Introduction
Tissue factor (TF) is a 47-kDa transmembrane
glycoprotein receptor regarded for its role as the initiator of the
extrinsic coagulation pathway (1–3).
Additionally, TF has non-haemostatic functions that arise from its
ability to activate various intracellular signalling pathways
including PKC, MAPK and AKT pathways (4). Cells typically come into contact with
TF following injury and inflammation (3,5–7).
However, prolonged TF-signalling alters the behaviour of cells
impacting the progression of chronic diseases including malignancy
(8–12). The cellular signals arising from TF
are regulated by proteases and cell-surface receptors that interact
with TF, and appear to be dependent on the concentration of TF
(4,13–17).
These signals also appear to be a determinant of the fate of the
cell, through controlling proliferative and pro-apoptotic mediators
(18). It was previously shown
that exposure of cells to lower concentrations of TF, promoted the
passage through the cell cycle, by upregulating proliferative
mediators including Cyclin D, and downregulating pro-apoptotic
factors (19). By contrast,
exposure of cells to high levels of TF, or the inability to release
excess TF promoted cellular apoptosis (20). This was initiated through
over-activation of steroid receptor coactivator-1 (Src1) by
β1-integrin, leading to prolonged p38-MAPK activation, and
subsequent increase in p53 nuclear localisation and Bax expression
(20,21). Consequently, the precise regulation
of cell-surface TF, permits the achievement of the optimal
proliferative and pro-survival signals, which may be exploited by
cancer cells for maximal growth (22). Highly proliferative tumours appear
to express (14,23,24)
and moderate (25) cell-surface TF
through different mechanisms in order to achieve the optimal cell
growth.
The transit of cells through the G1/S checkpoint is
mainly regulated by the inhibitors of CDK (INK) and the CDK
interacting protein (CIP)/wildtype p53-activated fragment (WAF)
family of tumour suppressors (26–29).
These proteins act as gatekeepers by inhibiting the kinase function
of Cyclin/Cyclin dependant kinases (Cdk) complexes (29,30).
The INK family of proteins (p16INKa, p15INKb,
p18INKc and p19INKd) are activated in
response to different stimuli, and inhibit Cyclin D in the Cyclin
D/Cdk-4 or Cdk-6 complexes (29,30).
Inhibition of the Cyclin D/Cdk-4/6 complex suppresses the
phosphorylation of retinoblastoma protein, which in turn suppresses
the expression of the genes essential for the S-phase of the cell
cycle by Early region 2 binding factor (E2F) transcription factor
(29,30). This constitutes the final
pro-mitogenic regulatory step in the progression of cell division
(31). The second stage of the
G1/S checkpoint is mitogen-independent and is promoted by the
Cyclin E/Cdk-2 complex formation and regulated by
p21CIP1/WAF1 and p27KIP1 (28–29).
Notably, at low concentrations, p21CIP1/WAF1 and
p27KIP1 proteins act as essential promoters of the
progression through the G1 phase by competing with
p16INKa, to facilitate the association between Cyclin D
and Cdk-4 or Cdk-6 (32–34). As such, p21CIP1/WAF1 has
been classified as a dual-function tumour suppressor and an
oncogene, simultaneously (35).
Treatment of cells with TF has been shown to impact the expression
of the regulatory proteins at both of the stages of the G1/S
checkpoint in various cell types (19,20).
For example, exposure of human umbilical vein endothelial cells
(HUVECs) to TF resulted in the upregulation of a number of
proliferative mediators including Cyclin D (19). TF was also reported to suppress the
expression of p21CIP1/WAF1 and p27KIP1 in
HUVECs (19). Furthermore,
preventing the release of TF within microvesicles (MVs) resulting
in an accumulation of the protein within human coronary artery
endothelial cells, resulted in increased expression of
p21CIP1/WAF1, while enhancing the release of TF lowered
the expression of p21CIP1/WAF1 (20). It has also been shown that MVs
isolated from patients with coronary artery syndrome promote the
upregulation of p16INKa and p21CIP1/WAF1 in
cultured endothelial cells (36,37).
Although the proliferative influences of TF have
been reported, the exact mechanism of signalling pathways has not
yet been elucidated. TF initiates cellular signalling by both
protease-dependent mechanisms involving the activation of
protease-activated receptor (PAR) 2 (38–41),
and protease-independent mechanisms involving the interaction of TF
with β1-integrin (42–44). Activation of PAR2 has been reported
to induce cell proliferation in a wide range of cell types
including vascular endothelial cells, pancreatic cancer cells and
colon cancer cell line SW620 (45–47).
However, induction of proliferation in HUVECs following TF
treatment was independent of coagulation factor VIIa (fVIIa)
(19) and disrupting
TF-β1-integrin interactions with a β1-integrin fragment peptide
prevented TF induced proliferation in human endothelial cells
(44). Furthermore, TF-containing
MVs (TF + MVs) derived from different cell lines appear to have
differential outcomes on endothelial cells, which was dependent on
the presence of other proteins carried on the MVs including
protease activated receptors and integrins (44). Therefore, to further decode the
underlying mechanisms, recombinant TF, together with antibodies
against TF, β1-integrin and PAR2 were used in the
present study, and the contribution of TF signals to regulation of
the cell cycle was examined. The present study hypothesised that
signalling from TF differentially regulates the G1/S cell cycle
checkpoint, and modulates the fate of cells by inducing apoptosis
and/or proliferation. In addition, the outcomes of prolonged
exposure of cells to TF as an environmental contributor to disease
were explored. It was hypothesised that protracted exposure to TF,
such as observed during prolonged inflammation, results in adaptive
alterations in these mechanisms which in turn leads to aberrations
detectable during chronic conditions.
Materials and methods
Cell culture and treatment
Human dermal blood endothelial cells (HDBECs;
http://promocell.com/uk_en/human-dermal-blood-endothelial-cells-hdbec.html)
and HUVECs (https://promocell.com/uk_en/human-umbilical-vein-endothelial-cells-huvec.html)
were purchased from PromoCell GmbH and cultured at 37°C in MV
medium (PromoCell GmbH) or M199 medium (Lonza Group, Ltd.)
containing 10% (v/v) foetal calf serum (FCS) (Gibco; Invitrogen;
Thermo Fisher Scientific, Inc.) until 80% confluent (19). These are primary cells which were
used at ~9 divisions (cells were guaranteed for at least 15
divisions). The immortalised pancreatic epithelial cells [human
telomerase reverse transcriptase (hTERT)-human pancreatic
nestin-expressing ductal cells (HPNE)] were purchased from American
Type Culture Collection (ATCC). The hTERT-HPNE (https://www.atcc.org/products/crl-4023)
were expanded at 37°C in DMEM (Lonza Group, Ltd.):M3 Base medium
(INCELL Corporation LLC; 75:25%) containing 5% (v/v) FCS, 10 ng/ml
human epidermal growth factor (hEGF; Sino Biological/Stratech
Scientific Ltd.) and 100 U/ml Penicillin-100 µg/ml Streptomycin
antibiotics (Lonza Group, Ltd.) until 80% confluent. The pancreatic
cancer cell line, AsPC-1 (https://www.atcc.org/products/crl-1682) was purchased
from ATCC and cultured at 37°C in RPMI-1640 medium (Lonza Group,
Ltd.), containing 10% (v/v) FCS until 80% confluent. The cell lines
were used after 3–4 passages from the time of purchase. Sets of
cells (2×105) were seeded out in 12-well plates and
adapted to serum-free medium prior to treatment with combinations
of agents as is described below, and is also stated for each figure
in the results section, and used in experiments once the cells had
reached ~85% confluence. The cells were activated by the addition
of recombinant relipidated Innovin TF (stock=0.13 µg/ml=1,000 U/ml;
Dade Behring, Inc.), or PAR2-agonsit peptide (PAR2-AP) SLIGKV (20
µM; Sigma-Aldrich; Merck KGaA) and incubated at 37°C overnight, or
for the durations described in the results section. The two
concentrations of recombinant relipidated TF utilised throughout
the present study were selected to mimic mild physiological
inflammation or severe pathological disease. The lower
concentration of TF (0.5 U/ml) was comparable to, but higher than
that detected in healthy plasma as determined using the Quantikine
ELISA kit (Human Coagulation Factor III/Tissue Factor; cat. no.
DCF300; R&D Systems Europe, Ltd.) (48) and represents mild
inflammation/injury. The higher concentration (2 U/ml) was in line
with the amount of TF released by the U87 cell line (48) but below some of the values reported
in the plasma of patients with severe cancer (49). In some experiments the cells were
incubated with the CDK4/6 inhibitor ribociclib (10 nM) in the
presence or absence of TF (0.5 U/ml) at 37°C, overnight.
