophagy
disrupts myoblast differentiation and promotes apoptosis (37). Increased autophagosome formation or
impaired lysosome-autophagosome fusion can cause myopathy (35). Mutations in autophagy genes and
dysregulation of the autophagic pathway can substantially
contribute to various muscle disorders (35). GA has been shown to activate
autophagy, which suppresses cancer cell growth, migration and
invasion while triggering cancer cell death (38). As indicated by elevated LC3 II
levels, the present study demonstrated that GA could induce
autophagy in differentiated C2C12 cells, thereby inhibiting
differentiation. Therefore, although optimal autophagy levels are
crucial for muscle health and the prevention of debilitating
conditions, determining the potential of GA for myoblast treatment
as a model of muscle pathology warrants further investigation at
functional, histological and molecular levels.
Regarding skeletal muscle cells, GA has been shown
to markedly enhance glucose uptake in 3T3-L1 adipocytes and C2C12
muscle cells by activating AMPK signaling (39). Accordingly, GA is a promising
therapeutic agent for type 2 diabetes. The present study focused on
the effects of GA on the proliferation, differentiation, apoptosis
and autophagy of C2C12 cells. Consistent with previous studies, the
data showed that in vitro, GA regulated not only glucose
uptake but also muscle cell proliferation and differentiation.
GA reportedly inhibits SUMOylation by blocking the
formation of an E1-SUMO intermediate and directly binding to E1
(21). SUMOylation, a protein
modification involving the addition of SUMO molecules, is a key
process in various biological and disease contexts (40). SUMOylation involves a cascade of
enzymatic reactions catalyzed by E1, E2 and E3 ligases (41). TAK-981 is a novel and selective
SUMO E1 inhibitor that affects several cancer cell lines (42,43).
Treating multiple myeloma cell lines with TAK-981 and lenalidomide
elicited potent synergistic anti-MM activity (44). Moreover, TAK-981 was found to exert
anti-leukemic effects mediated via apoptosis induction, cell cycle
arrest and immune-independent anti-acute myeloid leukemia activity
at nanomolar concentrations (45).
Thus, SUMO signaling inhibitors may be beneficial in treating
various diseases, including cancer. However, given the potential
side effects, such as muscle loss, caution is necessary when using
such medications.
Nevertheless, the limitations of the present study
need to be addressed. First, it focused solely on C2C12 cells
without evaluating other myogenic cell types, such as primary
myoblasts or human skeletal myoblast cells. Although C2C12 cells
are widely used as models for studying myogenesis, caution is
required when extrapolating these results to other cell types or
human physiology. Second, although the results suggested that GA
treatment affects the proliferation and myogenesis of C2C12 cells,
its broad physiological relevance remains uncertain. It is crucial
to conduct further studies using diverse myogenic cell types and,
more importantly, validate these findings in vivo.
Additional in vivo studies are necessary to fully understand
the relevance of the results of the present study in more complex
biological contexts, along with their potential therapeutic
implications.
In summary, the present study showed that GA
inhibited C2C12 cell proliferation by suppressing ERK signaling and
reduced myotube formation by inhibiting myogenesis and activating
autophagy. Due to its pharmacological effects, GA has therapeutic
potential against various diseases, including cancer. Despite its
potential for future drug development and as an alternative
treatment in humans, GA causes muscle loss by reducing muscle
protein synthesis and enhancing muscle protein breakdown. The
present study offered new evidence regarding the molecular
mechanisms of GA in C2C12 cells, although this requires further
validation.
Supplementary Material
Supporting Data
Acknowledgements
Not applicable.
Funding
The present study was supported by a research fund from Chosun
University (grant no. K208554002).
Availability of data and materials
The data generated in the present study may be
requested from the corresponding author.
Authors' contributions
HL designed the experiments and revised the
manuscript accordingly. HL and HJ conducted experiments, analyzed
the data and wrote the manuscript. HL and HJ confirm the
authenticity of all the raw data. Both authors have read and
approved the final version of the manuscript.
