Introduction
Atrial fibrillation (AF), which affects >37.5
million individuals worldwide, is the most common and persistent
type of arrhythmia and AF is also the second most common cause of
mortality (1,2). The main mechanism involved in AF is
structural remodeling (3). AF
places a burden on families and society; however, its pathogenesis
remains unclear.
Previous studies have identified an association
between inflammation and the occurrence/maintenance of AF (2,4).
Inflammation is a protective biological response of a host to
infection-induced damage; when such changes occur in the atrium,
inflammation induces atrial fibrosis, which predisposes individuals
to AF. Notably, fibrosis is considered the basis for the structural
remodeling of the atrium (5). In
addition, AF induces atrial inflammation, which activates cardiac
fibroblasts to cause atrial fibrotic remodeling, thus generating a
vicious cycle; that is, AF-inflammation-fibrosis (5). Fibrosis and inflammation directly or
indirectly promote the structural remodeling of AF (5–7). In
addition, inflammation and fibrosis not only function in the
occurrence and development of AF, but also serve etiological roles
in its pathogenesis.
The amino-terminal sequence which is encoded by
programmed cell death factor 4 (PDCD4) is highly conserved among
species and freely shuttles within and out of the nucleus. Notably,
PDCD4 is enriched in the inflammatory process of AF and is
associated with a high risk of developing stroke in patients with
AF (8). PDCD4 regulates the
pathophysiological processes of fibrosis and inflammation (9,10);
the silencing or knockdown of PDCD4 has been shown to reduce
inflammation in human dermal papillary cells (11), and to improve ventricular
remodeling, oral mucosal fibrosis and liver fibrosis (12–14).
However, the mechanisms by which PDCD4 affects AF, inflammation and
fibrosis remain to be elucidated.
As a transcription factor in the nuclear receptor
superfamily, peroxisome proliferator-activated receptor γ (PPARγ)
is deemed to be a prospective target for treating inflammatory
diseases (15). The PPARγ agonist
pioglitazone hydrochloride (Pio) can reduce TNF-α expression,
thereby attenuating the inflammation of bronchial occlusion
following lung transplantation (16). NF-κB is an inflammatory
transcription factor, the activation of which is caused by a
variety of external stimuli, such as inflammation and immune
responses (17). During
inflammation, the activation of PPARγ inactivates NF-κB by direct
binding to NF-κB p65, resulting in a diminished inflammatory
response after ubiquitin degradation (18).
The present study aimed to determine whether PDCD4
mediates PPARγ-mediated inflammation via the NF-κB pathway,
inducing the structural remodeling of mouse atrial myocytes (HL-1
cells).
Materials and methods
Cells, cell culture, transfection and
treatment
Mouse atrial myocytes (HL-1; cat. no. iCell-m077;
iCell Bioscience, Inc.) were grown in minimum essential medium
containing 1% streptomycin/penicillin plus 10% fetal calf serum
(Thermo Fisher Scientific, Inc.) in an incubator at 37°C and 5%
CO2.
Overexpression negative control plasmid pcDNA3.1
(oeNC; 2 µg; GenScript), PDCD overexpression plasmid (oePDCD4; 2
µg; GenScript), or small interfering (si)RNA NC (siNC; 10 nM;
Guangzhou RiboBio Co., Ltd.) and PDCD4 siRNAs (siPDCD4-1, 2 and 3;
10 nM; Guangzhou RiboBio Co., Ltd.) were transfected into HL-1
cells (3×105 cells/well) using Lipofectamine®
2000 (Thermo Fisher Scientific, Inc.) for 48 h at 37°C. At 48 h
after transfection, cells were harvested for the subsequent
experiments. The sequences of siNC, siPDCD4-1, siPDCD4-2 and
siPDCD4-3 are provided in Table
I.
 | Table I.Sequences of siPDCD4-1, siPDCD4-2 and
siPDCD4-3. |
Table I.
Sequences of siPDCD4-1, siPDCD4-2 and
siPDCD4-3.
| siRNA | Sequence,
5′-3′ |
|---|
| siNC |
UUCUCCGAACGUGUCACGU |
| siPDCD4-1 |
CCAGGAGAACUGTGUUUAU |
| siPDCD4-2 |
GCUCCUGAGUAUGUCCAAA |
| siPDCD4-3 |
CCCACACUCAUACUCUGUU |
The HL-1 cells in the overexpression group were
further divided into four groups as follows: i) oeNC; ii) oePDCD4;
iii) oePDCD4 + PPARγ agonist (Pio; 0, 1.25, 2.5, 5, 10, 20, 40 and
80 µM, cat. no. HY-14601; MedChemExpress); and iv) oePDCD4 + NF-κB
inhibitor (CBL0137; 0, 0.125, 0.25, 0.5, 1, 2, 4 and 8 µM, cat. no.
HY-18935A; MedChemExpress). The HL-1 cells in the silencing group
were further divided into the following four groups: i) siNC; ii)
siPDCD4; iii) siPDCD4 + PPARγ inhibitor (GW9962; 0, 1.25, 2.5, 5,
10, 20, 40 and 80 µM, cat. no. M6191-5MG; Merck KGaA); and iv)
siPDCD4 + NF-κB agonist (betulinic acid; 0, 1.25, 2.5, 5, 10, 20,
40 and 80 µM, cat. no. HY-10529; MedChemExpress). The medium was
changed every day. The cells were harvested following 48 h of
transfection and 24 h of drug treatment at 37°C.
Reverse transcription-quantitative PCR
(RT-qPCR)
Total RNA was extracted from the HL-1 cells using
TRIzol® reagent (Thermo Fisher Scientific, Inc.) and
cDNA was generated using a RevertAid First Strand CDNA Synthesis
Kit (Thermo Fisher Scientific, Inc.) according to manufacturer's
protocol. Subsequently, cDNA (2 µl) was used for qPCR (20 µl
reaction volume), which was performed using 2X SYBR-Green qPCR
MasterMix (Thermo Fisher Scientific, Inc.) on A 7500HT Fast
real-time PCR System (Applied Biosystems; Thermo Fisher Scientific,
Inc.). The cycling conditions were as follows: initial denaturation
at 94°C for 10 min, 94°C for 20 sec, 55°C for 20 sec and 72°C for
20 sec, for a total of 40 cycles, followed by a final extension at
72°C. The mRNA expression levels of PDCD4 were normalized to GAPDH
using the 2−ΔΔCq method (19). GAPDH served as an internal control.
The sequences of the primers used are given in Table II.
| Table II.Primer sequences. |
Table II.
Primer sequences.
| Gene | Forward
(5′-3′) | Reverse
(5′-3′) |
|---|
| PDCD4 |
CCTGGATGAGACCGCATTTG |
GCAAAGGTCAGAAAGCAGCT |
| GAPDH |
GGTGAAGGTCGGTGTGAACG |
CTCGCTCCTGGAAGATGGTG |
Western blot analysis
Nuclear (Nu) and cytoplasmic (Cy) proteins from HL-1
cardiomyocytes were isolated using NE-PER™ (Thermo
Fisher Scientific, Inc.). Total proteins were extracted from the
HL-1 cardiomyocytes utilizing RIPA lysis buffer (Beijing Solarbio
Science & Technology Co., Ltd.) containing PMSF and protease
inhibitor mixture (Roche Diagnostics). The protein concentration
was assessed using a BCA protein concentration assay kit (Biosharp
Life Sciences). The proteins (15 µg/lane) were then separated by
sodium dodecyl-sulfate polyacrylamide gel electrophoresis on 10%
gels and were transferred onto polyvinylidene fluoride (PVDF)
membranes (MilliporeSigma). Then the PVDF membranes were blocked by
5% non-fat milk for 2 h at room temperature. Thereafter, the PVDF
membranes were incubated with primary antibodies against PDCD4
(cat. no. bs-1608R; 1:1,000; BIOSS), collagen I (cat. no. AF7001;
1:1,000; Affinity Biosciences), collagen III (cat. no. AF0136;
1:1,000; Affinity Biosciences), fibronectin (cat. no. bs-13455R;
1:1,000; BIOSS), α-smooth muscle actin (α-SMA; cat. no. bsm-33187M;
1:1,000; BIOSS), matrix metalloproteinase (MMP) 2 (cat. no. AF0577;
1:1,000; Affinity Biosciences), PPARγ (cat. no. bs-0530R; 1:1,000;
BIOSS), NF-κB p65 (cat. no. bs-0465R; 1:1,000; BIOSS), NF-κB
phosphorylated (p-)p65 (cat. no. bs-0982R; 1:1,000; BIOSS), NF-κB
p50 (cat. no. bs-1194R; 1:1,000; BIOSS), proliferating cell nuclear
antigen (PCNA; cat. no. 10205-2-AP; 1:1,000; Proteintech Group,
Inc.) and GAPDH (cat. no. 60004-1-Ig; 1:1,000; Proteintech Group,
Inc.) at 4°C overnight. Subsequently, the membranes were incubated
with an HRP-labeled goat anti-rabbit (cat. no. A0208; 1:1,000;
Beyotime Institute of Biotechnology) or goat anti-mouse secondary
antibody (cat. no. A0216; 1:1,000; Beyotime Institute of
Biotechnology) at room temperature for 1 h. ECL solution was then
added (Beijing Dingguo Changsheng Biotechnology Co., Ltd.). Images
were saved using an integrated chemiluminescence instrument (Clinx
Science Instruments Co., Ltd.). PCNA served as an internal control
for Nu p50, p-p65, and p65. GAPDH served as an internal control for
Cy p50, p-p65, p65, and the remaining proteins. Image J (Version
1.8.0; National Institutes of Health) was used for
densitometry.