In some experiments, TF was pre-incubated at 37°C
for 60 min with 10H10 antibody (20 µg/ml; cat. no. 9010-5059;
Bio-Rad Laboratories, Inc.) to block TF proliferative signalling
via the TF exosite (50) or HTF1
antibody (20 µg/ml; cat. no. 16-1429-85; eBioscience; Thermo Fisher
Scientific, Inc.) to block the protease activity of the TF-fVIIa
complex (51). In other
experiments, the cells were pre-incubated at 37°C for 60 min with
AIIB2 antibody (20 µg/ml; cat. no. AIIB2-c; Merck KGaA) to block
β1-integrin signalling, or with SAM11 antibody (20 µg/ml; cat. no.
sc-13504; Santa Cruz Biotechnology, Inc.) to block PAR2 activation
prior to the addition of TF. The cells were incubated at 37°C for
24 h and samples were separated for mRNA and protein isolation as
described below. Finally, sets of HDBECs and HUVECs were incubated
with TF (in the presence and absence of the inhibitory antibodies,
as aforementioned) or PAR2-AP (20 µM) for 24 h at 37°C. The cells
were then fixed with glutaraldehyde (3% v/v) at room temperature
for 10 min, washed 3 times with PBS and cell numbers were assessed
using crystal violet (0.02% w/v) staining (Sigma-Aldrich; Merck
KGaA) as described previously at room temperature for 30 min
(52). The stain was then eluted
in 1% (w/v) sodium dodecyl sulphate solution at room temperature
for 10 min, and absorptions measured at 590 nm as described
previously (52).
For prolonged treatment, epithelial cells were
cultured at 37°C in 25 cm2 flasks and repetitively
supplemented with recombinant TF (0.5 U/ml) every 2–3 days. The
cells were passaged every 7 days, at which time the cells had reach
~90% confluence. The cells were counted manually using a
haemocytometer and samples collected for mRNA and protein isolation
as described below.
RNA isolation and RT-PCR
Total RNA was isolated using the Monarch total RNA
extraction kit (New England BioLabs, Inc.) from 1×105
cells. Samples of the extracted RNA (100 ng) were amplified using
the primer sets shown in Table
SI. The relative amount of each mRNA was determined against
β-actin using QuantiTect primer set (Qiagen AB; sequence not
disclosed by the company). Reverse transcription (RT) and qPCR were
carried-out sequentially using the GoTaq 1-Step RT-qPCR System
(cat. no. A6020; Promega Corporation). GoTaq 1-Step RT-qPCR System
contained GoScript Reverse Transcriptase and RNasin Plus RNase
Inhibitor. RT was performed at 48°C for 30 min. The GoTaq 1-Step
RT-qPCR System also contained GoTaq Hot Start Polymerase, BRYT
Green fluorescent dye, MgCl2, dNTPs and a proprietary
reaction buffer. The qPCR reactions consisted of a denaturing step
at 95°C for 15 sec and an annealing and extending step at 60°C for
1 min. The reactions were performed using an iCycler thermal cycler
(Bio-Rad Laboratories, Inc.) for 40 cycles. Following
amplification, the relative amounts of target mRNA were determined
using the 2−ΔΔCq method (53). In some experiments and for
illustrative purposes, end-point RT-PCR amplifications were carried
out using the primers shown in Table
SI for the number of cycles shown and the products were
analysed by 2% (w/v) agarose gel (Thermo Fisher Scientific, Inc.)
electrophoresis.
Western blot analysis
Cells (1×105) were lysed in Cell culture
lysis reagent (cat. no. E1531; Promega) at 4°C for 30 min on a
rotator. The protein content of samples was assessed using Pierce
BCA protein assay (cat. no. 23227; Thermo Fisher Scientific, Inc.)
in accordance with the manufacturer's instructions. Samples were
added to Laemmli Buffer (cat. no. S3401-10VL; Sigma-Aldrich; Merck
KGaA; solution contains 4% SDS, 20% glycerol, 10%
2-mercaptoethanol, 0.004% bromphenol blue and 0.125 M Tris HCl; pH
~6.8). Aliquots (10 µg protein) of the lysates were separated by
electrophoresis carried out on a denaturing 14% (w/v)
polyacrylamide gel (Flowgen). The separated proteins were then
transferred to a nitrocellulose membrane (GE Healthcare) and
blocked with Tris-buffered saline Tween 0.01% (v/v) (TBST;
Sigma-Aldrich; Merck KGaA; pH 8) at room temperature for 60 min.
The membranes were probed overnight at 4°C with either a goat
anti-human p16 antibody (1:2,000 v/v; cat. no. AF5779; R&D
Systems Europe, Ltd.), a mouse anti-human p21 antibody (WA-1; cat.
no. MCA2325; Bio-Rad Laboratories, Inc.), a rabbit anti-human p14
antibody (cat. no. abx013162; Abbexa, Ltd.) or a rabbit anti-human
Cyclin E1 antibody (cat. no. abx012757; Abbexa, Ltd.), each diluted
1:3,000 (v/v) in TBST. Membranes were then washed and developed at
room temperature for 60 min with goat anti-mouse IgG (cat. no.
sc-2008), goat anti-rabbit IgG (cat. no. sc-2007) or donkey
anti-goat IgG (cat. no. sc-2022) alkaline phosphatase-conjugated
antibodies (Santa Cruz Biotechnology, Inc.), diluted 1:3,000 (v/v)
in TBST, and visualised using the Western Blue stabilised alkaline
phosphatase-substrate (Promega Corporation). All measurements were
normalised against the respective GAPDH band probed using an
HRP-conjugated rabbit anti-human GAPDH (W17079A; cat. no. 607901;
BioLegend, Inc.), or a goat anti-human GAPDH antibody (V18; cat.
no. sc-20357; Santa Cruz Biotechnology, Inc.) diluted 1:3,000 (v/v)
in TBST. Band densities were analysed using the ImageJ 1.53t
Software (National Institutes of Health).
Construction of E2F reporter vector
and measurement of transcriptional activity
The preferred consensus sequence for binding of E2F
transcription factor has previously been demonstrated to include
the sequence 5′-TTTCGCGC-3′ (54–58).
The double-stranded consensus binding DNA for E2F-1 transcription
factor (5′-ATTTAAGTTTCGCGCCCTTTCTCA-3′) was
synthesised with Mlu I and Bgl II restriction sites at the two ends
of the underlined preferred sequence, to be unidirectionally cloned
into the pGL3-promoter vector (Promega Corporation), and successful
clones identified by sequencing (Eurofins Scientific). The
pGL3-promoter vector contains a SV40 promoter upstream of the
luciferase gene, but does not include an enhancer element required
for efficient transcription. The plasmid DNA-construct (1 µg) was
transfected into HDBECs (1×105) using TransIT-2020 (3
µl; Geneflow, Ltd.) according to the manufacturer's instructions.
The cells were incubated at 37°C for 24 h to allow the plasmid to
be expressed. The cells were then treated with recombinant TF (0–2
U/ml) as described below for a further 24 h, and the luciferase
activity was measured using Nano-Glo® Luciferase Assay
Substrate (Promega Corporation) and a luminometer (Junior LB 9509;
Titertek-Berthold). The measurements were carried out alongside
appropriate positive and negative controls which were treated with
20% (v/v) FCS or were serum-starved, respectively.
In vitro measurement of retinoblastoma
protein phosphorylation at Thr821/826
Sets of endothelial cells (5×104) were
seeded out in 96-well plates and treated with recombinant TF (0–2
U/ml) at 37°C, overnight. The cells were then fixed at room
temperature for 10 min using glutaraldehyde (3% v/v), permeabilised
and incubated at room temperature with a goat
anti-phospho-Thr821/826 human retinoblastoma protein antibody
(1:1,000 v/v; cat. no. sc-16669; Santa Cruz Biotechnology, Inc.) in
TBST overnight. The cell samples were then washed and probed with
an HRP-conjugated donkey anti-goat IgG (1:3,000 v/v; cat. no.
sc-2020; Santa Cruz Biotechnology, Inc.) at room temperature for 1
h, and developed with TMB substrate (Promega Corporation). The
relative amounts of the phosphorylated protein were determined by
measuring the absorptions at 450 nm (PolarStar Optima plate reader,
BMG Labtech GmbH). This procedure was only used qualitatively since
both dephosphorylation and hyperphosphorylation of retinoblastoma
protein influence its function.
Analysis of DNA methylation by
bisulphite conversion
Genomic DNA (gDNA) was extracted from HPNE cells
(3×104 cells) using the Monarch Genomic DNA purification
kit (New England BioLabs, Inc.) according to manufacturer's
instructions. Bisulphite conversion of the gDNA (750 ng) was
carried out using the MethylDetector Bisulfite Modification Kit
(Active Motif, Inc.) according to manufacturer's instructions. To
assess the methylation state of the extracted gDNA, bisulphite
modified DNA was used in nested methylation specific (MS)-PCR
experiments with MS-primers to the p16 gene promoter region,
provided with the MethylDetector kit. The PCR amplification was
carried out using 10 ng bisulphite modified gDNA, with Taq DNA
polymerase (1U) in the supplied reaction buffer (Invitrogen; Thermo
Fisher Scientific, Inc.), 200 µM of each primer, 0.2 mM dNTPs, 1.5
mM MgCl2 and 5% (v/v) DMSO (Sigma-Aldrich; Merck KGaA).