Ethics approval and consent to
participate
Not applicable.
Patient consent for publication
Not applicable.
Competing interests
The authors declare no conflict of interest.
Glossary
Abbreviations
Abbreviations:
|
GA
|
ginkgolic acid
|
|
GM
|
growth medium
|
|
DM
|
differentiation medium
|
|
MHC
|
myosin heavy chain
|
|
MEK
|
mitogen-activated protein kinase
kinase
|
|
ERK
|
extracellular signal-regulated
kinase
|
|
LC3
|
microtubule-associated protein 1 light
chain 3
|
|
SD
|
standard deviation
|
References
|
1
|
Tajbakhsh S: Skeletal muscle stem cells in
developmental versus regenerative myogenesis. J Intern Med.
266:372–389. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Burattini S, Ferri P, Battistelli M, Curci
R, Luchetti F and Falcieri E: C2C12 murine myoblasts as a model of
skeletal muscle development: Morpho-functional characterization.
Eur J Histochem. 48:223–233. 2004.PubMed/NCBI
|
|
3
|
Lyu P and Jiang H: RNA-sequencing reveals
upregulation and a beneficial role of autophagy in myoblast
differentiation and fusion. Cells. 11:35482022. View Article : Google Scholar
|
|
4
|
Cao RY, Li J, Dai Q, Li Q and Yang J:
Muscle atrophy: Present and future. Adv Exp Med Biol. 1088:605–624.
2018. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Yang J, Cao RY, Li Q and Zhu F: Muscle
atrophy in cancer. Adv Exp Med Biol. 1088:329–346. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Cao YY, Wang Z, Yu T, Zhang Y, Wang ZH, Lu
ZM, Lu WH and Yu JB: Sepsis induces muscle atrophy by inhibiting
proliferation and promoting apoptosis via PLK1-AKT signalling. J
Cell Mol Med. 25:9724–9739. 2021. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Sandri M: Protein breakdown in muscle
wasting: Role of autophagy-lysosome and ubiquitin-proteasome. Int J
Biochem Cell Biol. 45:2121–2129. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Bodine SC and Baehr LM: Skeletal muscle
atrophy and the E3 ubiquitin ligases MuRF1 and MAFbx/atrogin-1. Am
J Physiol Endocrinol Metab. 307:E469–E484. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Schuelke M, Wagner KR, Stolz LE, Hübner C,
Riebel T, Kömen W, Braun T, Tobin JF and Lee SJ: Myostatin mutation
associated with gross muscle hypertrophy in a child. N Engl J Med.
350:2682–2688. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Zarrabi A, Perrin D, Kavoosi M, Sommer M,
Sezen S, Mehrbod P, Bhushan B, Machaj F, Rosik J, Kawalec P, et al:
Rhabdomyosarcoma: Current therapy, challenges, and future
approaches to treatment strategies. Cancers (Basel). 15:52692023.
View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Ndjoko K, Wolfender JL and Hostettmann K:
Determination of trace amounts of ginkgolic acids in Ginkgo
biloba L. leaf extracts and phytopharmaceuticals by liquid
chromatography-electrospray mass spectrometry. J Chromatogr B
Biomed Sci Appl. 744:249–255. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Zhou C, Li X, Du W, Feng Y, Kong X, Li Y,
Xiao L and Zhang P: Antitumor effects of ginkgolic acid in human
cancer cell occur via cell cycle arrest and decrease the Bcl-2/Bax
ratio to induce apoptosis. Chemotherapy. 56:393–402. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Lü JM, Yan S, Jamaluddin S, Weakley SM,
Liang Z, Siwak EB, Yao Q and Chen C: Ginkgolic acid inhibits HIV
protease activity and HIV infection in vitro. Med Sci Monit.