Cell Counting Kit-8 (CCK-8) assay
HL-1 cell viability was assessed using a CCK-8 kit
(Beyotime Institute of Biotechnology). The HL-1 cells were
incubated in 96-well plates for 24 h, followed by incubation with
various concentrations of Pio (0, 1.25, 2.5, 5, 10, 20, 40 and 80
µM), CBL0137 (0, 0.125, 0.25, 0.5, 1, 2, 4 and 8 µM), GW9962 (0,
1.25, 2.5, 5, 10, 20, 40 and 80 µM) and betulinic acid (0, 1.25,
2.5, 5, 10, 20, 40 and 80 µM) for 24 h at 37°C. Different
concentrations were selected for CBL0137 compared with the other
three drugs, according to the recommendations of a previous study
(20). Subsequently, CCK-8 (10 µl,
Dojindo Laboratories, Inc.) was added to each well followed by
incubation in the dark for 2 h. The optical density (450 nm) was
measured using an automatic microplate reader (Thermo Fisher
Scientific, Inc.).
Enzyme-linked immunosorbent assays
(ELISAs)
The HL-1 cells were cultured until they reached the
logarithmic growth phase; subsequently, the cell concentration was
adjusted to 2×105 cells/ml. After seeding the cells in a
6-well plate (1 ml/well), the cells were transfected for 48 h and
treated with the different drugs for 24 h. Thereafter, the HL-1
cells were collected, and the Mouse ELISA kits (IFN-γ, cat. no.
EM007; IL-4, cat. no. EM003; IL-6, cat. no. EM004; IL-17A, cat. no.
EM015; and TNF-α, cat. no. EM008; Suzhou ExCell Biotechnology Co.,
Ltd.) were used, according to the manufacturer's instructions.
Immunofluorescence analysis
The HL-1 cells (2×105 cells/well) were
grown on slides, fixed in 4% paraformaldehyde for 15 min at 4°C and
incubated in 5% FBS (Cytiva) at 37°C for 2 h. The slides were then
incubated with primary antibodies against NF-κB p65 (cat. no.
bs-0465R; 1:1,000; BIOSS) and NF-κB p-p65 (cat. no. bs-0982R;
1:1,000 BIOSS) at 4°C overnight. The following day, the slides were
incubated with an Alexa Fluor 488-labeled Goat Anti-Rabbit IgG(H+L)
secondary antibody (cat. no. A0423, 1:1,000; Beyotime Institute of
Biotechnology) and an Alexa Fluor 555-labeled Donkey Anti-Rabbit
IgG(H+L) secondary antibody (cat. no. A0453, 1:1,000; Beyotime
Institute of Biotechnology) in the dark at room temperature for 1
h. The cells were then incubated with Hoechst 33342 (cat. no.
875756-97-1; MilliporeSigma) in the dark for 15 min at room
temperature. Images were captured under a fluorescence microscope
(Olympus Corporation) and semi-quantitative analyses were conducted
using ImageJ v1.6 software (National Institutes of Health).
Statistical analyses
The experiments were conducted three times. The
results are presented as the mean ± SD. An unpaired Student's
t-test or one-way analysis of variance followed by the Bonferroni
post hoc test was carried out using GraphPad Prism 9 software
(Dotmatics). P<0.05 was considered to indicate a statistically
significant difference.
Results
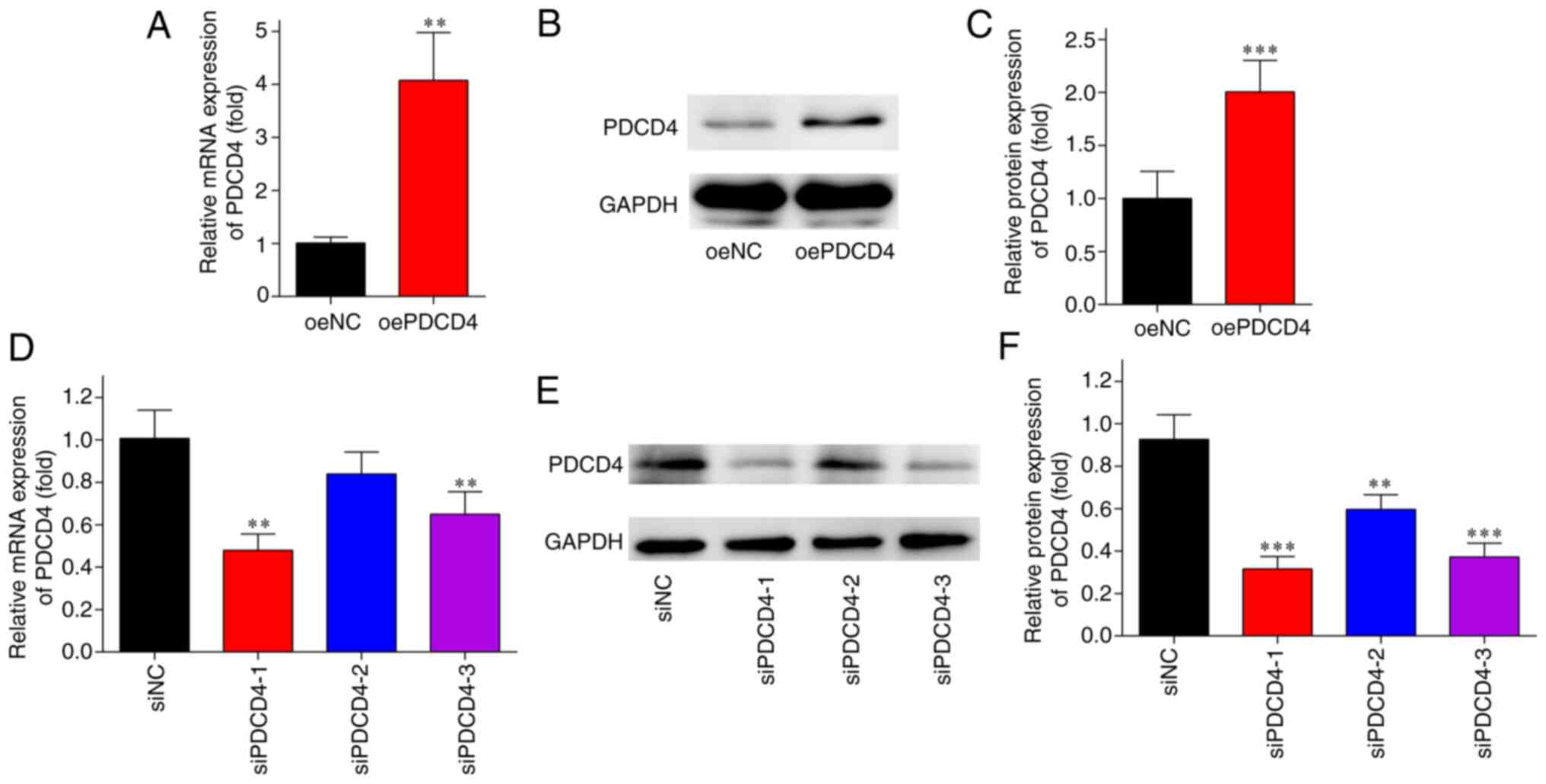
Transfection with oe-PDCD4-HL-1 and
si-PDCD4-HL-1
The results of RT-qPCR and western blot analysis
revealed that the mRNA and protein expression levels of PDCD4 were
significantly higher in the oePDCD4 group than those in the oeNC
group (Fig. 1A-C). By contrast,
the expression levels of PDCD4 were markedly lower in the siPDCD4
groups compared with those in the siNC group (Fig. 1D-F). siPDCD4-1 exerted the most
notable knockdown effect and was thus used in subsequent
experiments. These findings indicated the successful transfection
of oe-PDCD4 and si-PDCD4 into HL-1 cells.