Each of the nested amplification steps was carried out for 35
cycles at an annealing temperature of 60°C. The outer reaction was
carried out using primers specific for methylated DNA;
5′-TTATTAGAGGGTGGGGCGGATCGC-3′ (forward) and
5′-CCACCTAAATCGACCTCCGACCG-3′ (reverse), and also using synthesised
primers, specific for unmethylated DNA;
5′-TTATTAGAGGGTGGGGTGGATTGT-3′ (forward) and
5′-CCACCTAAATCAACCTCCAACCA-3′ (reverse). Aliquots (4 µl) of the
outer PCR reactions were then used as the template for the inner
PCR reactions using primers specific for methylated DNA;
5′-TTATTAGAGGGTGGGGCGGATCGC-3′ (forward) and
5′-GACCCCGAACCGCGACCGTAA-3′ (reverse), and also synthesised primers
specific for unmethylated DNA; 5′-TTATTAGAGGGTGGGGTGGATTGT-3′
(forward) and 5′-CAACCCCAAACCACAACCATAA-3′ (reverse). The products
(149 bp) were then examined by 2% (w/v) agarose gel
electrophoresis. Samples of the gDNA were also amplified by PCR
using primers for β-actin (forward 5′-TGATGGTGGGCATGGGTCAGA-3′ and
reverse 5′-CTGTGGTGGTGAAGCTGTAG-3′) and the products were examined
in parallel, as loading control.
Statistical analysis
Presented data include the calculated mean values ±
the calculated standard error of the mean from the number of
experiments indicated in each figure. Statistical analysis was
carried out using the GraphPad Prism version 9.0 (Dotmatics).
Significance was determined using one-way ANOVA and Tukey's post
hoc test. P<0.05 was considered to indicate a statistically
significant difference.
Results
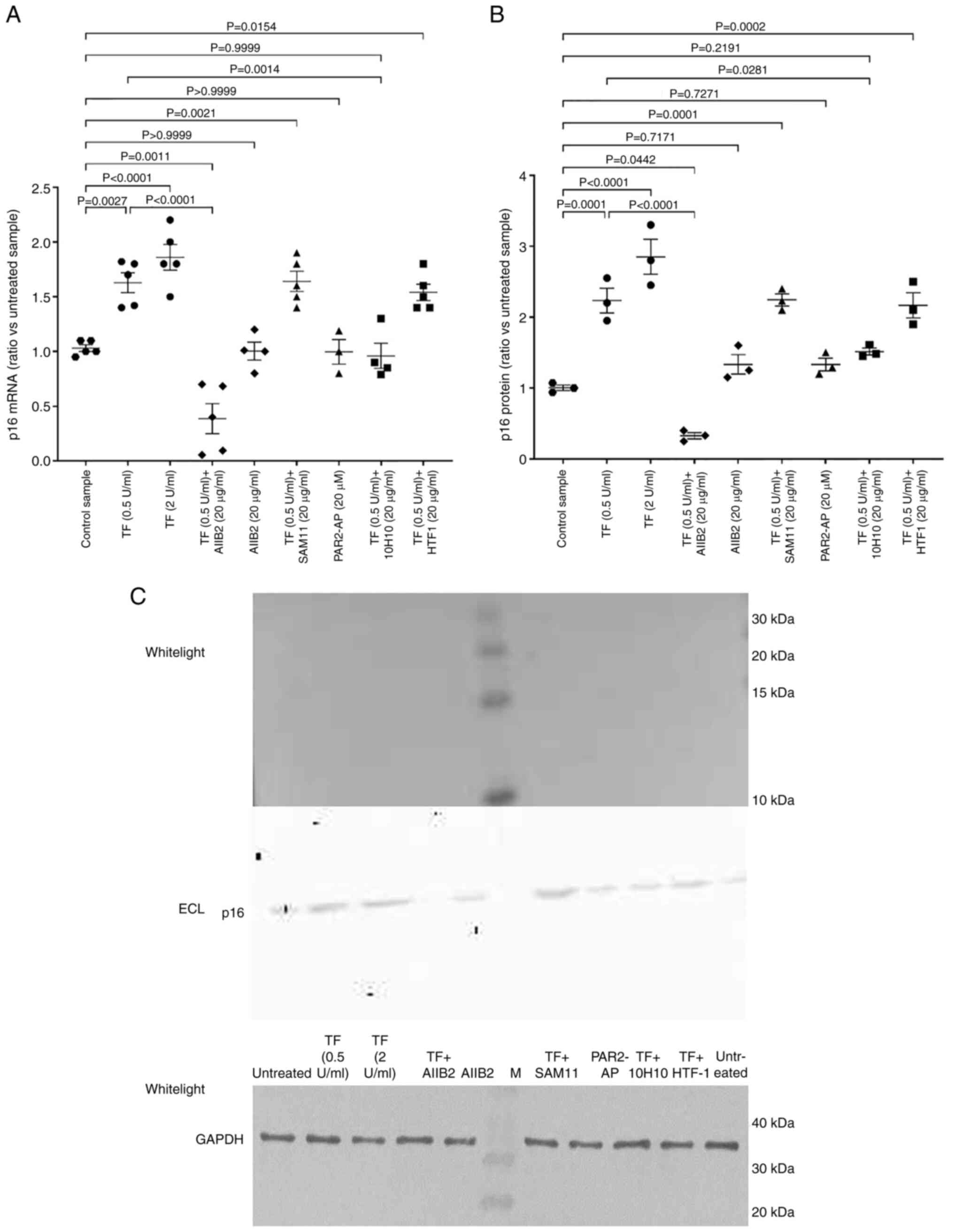
Exposure of cells to exogenous TF
modulates the expression of p16INKa
The experiments in this part of the present study
were carried out in HDBECs and HUVECs, which have previously been
shown to have similar properties and respond comparably to
inflammatory stimuli (59).
Incubation of endothelial cells with either 0.5 or 2 U/ml
recombinant TF resulted in increased expression of
p16INKa mRNA in HDBECs (Fig. 1A) and HUVECs (Fig. S1A) when compared with the control
cells which were untreated. In order to explore the mechanism of
TF-mediated regulation of p16INKa, cells were
pre-incubated for 60 min with antibodies which block the signalling
by β1-integrin (AIIB2) or PAR2 (SAM11). Alternatively, the
recombinant TF was pre-incubated for 60 min with 10H10 antibody to
block TF direct signalling (50),
or HTF1 antibody to block the protease activity of the TF-fVIIa
complex (51). Finally, groups of
cells were incubated with the PAR2-activating peptide (SLIGKV). The
TF-induced increase in p16INKa-mRNA expression was
suppressed following the inhibition of β1-integrin signalling
(using AIIB2), or by blocking of the exosite on TF using 10H10
antibody when compared with cells supplemented with recombinant TF
only (Fig. 1A). The blocking of
PAR2 activation using SAM11 antibody, or pre-incubation of TF with
HTF1, did not influence the upregulation of p16INKa mRNA
expression by TF. Moreover, activation of PAR2 using the agonist
peptide alone did not alter the p16INKa expression.
Analysis of p16INKa protein by western blotting
confirmed the observed alterations in mRNA expression (Fig. 1B and C). Collectively these data
indicate that the expression of p16INKa was dependent on
the interaction of TF with β1-integrin and was unaltered on
prevention of the activation of PAR2. In agreement with the data
obtained using the endothelial cells, supplementation of hTERT-HPNE
and AsPC-1 cells with recombinant TF (0.5 and 2 U/ml) resulted in
50 and 60% increases in p16INKa mRNA expression
respectively in both cell types (data not shown). These cells were
not examined in the presence of any of the inhibitory
antibodies.