18:BR293–BR298. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Hua Z, Wu C, Fan G, Tang Z and Cao F: The
antibacterial activity and mechanism of ginkgolic acid C15:1. BMC
Biotechnol. 17:52017. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Li J, Li A, Li M, Liu Y, Zhao W and Gao D:
Ginkgolic acid exerts an anti-inflammatory effect in human
umbilical vein endothelial cells induced by ox-LDL. Pharmazie.
73:408–412. 2018.PubMed/NCBI
|
|
16
|
Ma J, Duan W, Han S, Lei J, Xu Q, Chen X,
Jiang Z, Nan L, Li J, Chen K, et al: Ginkgolic acid suppresses the
development of pancreatic cancer by inhibiting pathways driving
lipogenesis. Oncotarget. 6:20993–21003. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Qiao L, Zheng J, Jin X, Wei G, Wang G, Sun
X and Li X: Ginkgolic acid inhibits the invasiveness of colon
cancer cells through AMPK activation. Oncol Lett. 14:5831–5838.
2017.PubMed/NCBI
|
|
18
|
Baek SH, Ko JH, Lee JH, Kim C, Lee H, Nam
D, Lee J, Lee SG, Yang WM, Um JY, et al: Ginkgolic acid inhibits
invasion and migration and TGF-β-induced EMT of lung cancer cells
through PI3K/Akt/mTOR inactivation. J Cell Physiol. 232:346–354.
2017. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Hamdoun S and Efferth T: Ginkgolic acids
inhibit migration in breast cancer cells by inhibition of NEMO
sumoylation and NF-κB activity. Oncotarget. 8:35103–35115. 2017.
View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Liu D, Li Z, Yang Z, Ma J and Mai S:
Ginkgoic acid impedes gastric cancer cell proliferation, migration
and EMT through inhibiting the SUMOylation of IGF-1R. Chem Biol
Interact. 337:1093942021. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Fukuda I, Ito A, Hirai G, Nishimura S,
Kawasaki H, Saitoh H, Kimura K, Sodeoka M and Yoshida M: Ginkgolic
acid inhibits protein SUMOylation by blocking formation of the
E1-SUMO intermediate. Chem Biol. 16:133–140. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Liu H, Lee SM and Joung H: 2-D08 treatment
regulates C2C12 myoblast proliferation and differentiation via the
Erk1/2 and proteasome signaling pathways. J Muscle Res Cell Motil.
42:193–202. 2021. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Miller JB: Myogenic programs of mouse
muscle cell lines: Expression of myosin heavy chain isoforms,
MyoD1, and myogenin. J Cell Biol. 111:1149–1159. 1990. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Jones NC, Fedorov YV, Rosenthal RS and
Olwin BB: ERK1/2 is required for myoblast proliferation but is
dispensable for muscle gene expression and cell fusion. J Cell
Physiol. 186:104–115. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Aman Y, Schmauck-Medina T, Hansen M,
Morimoto RI, Simon AK, Bjedov I, Palikaras K, Simonsen A, Johansen
T, Tavernarakis N, et al: Autophagy in healthy aging and disease.
Nat Aging. 1:634–650. 2021. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Yin H, Price F and Rudnicki MA: Satellite
cells and the muscle stem cell niche. Physiol Rev. 93:23–67. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Zhang W and Liu HT: MAPK signal pathways
in the regulation of cell proliferation in mammalian cells. Cell
Res. 12:9–18. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Cao Y, Kumar RM, Penn BH, Berkes CA,
Kooperberg C, Boyer LA, Young RA and Tapscott SJ: Global and
gene-specific analyses show distinct roles for Myod and Myog at a
common set of promoters. EMBO J. 25:502–511. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Andrés V and Walsh K: Myogenin expression,
cell cycle withdrawal, and phenotypic differentiation are
temporally separable events that precede cell fusion upon
myogenesis. J Cell Biol. 132:657–666. 1996. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Massaccesi L, Goi G, Tringali C, Barassi
A, Venerando B and Papini N: Dexamethasone-induced skeletal muscle
atrophy increases O-GlcNAcylation in C2C12 cells. J Cell Biochem.