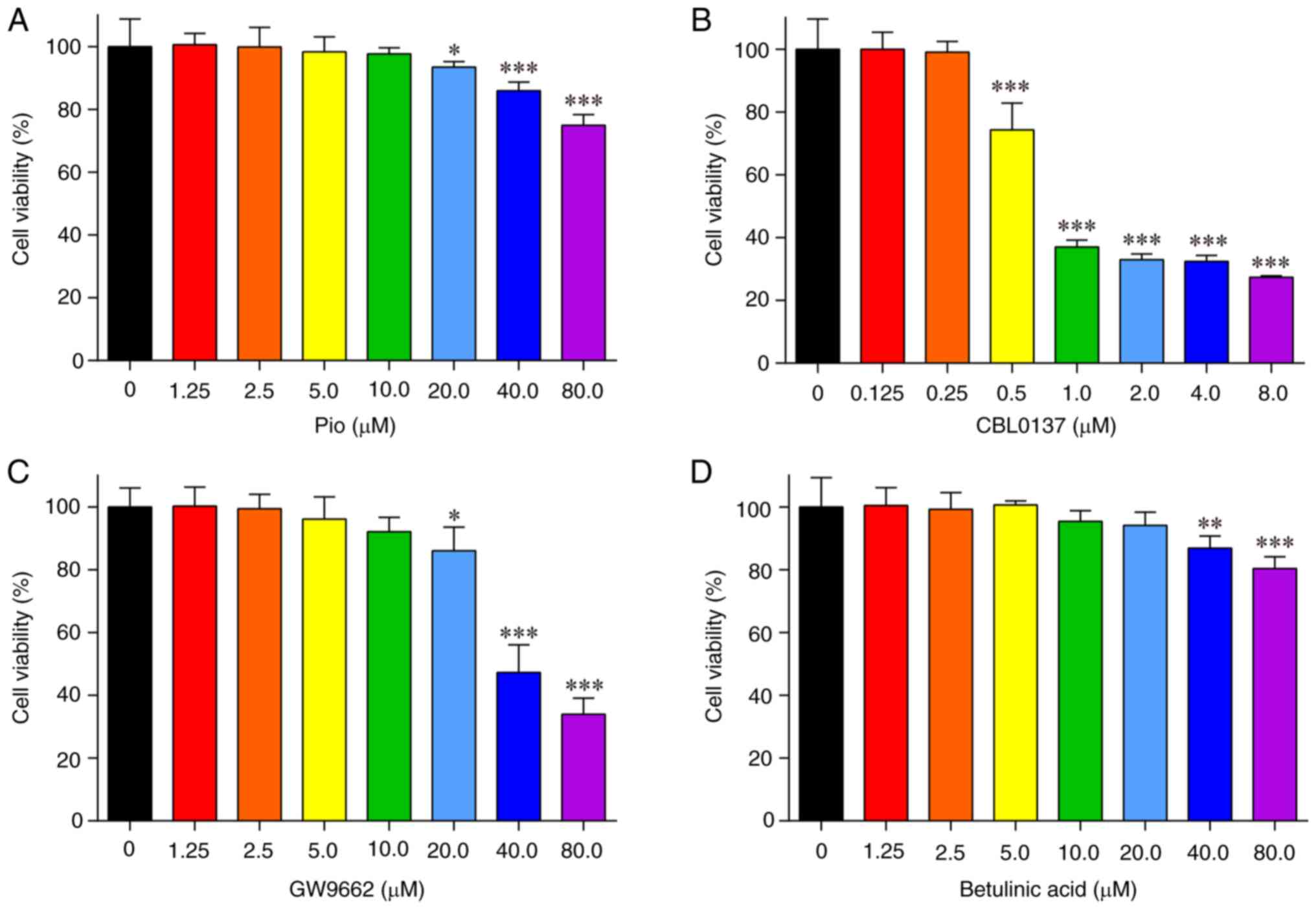
Viability of HL-1 cells treated with
Pio, CBL0137, GW9662 and betulinic acid
Different concentrations of Pio, CBL0137, GW9662 and
betulinic acid were administered to HL-1 cells for 24 h, to screen
the optimal drug concentrations using the CCK-8 assay.
Compared with in the untreated cells, HL-1 cell
viability was significantly inhibited by treatment with ≥20 µM Pio
(Fig. 2A), ≥0.5 µM CBL0137
(Fig. 2B), ≥20 µM GW9662 (Fig. 2C) and ≥40 µM betulinic acid
(Fig. 2D). Consequently, the
concentrations of the drugs used in the subsequent experiments were
as follows: Pio, 10 µM; CBL0137, 0.25 µM; GW9662, 10 µM; and
betulinic acid, 20 µM.
oePDCD4-induced inflammation and
fibrosis are mitigated by PPARγ agonist and NF-κB inhibitor
treatment in oePDCD4-HL-1 cells
The results of western blot analysis demonstrated
that, compared with in the oeNC group, the expression levels of
fibrosis-related proteins (collagen I, collagen III, fibronectin,
α-SMA and MMP2) were significantly higher in the oePDCD4 group;
these effects were significantly attenuated by treatment with the
PPARγ agonist (Pio) and NF-κB inhibitor (CBL0137) (Fig. 3A and B).
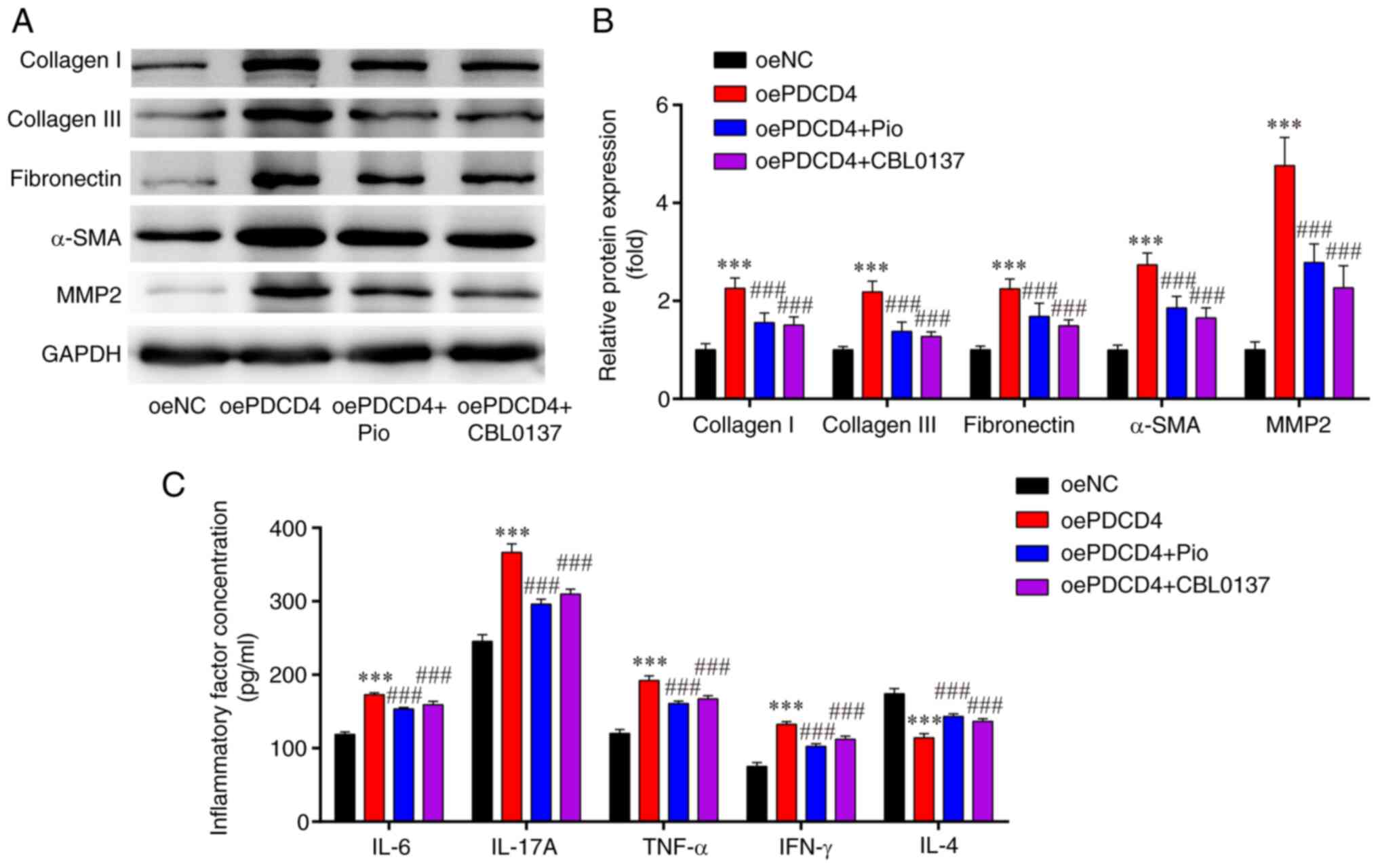 | Figure 3.Levels of fibrosis-associated
proteins and inflammatory-related cytokines in oePDCD4-HL-1 cells.