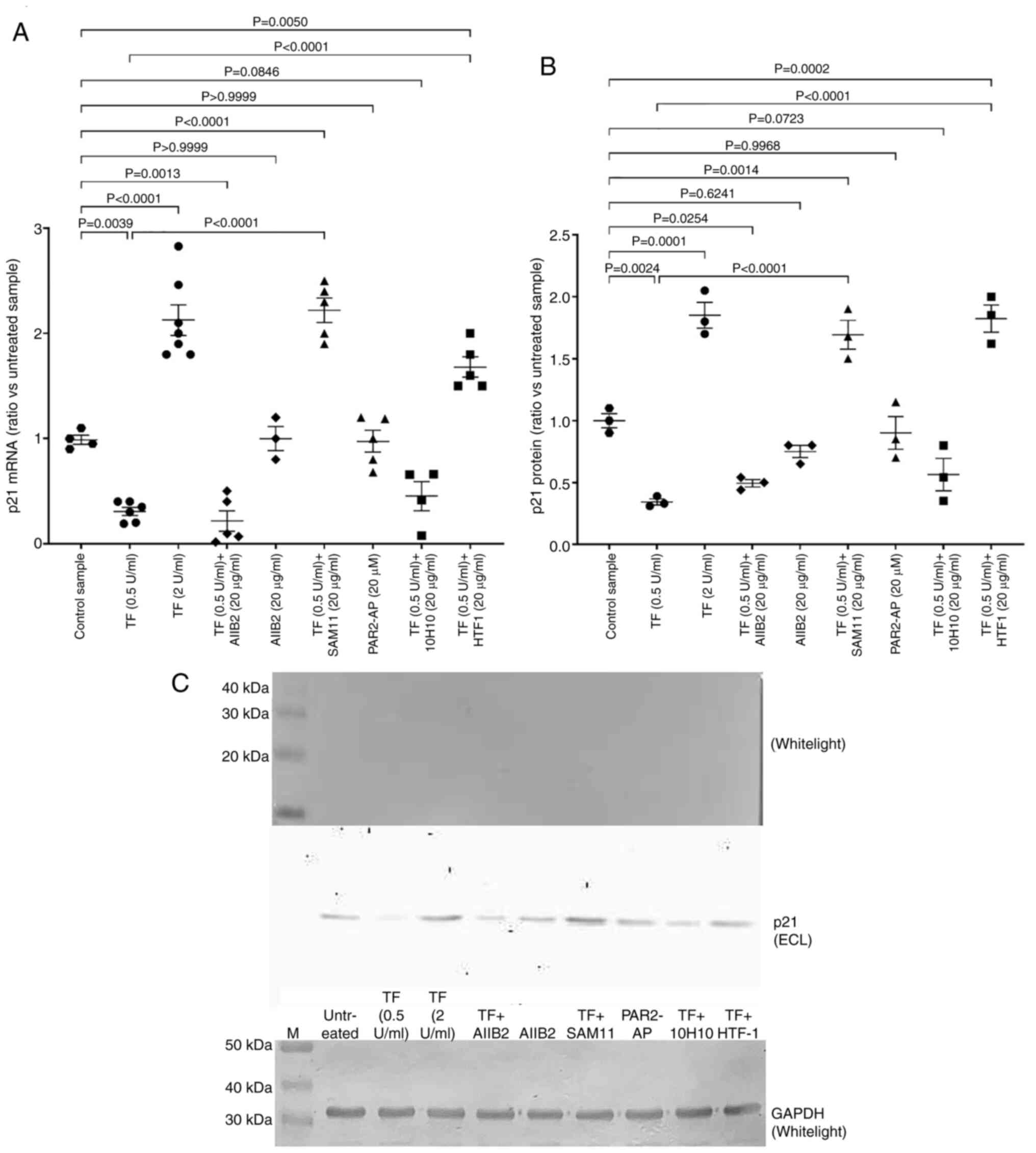
Exposure of cells to exogenous TF
modulates the expression of p21CIP1/WAF1
Incubation of endothelial cells with the lower
concentration of recombinant TF (0.5 U/ml) reduced the expression
of p21CIP1/WAF1 mRNA, whilst treatment with the higher
concentration of TF (2 U/ml) significantly increased
p21CIP1/WAF1 mRNA expression when compared with
untreated control cells (Figs. 2A
and S1B and C). Inhibition of
β1-integrin signalling (AIIB2) on cells prior to the addition of
recombinant TF (0.5 U/ml) marginally reduced the expression of
p21CIP1/WAF1 when compared with cells supplemented with
TF only (Fig. 2A). Notably,
inhibiting PAR2 activation using SAM11 antibody on cells, and to a
lesser extent blocking TF proteolytic activity using the HTF1
antibody, reversed the reduction in p21CIP1/WAF1
expression. These increases in p21CIP1/WAF1 expression
were comparable with those observed with the higher TF
concentration (2 U/ml). By contrast, induction of
p21CIP1/WAF1 expression was not significantly influenced
by the blocking of the TF exosite, and also appeared to be
unaffected by the direct activation of PAR2 alone. Analysis of
p21CIP1/WAF1 protein by western blotting further
confirmed the observed alterations in mRNA expression (Fig. 2B and C). These data indicate the
possible involvement of β1-integrin signalling in TF-induced
regulation of p21CIP1/WAF1, as well as highlighting the
exaggerated enhancement of p21CIP1/WAF1 expression on
prevention of PAR2 activation.
Exposure of cells to exogenous TF
modulates the expression of cyclin D and E2F activity
Incubation of endothelial cells with the lower
concentration of recombinant TF (0.5 U/ml) resulted in increased
transcriptional activity of E2F, as measured using the luciferase
reporter when compared with control cells which were untreated
(Fig. 3A). By contrast, incubation
of the cells with the higher concentration of TF (2 U/ml) reduced
E2F activity. These values were in line with the relative amounts
of phosphorylated retinoblastoma protein (Thr821/826) (Fig. 3B). These measurements were used
semi-quantitatively as an indicator of the state of the protein,
since both dephosphorylation and hyperphosphorylation of
retinoblastoma protein reduce its function. Treatment of cells with
SAM11 to block PAR2, or pre-incubation of recombinant TF with HTF1
to inhibit the protease function prevented the change in E2F
activity. Moreover, activation of PAR2 on the cells induced E2F
activity. Incubation of cells with the CDK4/6 inhibitor ribociclib
(10 nM) suppressed E2F activity regardless of the presence of TF
(Fig. S2). Incubation of cells
with recombinant TF promotes the upregulation of cyclin D1 mRNA
(19), and was used as an
indicator of the entry into the G1-phase of the cell cycle. In the
present study, incubation of endothelial cells with recombinant TF
resulted in dose-dependent increases in cyclin D1 mRNA expression
(Figs. 3C and S1B and D). Entry into the G1-phase was
also promoted by the direct activation of PAR2, and was reduced by
either blocking of PAR2 activation with SAM11 antibody prior to
addition of TF, or by inhibition of protease function of the
TF-fVIIa complex using the HTF1 antibody (Fig. 3C). Neither blocking of β1-integrin
signalling, nor blocking of the exosite on TF using 10H10 antibody
had any significant influence on cyclin D1 mRNA expression. To
explore the potential outcome on cell proliferation, the treated
cells were incubated for 24 h and cell numbers were determined. In
agreement with the aforementioned findings, incubation of cells
with the lower concentrations of recombinant TF (0.5 /ml) or the
direct activation of PAR2 promoted increases in HDBEC numbers
(Figs. 3D and S3), and HUVEC numbers (Fig. S1E) as measured by crystal violet
assay. By contrast, treatment of cells with the higher
concentration of recombinant TF resulted in the reduction in cell
numbers (Figs. 3D and S3 and S1E).