117:1833–1842. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Wang LJ, Lu W and Zhou TY: Current
applications of dexamethasone for cancer treatment. Yao Xue Xue
Bao. 50:1217–1224. 2015.(In Chinese). PubMed/NCBI
|
|
32
|
Madamsetty VS, Mohammadinejad R, Uzieliene
I, Nabavi N, Dehshahri A, Garcia-Couce J, Tavakol S, Moghassemi S,
Dadashzadeh A, Makvandi P, et al: Dexamethasone: Insights into
pharmacological aspects, therapeutic mechanisms, and delivery
systems. ACS Biomater Sci Eng. 8:1763–1790. 2022. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
McLoughlin TJ, Smith SM, DeLong AD, Wang
H, Unterman TG and Esser KA: FoxO1 induces apoptosis in skeletal
myotubes in a DNA-binding-dependent manner. Am J Physiol Cell
Physiol. 297:C548–C555. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Fernando P, Kelly JF, Balazsi K, Slack RS
and Megeney LA: Caspase 3 activity is required for skeletal muscle
differentiation. Proc Natl Acad Sci USA. 99:11025–11030. 2002.
View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Xia Q, Huang X, Huang J, Zheng Y, March
ME, Li J and Wei Y: The role of autophagy in skeletal muscle
diseases. Front Physiol. 12:6389832021. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Tanida I, Wakabayashi M, Kanematsu T,
Minematsu-Ikeguchi N, Sou YS, Hirata M, Ueno T and Kominami E:
Lysosomal turnover of GABARAP-phospholipid conjugate is activated
during differentiation of C2C12 cells to myotubes without
inactivation of the mTor kinase-signaling pathway. Autophagy.
2:264–271. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
McMillan EM and Quadrilatero J: Autophagy
is required and protects against apoptosis during myoblast
differentiation. Biochem J. 462:267–277. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Ding Y, Ding Z, Xu J, Li Y and Chen M:
Pharmacological activities of ginkgolic acids in relation to
autophagy. Pharmaceuticals (Basel). 15:14692022. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Yoon SY, Lee JH, Kwon SJ, Kang HJ and
Chung SJ: Ginkgolic acid as a dual-targeting inhibitor for protein
tyrosine phosphatases relevant to insulin resistance. Bioorg Chem.
81:264–269. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Vertegaal ACO: Signalling mechanisms and
cellular functions of SUMO. Nat Rev Mol Cell Biol. 23:715–731.
2022. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Wilkinson KA and Henley JM: Mechanisms,
regulation and consequences of protein SUMOylation. Biochem J.
428:133–145. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Langston SP, Grossman S, England D, Afroze
R, Bence N, Bowman D, Bump N, Chau R, Chuang BC, Claiborne C, et
al: Discovery of TAK-981, a first-in-class inhibitor of
SUMO-activating enzyme for the treatment of cancer. J Med Chem.
64:2501–2520. 2021. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Kumar S, Schoonderwoerd MJA, Kroonen JS,
de Graaf IJ, Sluijter M, Ruano D, Gonzalez-Prieto R, Verlaan-de
Vries M, Rip J, Arens R, et al: Targeting pancreatic cancer by
TAK-981: A SUMOylation inhibitor that activates the immune system
and blocks cancer cell cycle progression in a preclinical model.
Gut. 71:2266–2283. 2022. View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Du L, Liu W, Pichiorri F and Rosen ST:
SUMOylation inhibition enhances multiple myeloma sensitivity to
lenalidomide. Cancer Gene Ther. 30:567–574. 2023. View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Kim HS, Kim BR, Dao TTP, Kim JM, Kim YJ,
Son H, Jo S, Kim D, Kim J, Suh YJ, et al: TAK-981, a SUMOylation
inhibitor, suppresses AML growth immune-independently. Blood Adv.
7:3155–3168. 2023. View Article : Google Scholar : PubMed/NCBI
|