(A) Western blotting indicated that the upregulated protein
expression levels of collagen I, collagen III, fibronectin, α-SMA
and MMP2 in oe-PDCD4-HL-1 cells were attenuated by Pio and CBL0137.
(B) Statistical analysis of western blotting. (C) Enzyme-linked
immunosorbent assays showed that the upregulated protein
concentrations of IL-6, IL-17A, IFN-γ and TNF-α, and downregulated
protein concentration of IL-4, in oe-PDCD4-HL-1 cells were reversed
by Pio and CBL0137. ***P<0.001 vs. oeNC;
###P<0.001 vs. oe-PDCD4. n=3. oe, overexpression;
PDCD4, programmed cell death factor 4; α-SMA, α-smooth muscle
actin; MMP, matrix metalloproteinase; Pio, pioglitazone
hydrochloride; NC, negative control. |
ELISAs indicated that, compared with those in the
oeNC group, the levels of pro-inflammatory cytokines (IFN-γ, IL-6,
IL-17A and TNF-α) were significantly increased, while the levels of
the anti-inflammatory factor IL-4 were markedly decreased in the
oe-PDCD4 group; these effects were significantly reversed by
treatment with the PPARγ agonist (Pio) and NF-κB inhibitor
(CBL0137) (Fig. 3C).
These findings demonstrated that the overexpression
of PDCD4 may induce fibrosis and inflammation in HL-1 cells, and
these effects could be reversed by a PPARγ agonist and NF-κB
inhibitor.
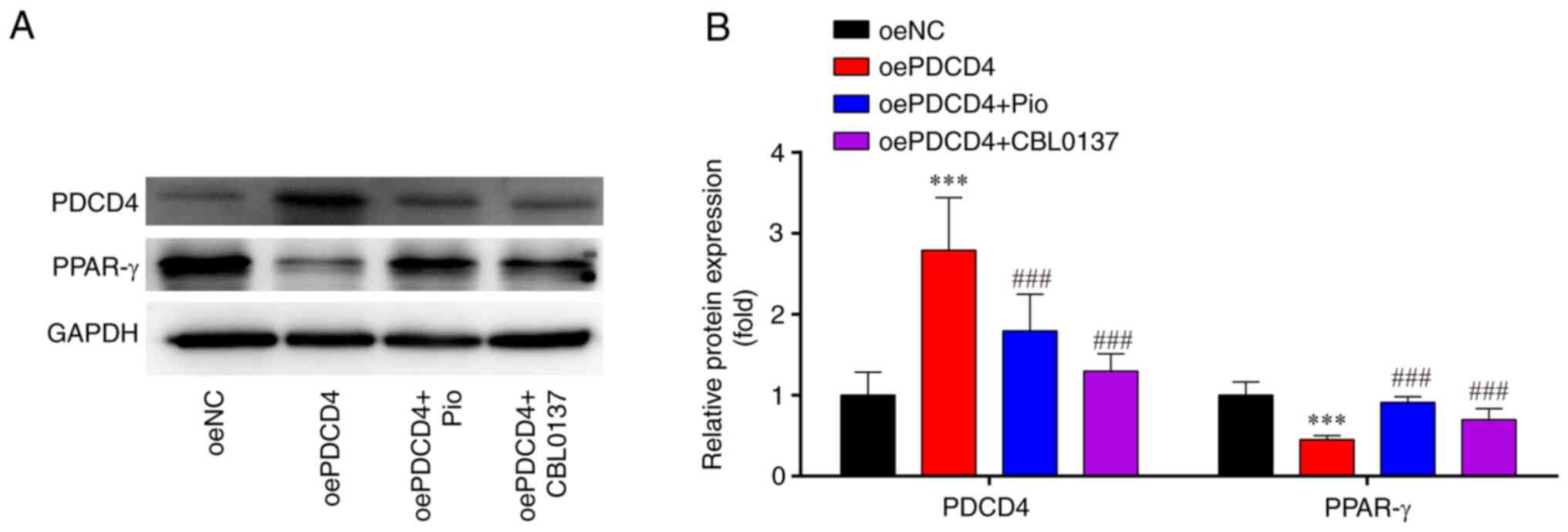
oePDCD4-induced activation of p65 is
mitigated by PPARγ agonist and NF-κB inhibitor treatment in
oePDCD4-HL-1 cells
Western blot analysis demonstrated that, compared
with those in the oeNC group, the expression levels of PDCD4 were
markedly higher, whereas the expression levels of PPAR-γ were
significantly lower in the oePDCD4 group; these effects were
significantly reversed by treatment with the PPARγ agonist (Pio)
and NF-κB inhibitor (CBL0137; Fig. 4A
and B).
In addition, western blot analysis demonstrated
that, compared with those in the oeNC group, the Nu p-p65/p65 ratio
and p-p50/PCNA ratio were increased in the oePDCD4 group, and these
effects were partially reversed by treatment with the PPARγ agonist
(Pio) and NF-κB inhibitor (CBL0137) (Fig. 5A and B). However, there was no
significant change in the Cy p-p65/p65 ratio or p-p50/GAPDH ratio
among the four groups (Fig. 5A and
B).
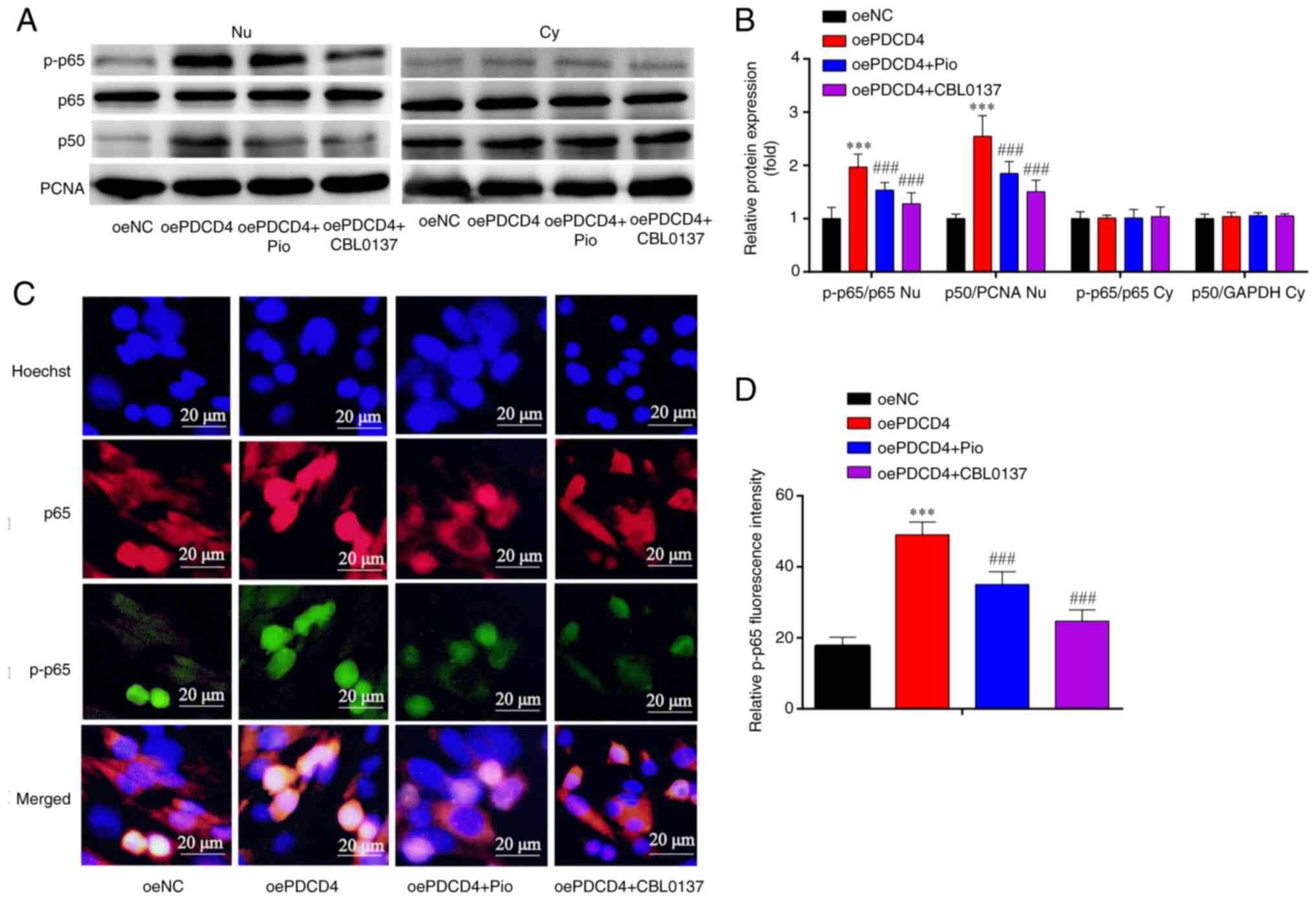 | Figure 5.Expression of NF-κB subunits in
oePDCD4-HL-1. (A) Western blotting indicated that, compared with in
the oeNC group, the Nu p-p65/p65 ratio and Nu p-p50/PCNA ratio were
increased in the oePDCD4 group, and these findings were partially
reversed by Pio and CBL0137. However, there was no significant
change in Cy p-p65/p65 ratio and Cy p-p50/GAPDH ratio among the
four groups. (B) Statistical analysis of western blotting. (C)
Immunofluorescence analysis showed that the increased protein
expression of p-p65 in oe-PDCD4-HL-1 cells was attenuated by Pio
and CBL0137. (D) Statistical analysis of fluorescence intensity.