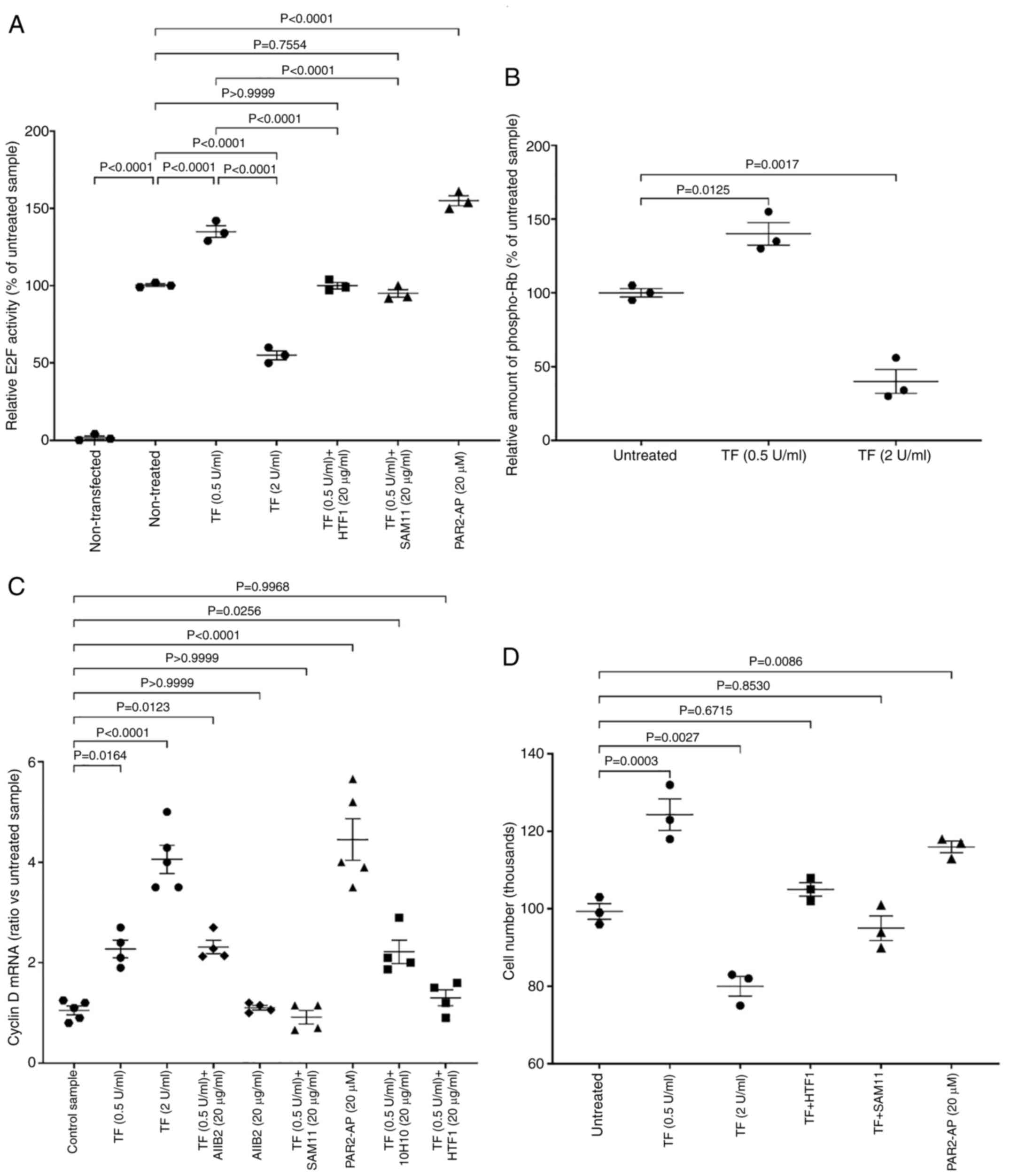 | Figure 3.Examination of the influence of TF on
cell cycle indicators. (A) Groups of HDBECs (1×105) were
transfected with the pGL3-promoter vector containing the E2F-1
enhancer sequence. The cells were then treated for 24 h with
recombinant TF (0, 0.5 and 2 U/ml), combinations of TF (0.5 U/ml)
with the shown antibodies, or with PAR2-AP (SLIGKV; 20 µM) and the
luciferase activity was measured within 24 h (n=3). (B) Groups of
cells (5×104) were seeded in 96-well plates and treated
with recombinant TF (0, 0.5 and 2 U/ml) for 24 h. The cells were
then fixed and probed with an anti-phospho-Thr821/826 human
retinoblastoma protein antibody (1:1,000 v/v) for 1 h. The samples
were then incubated with an HRP-conjugated donkey anti-goat IgG
diluted 1:3,000 v/v) for 1 h, developed with a TMB substrate and
the absorptions determined at 450 nm (n=3). (C) Groups of cells
(1×105) were incubated for 24 h with TF (0, 0.5 and 2
U/ml) or PAR2-AP (20 µM), or with recombinant TF (0.5 U/ml) that
was pre-incubated for 60 min with 10H10 or HTF1 antibodies (20
µg/ml). Groups of cells were also pre-incubated for 60 min with
AIIB2 or SAM11 antibodies (20 µg/ml), prior to addition of
recombinant TF. The cells were harvested after 24 h, total RNA was
isolated and the amount of cyclin D1 mRNA determined against that
of β-actin (n=4). (D) Groups of cells (1×105) were
treated as aforementioned and cell numbers were determined using
crystal violet staining (n=3). TF, tissue factor; PAR2,
protease-activated receptor 2; E2F-1; Early region 2 binding
factor. |
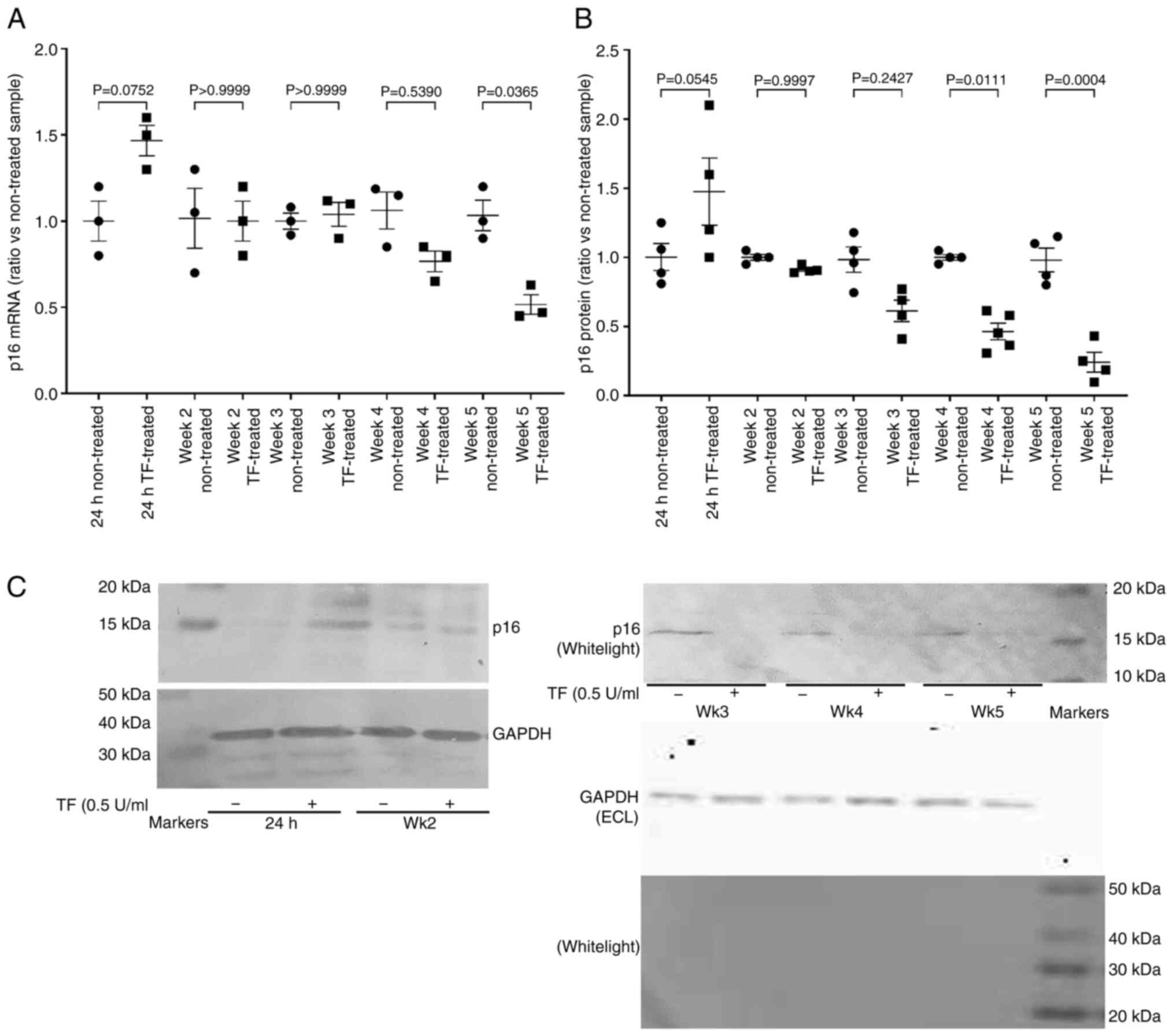
Prolonged exposure of cells to
exogenous TF alters the regulations of G1/S checkpoint
proteins
In order to assess the influence of prolonged
exposure to TF, hTERT-HPNE cells were supplemented with recombinant
TF (0.5 U/ml) every 2–3 days for up to 5 weeks, and the outcome on
p16INKa mRNA and protein levels examined. Similar
studies in primary endothelial cells were not feasible due to the
durations involved. As aforementioned, supplementation of
hTERT-HPNE with recombinant TF resulted in a 50% increase in
p16INKa mRNA expression within 24 h when compared with
untreated control cells (Fig. 4A).