***P<0.001 vs. oeNC; ###P<0.001 vs. oe-PDCD4. n=3.
Magnification, ×1,000. oe, overexpression; PDCD4, programmed cell
death factor 4; NC, negative control; p-, phosphorylated; PCNA,
proliferating cell nuclear antigen; Pio, pioglitazone
hydrochloride; Nu, nuclear; Cy, cytoplasmic; NC, negative
control. |
The results of immunofluorescence analysis revealed
that, compared with in the oeNC group, the expression of p-p65 was
increased in the oePDCD4 group; this effect was partially reversed
by treatment with the PPARγ agonist (Pio) and NF-κB inhibitor
(CBL0137) (Fig. 5C and D). These
findings suggested that the overexpression of PDCD4 activated NF-κB
p65 in HL-1 cells, and these effects were reversed by the PPARγ
agonist and NF-κB inhibitor.
siPDCD4-induced mitigation of fibrosis
and inflammation are aggravated by PPARγ inhibitor and NF-κB
agonist treatment in siPDCD4-HL-1 cells
Western blot analysis demonstrated that, compared
with those in the siNC group, the expression levels of
fibrosis-related proteins (collagen I, collagen III, fibronectin,
α-SMA and MMP2) were significantly lower in the siPDCD4 group;
these effects were partially reversed by treatment with the PPARγ
inhibitor (GW9662) and NF-κB agonist (betulinic acid) (Fig. 6A and B).
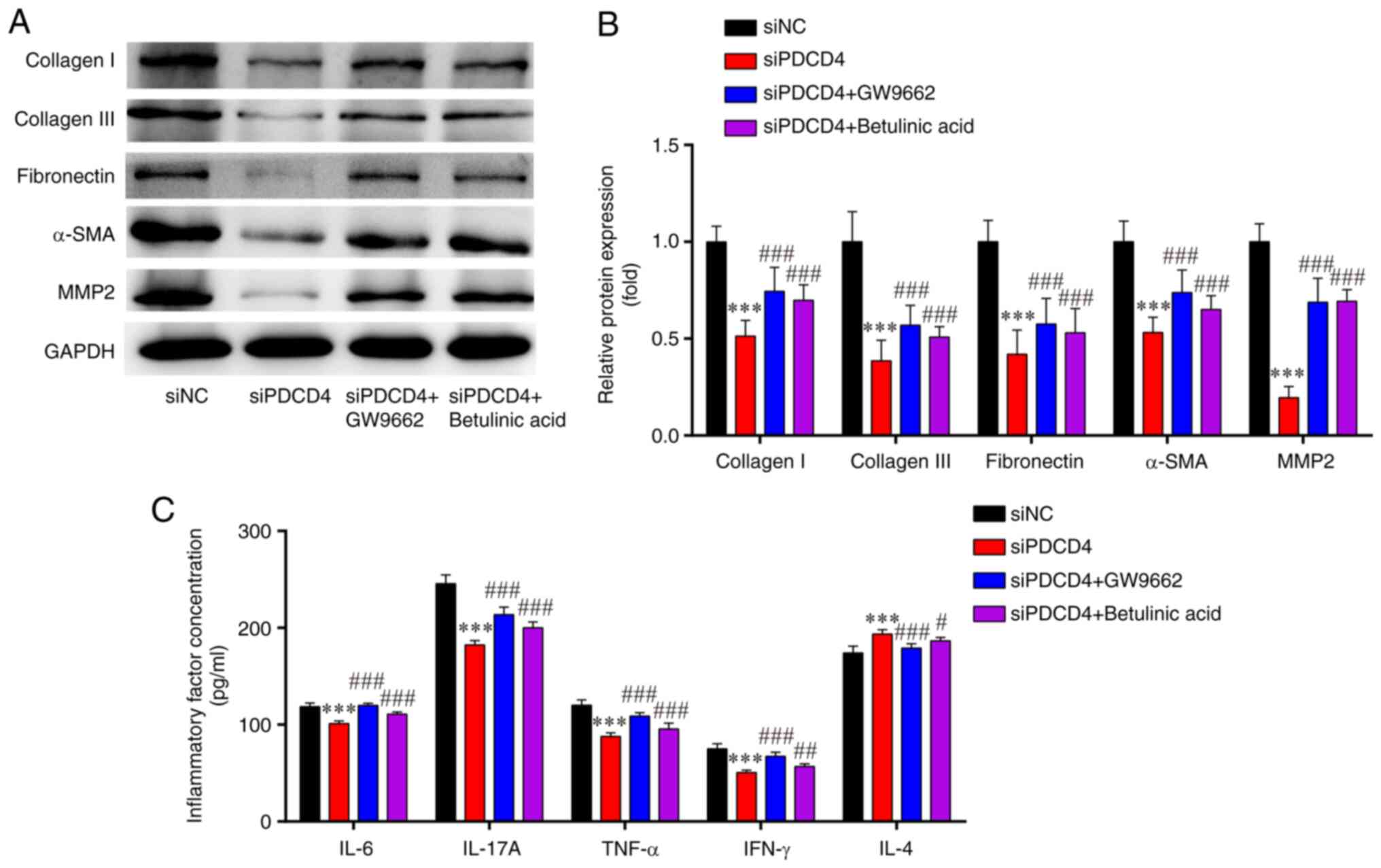 | Figure 6.Levels of fibrosis-associated
proteins and inflammatory-related cytokines in siPDCD4-HL-1 cells.
(A) Western blotting indicated that the downregulated protein
expression levels of collagen I, collagen III, fibronectin, α-SMA
and MMP2 in si-PDCD4-HL-1 cells were attenuated by GW9662 and
betulinic acid. (B) Statistical analysis of western blotting. (C)
Enzyme-linked immunosorbent assays showed that the downregulated
protein concentrations of IL-6, IL-17A, IFN-γ and TNF-α, and
upregulated protein concentration of IL-4, in si-PDCD4-HL-1 cells
were reversed by GW9662 and betulinic acid. ***P<0.001 vs. siNC;
#P<0.05, ##P<0.01,
###P<0.001, vs. si-PDCD4. n=3. si, small interfering;
PDCD4, programmed cell death factor 4; α-SMA, α-smooth muscle
actin; MMP, matrix metalloproteinase; NC, negative control. |
The results of ELISAs indicated that, compared with
those in the siNC group, the levels of pro-inflammatory cytokines
(IFN-γ, IL-6, IL-17A and TNF-α) were significantly lower, while the
levels of the anti-inflammatory factor IL-4 were markedly higher in
the siPDCD4 group; these effects were partially reversed by the
PPARγ inhibitor (GW9662) and NF-κB agonist (betulinic acid)
(Fig. 6C). These results indicated
that the silencing of PDCD4 may attenuate fibrosis and inflammatory
injury responses in HL-1 cells, and these effects could be reversed
by a PPARγ inhibitor and NF-κB agonist.
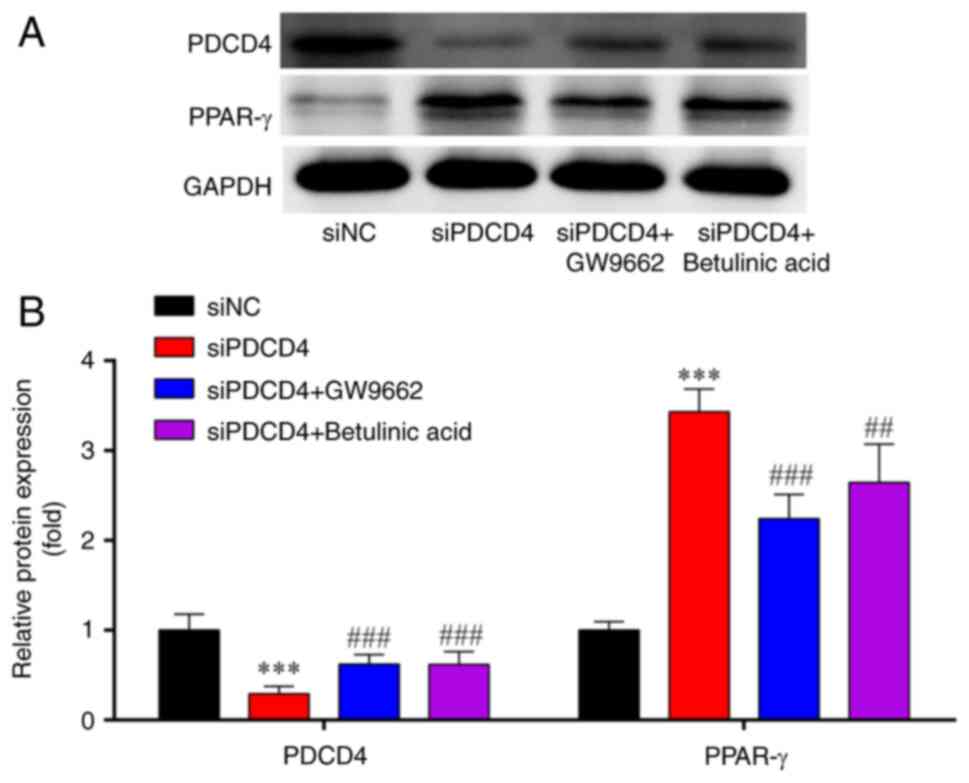
siPDCD4-induced inactivation of p65 is
reversed by PPARγ inhibitor and NF-κB agonist treatment in
siPDCD4-HL-1 cells
Western blot analysis demonstrated that, compared
with those in the siNC group, the expression levels of PDCD4 were
significantly lower, whereas PPAR-γ expression was significantly
higher in the siPDCD4 group; these effects were partially reversed
by treatment with the PPARγ inhibitor (GW9662) and NF-κB agonist
(betulinic acid) (Fig. 7A and
B).
The results of western blot analysis also
demonstrated that, compared with those in the siNC group, the Nu
p-p65/p65 ratio and p-p50/PCNA ratio were decreased in the siPDCD4
group; these effects were partially reversed by treatment with the
PPARγ inhibitor (GW9662) and NF-κB agonist (betulinic acid)
(Fig. 8A and B). However, there
was no significant change in the Cy p-p65/p65 ratio and p-p50/GAPDH
ratio among the four groups (Fig. 8A
and B).
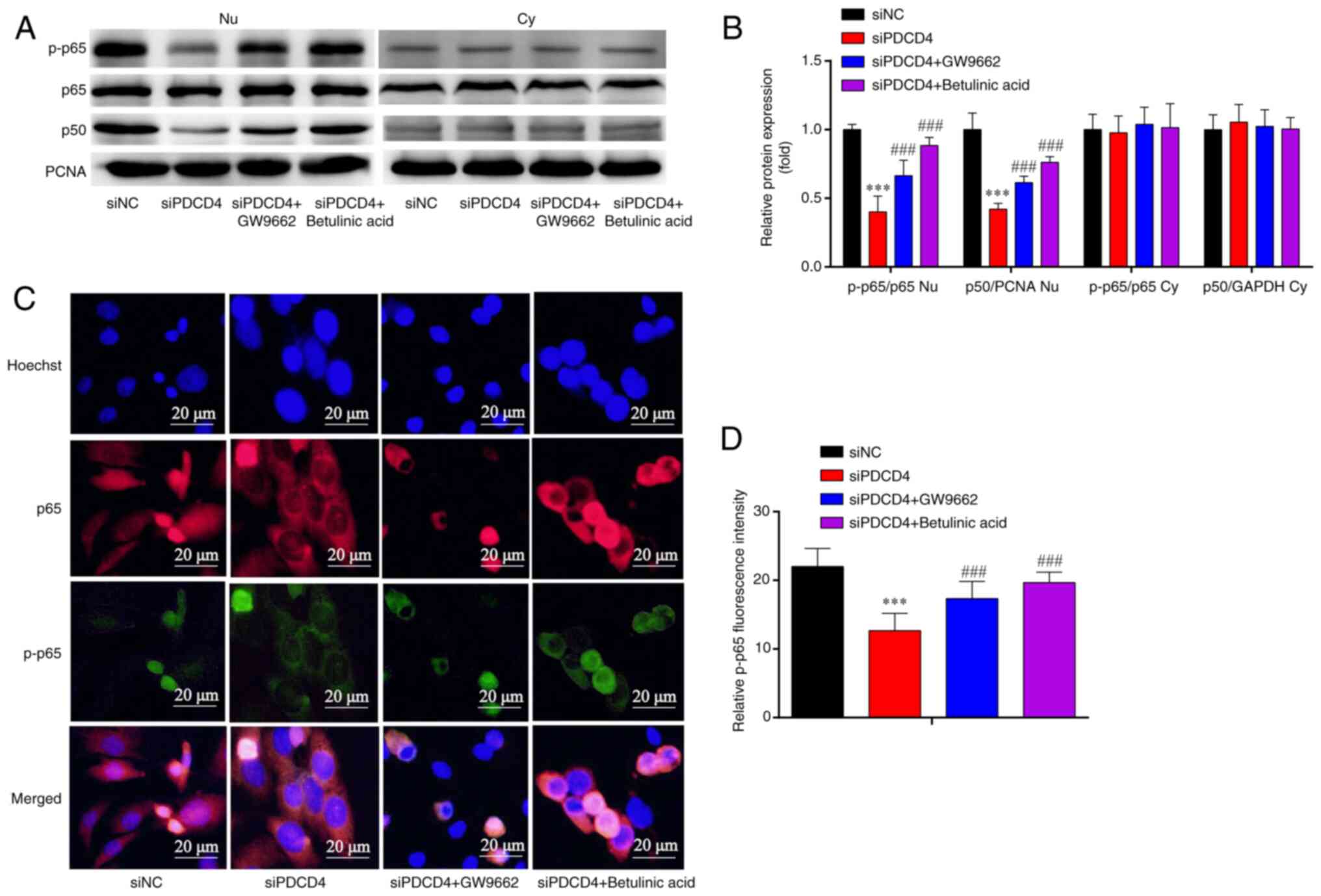 | Figure 8.Expression of NF-κB subunits in
siPDCD4-HL-1. (A) Western blotting showed that, compared with in
the siNC group, the Nu p-p65/p65 ratio and Nu p-p50/PCNA ratio were
decreased in the siPDCD4 group, and the results were partially
reversed by GW9662 and betulinic acid. However, there was no
significant change in Cy p-p65/p65 ratio and Cy p-p50/GAPDH ratio
among the four groups. (B) Statistical analysis of western
blotting. (C) Immunofluorescence analysis showed that the decreased
protein expression of p-p65 in si-PDCD4-HL-1 cells was attenuated
by GW9662 and betulinic acid. (D) Statistical analysis of
fluorescence intensity. Magnification, ×1,000. ***P<0.001 vs.
siNC; ###P<0.001 vs. si-PDCD4. n=3. si, small
interfering; PDCD4, programmed cell death factor 4; NC, negative
control; p-, phosphorylated; PCNA, proliferating cell nuclear
antigen; Nu, nuclear; Cy, cytoplasmic. |
Immunofluorescence analysis revealed that, compared
with that in the siNC group, p-p65 expression was decreased in the
siPDCD4 group; this effect was partially reversed by treatment with
the PPARγ inhibitor (GW9662) and NF-κB agonist (betulinic acid)
(Fig. 8C and D). These findings
suggested that the silencing of PDCD4 reduced the levels of p-p65,
and these effects were reversed by PPARγ inhibitor and NF-κB
agonist in HL-1 cells.