These values were normalised by week 2, but further decline only
became significant on week 5 when compared with untreated control
cells (Fig. 4A). Short-term
treatment (24 h) of AsPC-1 cells with 0.5 U/ml of TF also resulted
in a 50% increase in p16INKa mRNA, but was not analysed
further. In HPNE cells the level of cellular p16INKa
protein began to decline by week 3 of TF treatment when compared
with control cells grown for the same number of weeks in untreated
media (Fig. 4B and C). Examination
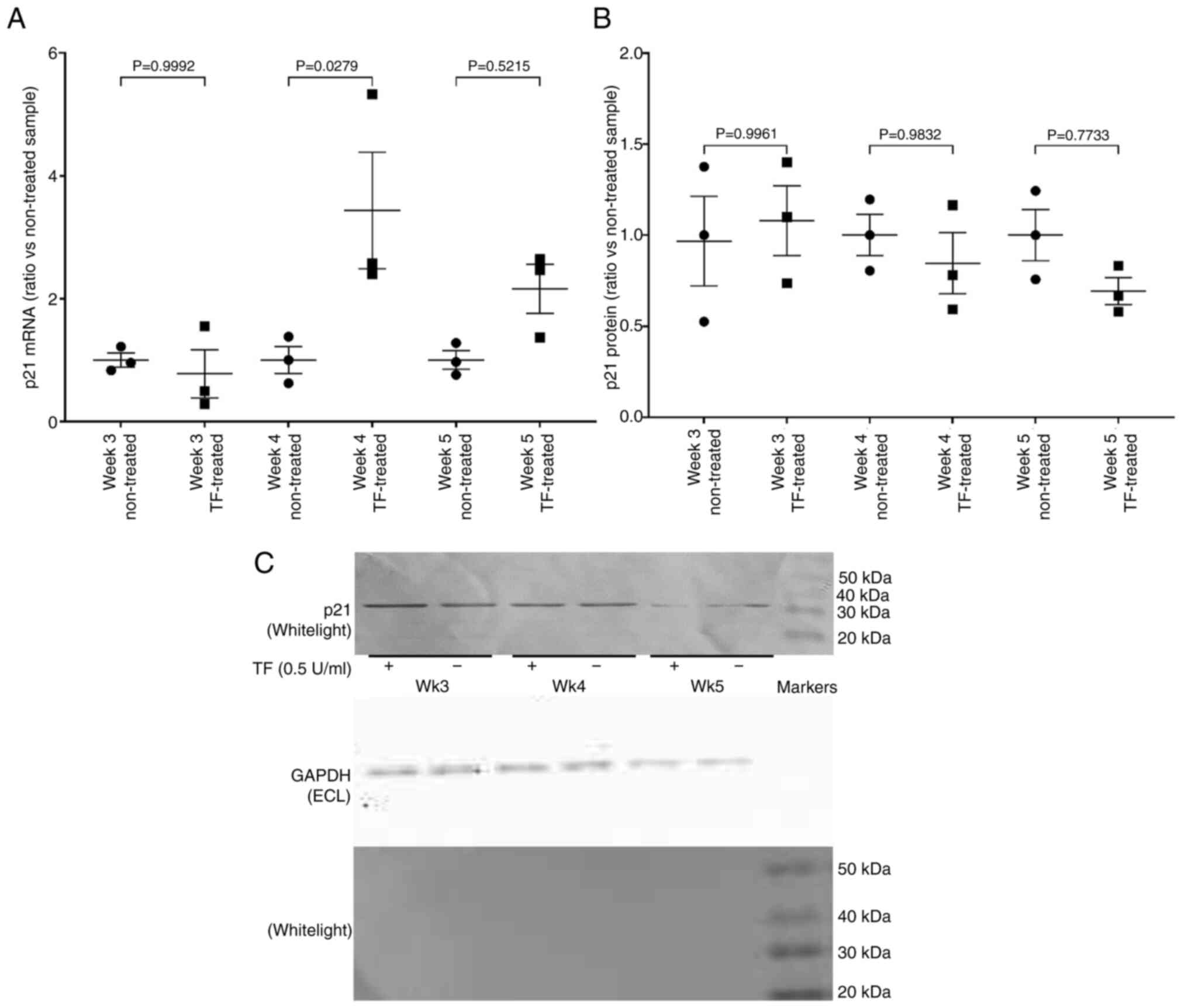
of the levels of p21CIP1/WAF1 mRNA over the same period,
indicated increased levels of mRNA expression on weeks 4 and 5
(Fig. 5A), but was not reflected
in increased cellular p21CIP1/WAF1 antigen (Fig. 5B and C).
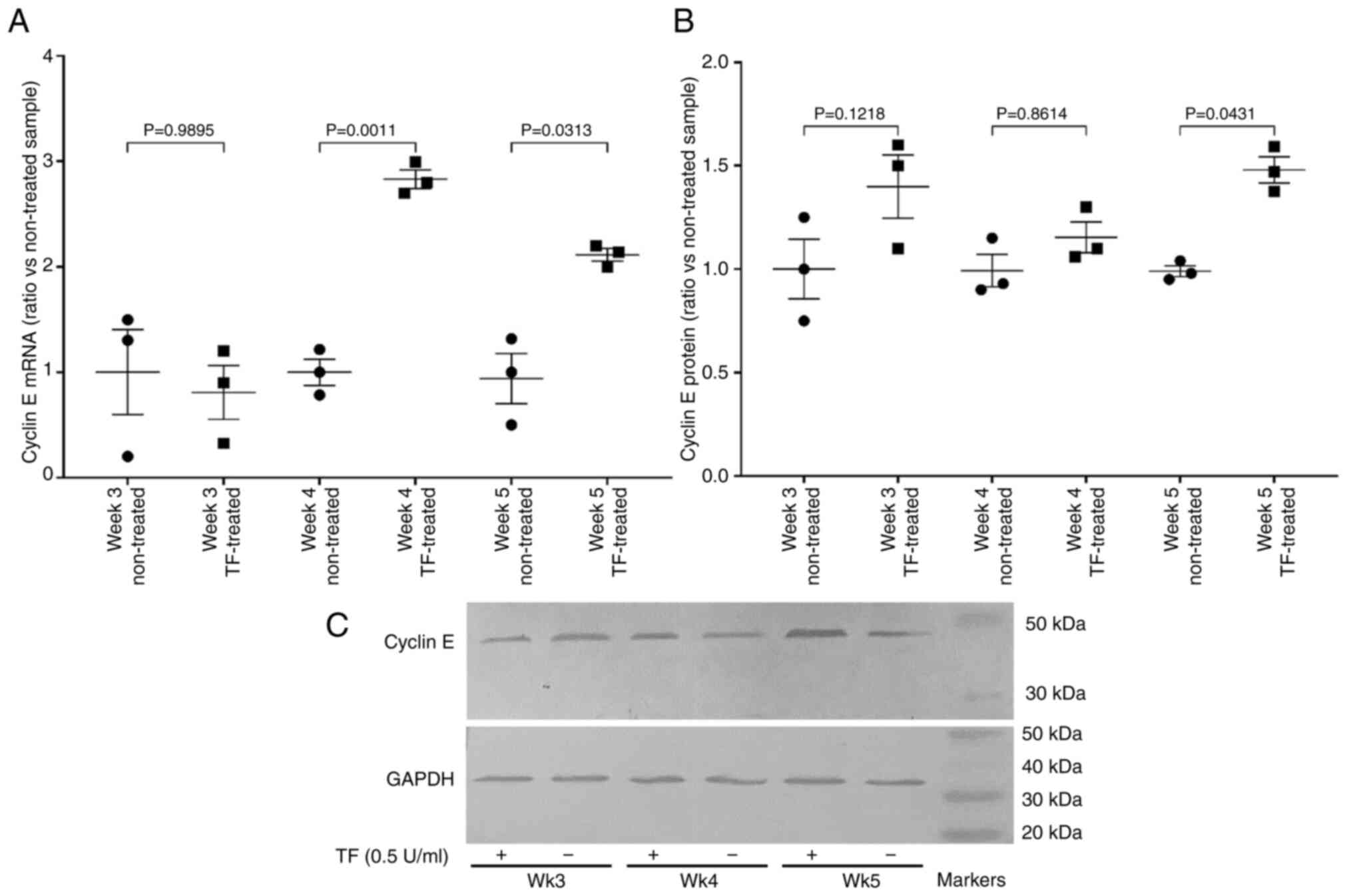
In order to assess the ability of the cells to
progress past the G1/S checkpoint, the expression of cyclin E mRNA
and the transcriptional activity of E2F were examined. Prolonged
treatment of cells with recombinant TF (0.5 U/ml) resulted in the
upregulation of cyclin E mRNA (Fig.
6A) and protein (Fig. 6B and
C). Additionally, measurement of E2F activity using the
luciferase reporter, indicated 3-fold increased activity on weeks
4, but the increases were not significant on week 5 (data not
shown). Collectively, these data indicate the permissive progress
of cells through the G1/S checkpoint.