Discussion
In patients with paroxysmal AF, AF is positively
associated with IL-6 and TNF-α expression (2). In addition, patients with AF who
received 12-month catheter ablation were shown to have higher
plasma levels of IFN-γ, IL-6 and TNF-α (21). TNF-α, an endogenous mediator of
inflammation, induces the transcription of MMPs (22). Notably, an increased level of
inflammatory factors, which promotes the expression of related
fibrotic proteins, leads to the development of AF (23). The fibrotic process and
inflammatory process influence each other, forming a vicious cycle,
which eventually promotes structural remodeling in AF.
PDCD4 is involved in the formation of coronary
atherosclerosis by upregulating the transcription of IL-6 (24). The downregulation of PDCD4 has been
reported to attenuate myocardial collagen deposition and fibrotic
remodeling in mice and rats (25,26).
IFN-γ, IL-4, IL-6, IL-17A and TNF-α are common markers of
inflammatory injury in myocardial cells (5,27).
In the present study, the levels of fibrotic proteins (α-SMA,
collagen I, collagen III, fibronectin and MMP2) and
pro-inflammatory factors (TNF-α, IL-6, IL-17A and IFN-γ) were
increased, whereas the levels of the anti-inflammatory factor IL-4
were decreased in response to the overexpression of PDCD4. By
contrast, silencing PDCD4 exerted the opposite effects. Therefore,
it was hypothesized that PDCD4 may possess a crucial function in AF
structural remodeling by regulating inflammation and fibrosis.
However, the mechanisms through which PDCD4 carries out its
functions in inflammation and fibrosis remain unclear.
PDCD4 has been shown to regulate fibrotic expression
by PPARγ (28). The upregulation
of PPAR-γ exerts its antifibrotic effects on liver fibrosis and
lung fibrosis by reducing the collagen I content in veins, and
exerts its anti-inflammatory effects by decreasing IL-17A and IFN-γ
expression (29–32). However, it is unclear whether PDCD4
regulates inflammation and fibrosis in HL-1 cells via PPARγ.
Therefore, oePDCD4-HL-1 cells were treated with a PPARγ agonist and
siPDCD4-HL-1 cells were treated with a PPARγ inhibitor. Notably,
there was a decrease in factors associated with inflammatory damage
and fibrosis in oePDCD4-HL-1 cells following treatment with the
PPARγ agonist; by contrast, there was an increase in factors
associated with inflammatory damage and fibrosis in siPDCD4-HL-1
cells following treatment with the PPARγ inhibitor. These findings
indicated that PDCD4 regulates inflammation and fibrosis in HL-1
cells via the PPARγ pathway and thus serves a role in structural
remodeling in AF.
PDCD4 can trigger local inflammation by activating
NF-κB (11), which promotes the
transcription and translation of inflammatory cytokines (11,33),
and induces inflammation and fibrosis in myocardial cells in mouse
atria (33,34). However, whether PDCD4 affects
fibrosis and inflammation in HL-1 cells via the NF-κB pathway
remains unclear. Therefore, in the present study, oePDCD4-HL-1
cells were treated with an NF-κB inhibitor, whereas siPDCD4-HL-1
cells were treated with an NF-κB agonist. There was a decrease in
factors associated with inflammatory damage and fibrosis in
oePDCD4-HL-1 cells following treatment with the NF-κB inhibitor; By
contrast, there was an increase in factors associated with
inflammatory damage and fibrosis in the siPDCD4-HL-1 cells
following treatment with the NF-κB agonist, suggesting that PDCD4
may regulate fibrosis and inflammation in HL-1 cells via the NF-κB
pathway and is crucial for AF structural remodeling. Furthermore,
the silencing of PDCD4 has been shown to improve left ventricular
structural remodeling in rabbits with type 2 diabetes (12). PDCD4 inhibition has also been shown
to decrease the expression of pro-inflammatory cytokines (IL-1β,
IL-6 and TNF-α) and increase the expression of the
anti-inflammatory factor IL-10 by inactivating NF-κB, thus
attenuating inflammatory injury in macrophage-mediated
inflammation, atherosclerosis and lipopolysaccharide-induced injury
(35–37). However, it is unclear whether the
function of PDCD4 was mediated by NF-κB in HL-1 cells.
NF-κB is a key molecule in inflammatory signaling
pathways; once activated, p65 and p50 heterodimers in the cytoplasm
are translocated to the nucleus, where they are responsible for the
transcription of target genes (38). In the present study,
immunofluorescence was performed and the results indicated that
p-p65 expression was increased in the oePDCD4 group, whereas this
effect was attenuated by treatment with the NF-κB inhibitor. In
addition, p-p65 expression was decreased in the siPDCD4 group and
this effect was reversed by treatment with the NF-κB agonist,
indicating that PDCD4 may regulate inflammation and fibrosis in
HL-1 cells by activating p65. These findings are consistent with
those of previous studies; for example, in a previous study, the
function of PPARγ in inflammation was reported to be inactivation
of NF-κB (18). Furthermore,
PPAR-γ upregulation has been shown to inactivate NF-κB p65 and
exert its antifibrotic effect by reducing type I collagen in veins
(29,30), and a PPARγ agonist (Pio) can
decrease the NF-κB level in the left atrium in rabbits (39).
The present study revealed the following: i) In HL-1
cells, the overexpression of PDCD4 promoted the secretion of
pro-inflammatory cytokines and fibrotic factors, whereas it
inhibited the secretion of anti-inflammatory factors; however,
silencing of PDCD4 exerted the opposite effects. ii) In
oePDCD4-HL-1 cells, PPARγ activation and NF-κB inhibition
attenuated the levels of factors associated with inflammatory
injury and fibrosis; however, in siPDCD4-HL-1 cells, PPARγ
inhibition and NF-κB activation aggravated the levels of factors
associated with inflammatory injury and fibrosis. iii) By PPARγ
indirectly or directly phosphorylating NF-κB p65, PDCD4 may
regulate the expression of inflammation- and fibrosis-related
factors, possibly by inducing the nuclear import of p-p65.
However, several limitations remain in the present
study. The in vitro experimental repeats should be
increased. The clinical safety and feasibility of PDCD4 still
require further animal experiments and clinical studies. Knockdown
of PDCD4 under pathological conditions needs to be performed to
further demonstrate its role in inflammation and fibrosis, which
are under investigation in our ongoing work.
In conclusion, the present study demonstrated that
PDCD4 may promote inflammatory damage and fibrosis by activating
the PPARγ/NF-κB pathway, which could induce the structural
remodeling of HL-1 cells. These findings provide a novel basic
understanding of AF and proposed PDCD4 as a potential therapeutic
target for AF structural remodeling. However, the clinical safety
and feasibility of PDCD4 require further animal experiments and
clinical studies.
Acknowledgements
Not applicable.
Funding
The present study was funded by the National Natural Science
Foundation of China (grant no. 82160067), and the Tianshan
Yingcai-Leading Talents in Scientific and Technological Innovation
Project (grant no. 2022TSYCLJ0065).
Availability of data and materials
The data generated in the present study may be
requested from the corresponding author.
Authors' contributions
LY, YY, JW and MW conceived the project, conducted
the experiments and analyzed the data. ZB, MZ, XW and YZ conducted
the experiments and analyzed the data. MW drafted the manuscript.
LY, YY, JW and MW confirm the authenticity of all the raw data. All
authors read and approved the final version of the manuscript.
Ethics approval and consent to
participate
Not applicable.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Lippi G, Sanchis-Gomar F and Cervellin G:
Global epidemiology of atrial fibrillation: An increasing epidemic
and public health challenge. Int J Stroke. 16:217–221. 2021.
View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Scott L Jr, Li N and Dobrev D: Role of
inflammatory signaling in atrial fibrillation. Int J Cardiol.
287:195–200. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Pozios I, Vouliotis AI, Dilaveris P and
Tsioufis C: Electro-mechanical alterations in atrial fibrillation:
Structural, electrical, and functional correlates. J Cardiovasc Dev
Dis. 10:1492023.PubMed/NCBI
|
|
4
|
Harada M, Van Wagoner DR and Nattel S:
Role of inflammation in atrial fibrillation pathophysiology and
management. Circ J. 79:495–502. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Harada M and Nattel S: Implications of
inflammation and fibrosis in atrial fibrillation pathophysiology.
Card Electrophysiol Clin. 13:25–35. 2021. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Sygitowicz G, Maciejak-Jastrzębska A and
Sitkiewicz D: A review of the molecular mechanisms underlying
cardiac fibrosis and atrial fibrillation. J Clin Med. 10:44302021.
View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Noubiap JJ, Sanders P, Nattel S and Lau
DH: Biomarkers in atrial fibrillation: pathogenesis and clinical
implications. Card Electrophysiol Clin. 13:221–233. 2021.