Prolonged exposure to TF leads to
p16INKa promoter methylation and p14ARF
upregulation
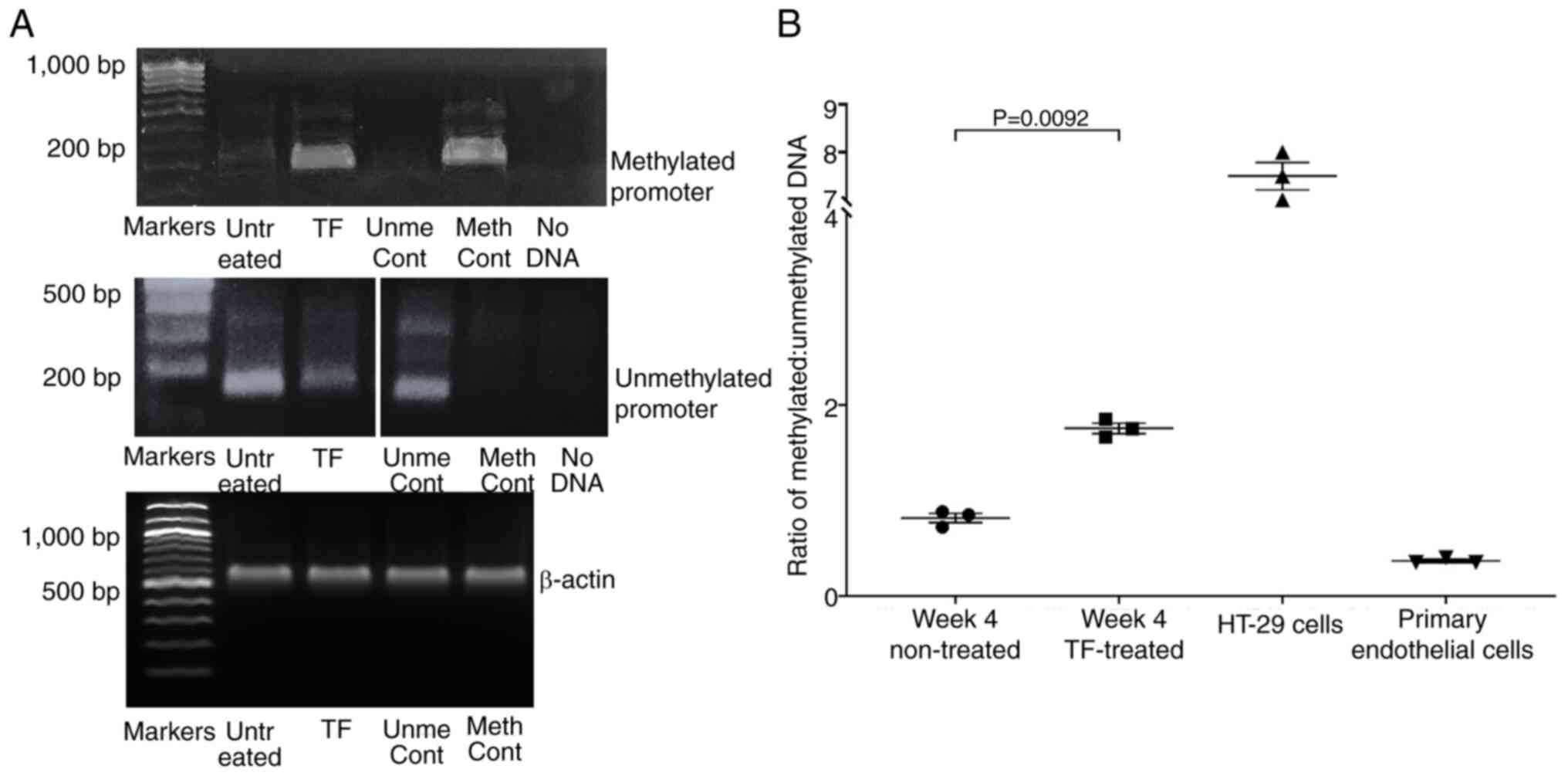
To investigate the mechanism of the reduction in
p16INKa, gDNA was extracted from TF-treated and
untreated cells, converted with bisulphite, and the status of the
p16INKa promoter was analysed by MS-PCR. Analysis of the
gDNA indicated the presence of both methylated and non-methylated
promoter sequences in the hTERT-HPNE cells. Moreover, the ratio of
methylated:unmethylated DNA increased significantly by week 4 of
supplementation with recombinant TF when compared with control
cells grown for 4 weeks in untreated media (Fig. 7A and B). It should be noted that
the untreated control hTERT-HPNE cells showed partial methylation
of the p16INKa promoter, which was expected since the
immortalisation of some cells by constitutive hTERT expression has
an impact on the regulation of p16INKa expression, but
not the function of this protein (60,61).
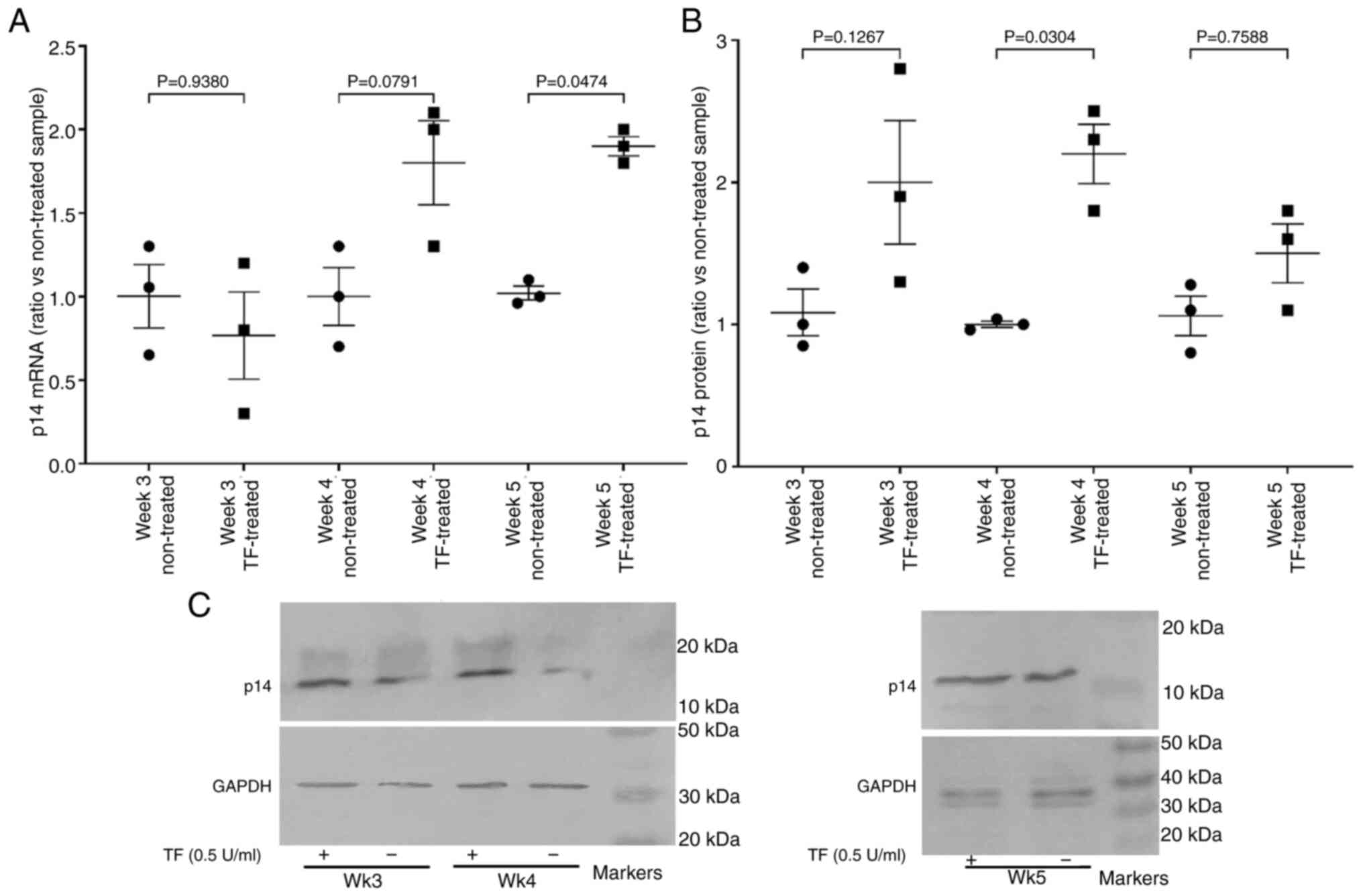
Since p16INKa protein levels may be
reduced by the expression of p14ARF, the expression of
p14ARF was also analysed. Examination of the hTERT-HPNE
cells showed increases in the levels of p14ARF mRNA and
protein expression in TF-treated cells compared with untreated
cells (Fig. 8A-C).
Discussion
TF is considered to have additional functions beyond
the initiation of coagulation within the body, and contributes to
the healing process by regulating cellular responses including
apoptosis of severely damage cells and proliferation of viable
cells (4,8–20).
The precise regulation of these processes and the distinction
between viable and damaged cells is imperative in order to prevent
the survival of aberrant cells, without unnecessary cell loss. The
rapid exposure of TF at the site of injury makes it an ideal
first-on the scene regulator, to initiate these clearance and
repair processes, as well as the immediate initiation of blood
coagulation (5–7). Additionally, as a protein present in
the sub-endothelial vasculature, it is hypothesized that the level
of TF exposure is likely to be determined by the amount of injury.
One of the mechanisms by which TF impacts clearance and repair
processes is likely to be by regulating cell cycle progression
within cells at the site of injury (19). In support of this, the ability of
TF to promote the entry of cells into cell cycle was previously
shown (19). However, progress
through to the end of the G1/S checkpoint was regulated by the
amount of TF, with either high levels of TF (19,22)
or inability of cells to dissipate any excess TF resulting in
cellular apoptosis (20,21).