View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Li Y, Tan W, Ye F, Wen S, Hu R, Cai X,
Wang K and Wang Z: Inflammation as a risk factor for stroke in
atrial fibrillation: Data from a microarray data analysis. J Int
Med Res. 48:3000605209216712020.PubMed/NCBI
|
|
9
|
Liao Y, Tsai L, Lee Y, Hsieh P, Yu C and
Lu M: miR-21 promotes the fibrotic properties in oral mucosa
through targeting PDCD4. J Dent Sci. 17:677–682. 2022. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Zhao M, Zhu N, Hao F, Song Y, Wang Z, Ni Y
and Ding L: The regulatory role of non-coding RNAs on programmed
cell death four in inflammation and cancer. Front Oncol. 9:9192019.
View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Nara K, Kawashima N, Noda S, Fujii M,
Hashimoto K, Tazawa K and Takashi Okiji: Anti-inflammatory roles of
microRNA 21 in lipopolysaccharide-stimulated human dental pulp
cells. J Cell Physiol. 234:21331–21341. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Zhang J, Zhang M, Yang Z, Huang S, Wu X,
Cao L, Wang X, Li Q, Li N and Gao F: PDCD4 deficiency ameliorates
left ventricular remodeling and insulin resistance in a rat model
of type 2 diabetic cardiomyopathy. BMJ Open Diabetes Res Care.
8:e0010812020. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Desai KM, Kale AD, Angadi PV, Datar UV,
Belaldavar C and Arany PR: Role of programmed cell death 4 in
myofibroblast differentiation in oral submucous fibrosis. J Oral
Maxillofac Pathol. 25:430–436. 2021. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Perveen R, Ozaki I, Manirujjaman M, Mine
K, Murata Y, Tanaka K, Xia J, Takahashi H, Anzai K and Matsuhashi
S: Induction of premature senescence and a less-fibrogenic
phenotype by programmed cell death 4 knockdown in the human hepatic
stellate cell line Lieming Xu-2. Hum Cell. 36:583–601. 2023.
View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Takada I and Makishima M: Peroxisome
proliferator-activated receptor agonists and antagonists: A patent
review (2014-present). Expert Opin Ther Pat. 30:1–13. 2020.
View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Gao Z, Xu X, Li Y, Sun K, Yang M, Zhang Q,
Wang S, Lin Y, Lou L, Wu A, et al: Mechanistic insight into PPARγ
and Tregs in atherosclerotic immune inflammation. Front Pharmacol.
12:7500782021. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Yu H, Lin L, Zhang Z, Zhang H and Hu H:
Targeting NF-κB pathway for the therapy of diseases: Mechanism and
clinical study. Signal Transduct Target Ther. 5:2092020. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Korbecki J, Bobiński R and Dutka M:
Self-regulation of the inflammatory response by peroxisome
proliferator-activated receptors. Inflamm Res. 68:443–458. 2019.
View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Livak KJ and Schmittgen TD: Analysis of
relative gene expression data using real-time quantitative PCR and
the 2(−Delta Delta C(T)) method. Methods. 25:402–408. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Lu X, He Y, Johnston RL, Nanayakarra D,
Sankarasubramanian S, Lopez JA, Friedlander M, Kalimutho M, Hooper
JD, Raninga PV and Khanna KK: CBL0137 impairs homologous
recombination repair and sensitizes high-grade serous ovarian
carcinoma to PARP inhibitors. J Exp Clin Cancer Res. 41:3552022.
View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Cabrera-Bueno F, Medina-Palomo C,
Ruiz-Salas A, Flores A, Rodríguez-Losada N, Barrera A,
Jiménez-Navarro M and Javier Alzueta J: Serum levels of
interleukin-2 predict the recurrence of atrial fibrillation after
pulmonary vein ablation. Cytokine. 73:74–78. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Diwan A, Dibbs Z, Nemoto S, DeFreitas G,
Carabello BA, Sivasubramanian N, Wilson EM, Spinale FG and Mann DL:
Targeted overexpression of noncleavable and secreted forms of tumor
necrosis factor provokes disparate cardiac phenotypes. Circulation.
109:262–268. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Chang X, Zhang T, Wang J, Liu Y, Yan P,
Meng Q, Yin Y and Wang S: SIRT5-related desuccinylation
modification contributes to quercetin-induced protection against
heart failure and high-glucose-prompted cardiomyocytes injured
through regulation of mitochondrial quality surveillance. Oxid Med
Cell Longev. 2021:58768412021. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Gao Y, Li H, Zhou Y, Lv H and Chen Y:
PDCD4 expression in coronary atherosclerosis rat models and its
mechanism. Exp Ther Med. 17:3150–3154. 2019.PubMed/NCBI
|
|
25
|
Watanabe K, Narumi T, Watanabe T, Otaki Y,
Takahashi T, Aono T, Goto J, Toshima T, Sugai T, Wanezaki M, et al:
The association between microRNA-21 and hypertension-induced
cardiac remodeling. PLoS One. 15:e02260532020. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Zhou X, Chai H, Bai M and Zhang Z:
LncRNA-GAS5 regulates PDCD4 expression and mediates myocardial
infarction-induced cardiomyocytes apoptosis via targeting MiR-21.
Cell Cycle. 19:1363–1377. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Dobrev D, Heijman J, Hiram R, Li N and
Nattel S: Inflammatory signalling in atrial cardiomyocytes: A novel
unifying principle in atrial fibrillation pathophysiology. Nat Rev
Cardiol. 20:145–167. 2023. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Lu K, Chen Q, Li M, He L, Riaz F, Zhang T
and Li D: Programmed cell death factor 4 (PDCD4), a novel therapy
target for metabolic diseases besides cancer. Free Radic Biol Med.
159:150–163. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Li J, Guo C and Wu J: The agonists of
peroxisome proliferator-activated receptor-γ for liver fibrosis.
Drug Des Devel Ther. 15:2619–2628. 2021. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Zulueta A, Colombo M, Peli V, Falleni M,
Tosi D, Ricciardi M, Baisi A, Bulfamante G, Chiaramonte R and
Caretti A: Lung mesenchymal stem cells-derived extracellular
vesicles attenuate the inflammatory profile of cystic fibrosis
epithelial cells. Cell Signal. 51:110–118. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Kökény G, Calvier L and Hansmann G: PPARγ
and TGFβ-major regulators of metabolism, inflammation, and fibrosis
in the lungs and kidneys. Int J Mol Sci. 22:104312021. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Luo J, Wang J, Zhang J, Sang A, Ye X,
Cheng Z and Li X: Nrf2 deficiency exacerbated CLP-induced pulmonary
injury and inflammation through autophagy- and NF-κB/PPARγ-mediated
macrophage polarization. Cells. 11:39272022. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Sun W, Wu Y, Gao M, Tian Y, Qi P, Shen Y,
Huang L, Shi L, Wang Y and Liu X: C-reactive protein promotes
inflammation through TLR4/NF-κB/TGF-β pathway in HL-1 cells. Biosci
Rep. 39:BSR201908882019. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Ren KW, Yu XH, Gu YH, Xie X, Wang Y, Wang
SH, Li HH and Bi HL: Cardiac-specific knockdown of Bhlhe40
attenuates angiotensin II (Ang II)-Induced atrial fibrillation in
mice. Front Cardiovasc Med. 9:9579032022. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Liu H, Sun J, Gao L, Fan L, Chen D and Wu
L: MicroRNA-421 attenuates macrophage-mediated inflammation by
inhibiting PDCD4 in vitro. Mol Med Rep. 24:5272021.
View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Liang X, Xu Z, Yuan M, Zhang Y, Zhao B,
Wang J, Zhang A and Li G: MicroRNA-16 suppresses the activation of
inflammatory macrophages in atherosclerosis by targeting PDCD4. Int
J Mol Med. 37:967–975. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Zhou G, Duan Y, Lu C and Wang W: Knockdown
of circ-UQCRC2 ameliorated lipopolysaccharide-induced injury in
MRC-5 cells by the miR-326/PDCD4/NF-κB pathway. Int
Immunopharmacol. 97:1076332021. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Wan F and Lenardo MJ: The nuclear
signaling of NF-kappaB: Current knowledge, new insights, and future
perspectives. Cell Res. 20:24–33. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Zhang Z, Zhang X, Meng L, Gong M, Li J,
Shi W, Qiu J, Yang Y, Zhao J, Suo Y, et al: Pioglitazone inhibits
diabetes-induced atrial mitochondrial oxidative stress and improves
mitochondrial biogenesis, dynamics, and function through the
PPAR-γ/PGC-1α signaling pathway. Front Pharmacol. 12:6583622021.
View Article : Google Scholar : PubMed/NCBI
|