The regulation of the transition through the G1/S
checkpoint is controlled at two stages. The earlier
mitogen-responsive stage is promoted by Cyclin D/Cdk 4/6 complex
formation and may be inhibited by p16INKa (30). p16INKa inhibits the
Cyclin D/Cdk 4/6 mediated phosphorylation of the retinoblastoma
protein, permitting E2F activity and the progression through the G1
phase (29,29). In the present study it was shown
that in HDBECs TF induces an upregulation in p16INKa
expression which appears to be mediated by TF-β1-integrin
signalling in a saturable manner, and independent of PAR2
activation. The second step in the G1/S checkpoint is
mitogen-independent and is promoted by the Cyclin E/Cdk 2 complex
formation and inhibited by p21CIP1/WAF1 and
p27KIP1 (31). TF has a
concentration dependant influence on p21CIP1/WAF1
expression in HDBECs, with low concentrations causing a decrease in
expression and high concentrations causing an increase. These data
agree with previous observations which suggest opposite outcomes on
p21CIP1/WAF1 expression, depending on the retention of
TF by the cell (18,20). However, in the present study
inhibition of the protease activity of TF-fVIIa partially augmented
the p21CIP1/WAF1 expression. Since PAR2 is a robust
promoter of the incorporation of TF and its release within
cell-derived MVs (62), it was
hypothesised that inhibition of TF-fVIIa prevents the PAR2-induced
release of TF + MVs, resulting in the accumulation of TF on the
surface of the cells, which amplifies the subsequent TF signalling
(Fig. 9). At lower TF
concentrations, p21CIP1/WAF1 facilitates progression
through the checkpoint by competing with p16INKa
(32–34) and explains the rapid alterations in
E2F activity, which is in turn responsible for the expression of
genes required for the S-phase. Finally, it should be noted that
the processes involved in clearance and healing include a
substantial number of proteins and mechanisms. A large amount of
further study is required to determine how the signalling arising
from TF interacts and synergises with mechanisms induced by other
mediators of healing and repair. Furthermore, how the culmination
of all of these processes over the long-term are likely to give
rise to chronic diseases requires extensive and diverse
investigations.
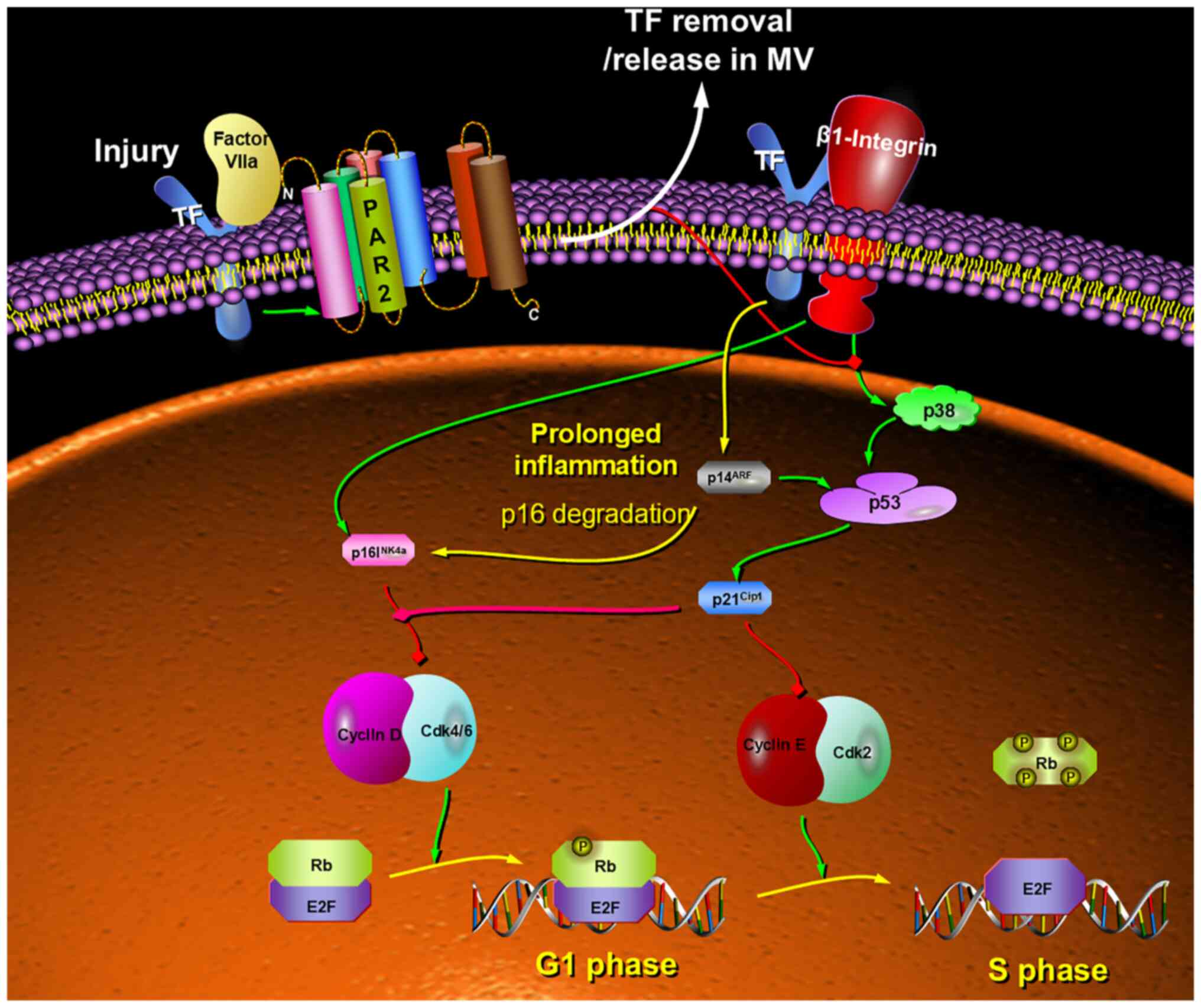 | Figure 9.Proposed model for the mechanism by
which the level of TF on the cell surface may have differential
outcomes on G1/S checkpoint regulation. The presence of TF on the
cell surface differentially upregulates the expression of Inhibitor
of CDK p16INKa, CDK interacting protein/Wildtype
p53-activated fragment p21CIP1/WAF1 and Alternative
reading frame p14ARF, which is dependent on the
concentration of TF and the ability of the cell to dissipate excess
TF. Therefore, alterations in p21CIP1/WAF1 are highly
effective in the regulation of the cellular response to acute
stress. The interplay between these proteins modulates the signal
permitting passage through the cell cycle, or alternatively its
arrest. Consequently, the concentration of TF may be an ideal gauge
for determining the level of cellular damage. However, the adaptive
loss of p16INK4a function may be promoted by prolonged
inflammation leading to permissive transition through the G1/S
checkpoint, even in the absence of mutational loss of
p16INKa. TF, tissue factor; PAR2, protease-activated
receptor 2, Rb, retinoblastoma protein; E2F, early region 2 binding
factor; p21CIP1/WAF1, CDK interacting protein/wildtype
p53-activated fragment; p16INK4a, inhibitor of CDK;
p14ARF, alternative reading frame. |
It has been proposed that the disruption of the
CDKN2A gene (encoding p16INKa) is one of the major
events in cancer development and progression to malignant phenotype
(63,64), as well as the onset of
chemoresistance (65). In the
present study, prolonged incubation of hTERT-HPNE cells with
recombinant TF increased methylation of the p16INKa
promoter region, together with a late-onset reduction in mRNA
expression which were comparable to previous observations reporting
the methylation of the proximal region of the oestrogen receptor on
prolonged exposure of cells to TF (66). However, the more substantial
reduction in p16INKa protein following long-term TF
treatment was also attributed to the expression of
p14ARF. The CDKN2A locus encodes for two separate
proteins, p16INKa and p14ARF, which are
expressed via separate first exons and by usage of alternative
reading frames (67).
p14ARF inhibits murine double minute 2 (mdm2)-mediated
removal of the tumour suppressor protein p53. However,
p14ARF may also induce the degradation of
p16INKa leading to the reduction of the latter protein,
without altering its mRNA expression (68). Overall, the data from the present
study suggest that prolonged exposure of cells to TF may be an
environmental factor that can confer an advantage to early
cancerous cells, even in the absence of mutational loss of
p16INKa expression by allowing accelerated growth and
transformation to the malignant phenotype.
In conclusion, the present study indicates that upon
injury or trauma, TF concentration acts as a cellular gauge for the
proximity/magnitude of injury sustained by cells. The interaction
between TF and β1-integrin differentially upregulates the
expression of p16INKa, p21CIP1/WAF1 and
p14ARF. Subsequently, the interplay between these
proteins translates the signal which depends on the concentration
of TF present on the cell surface. The alterations in
p21CIP1/WAF1 are highly effective in the regulation of
the response to acute stress and therefore, ideal for gauging the
level of cellular damage. However, the data also allude to an
adaptive mechanism by which the loss of p16INKa function
may be promoted by inflammatory factors, even in the absence of
loss of p16INKa expression.
Supplementary Material
Supporting Data
Supporting Data
Acknowledgements
Not applicable.
Funding
Funding: No funding was received.
Availability of data and materials
The data generated in the present study may be
requested from the corresponding author.
Authors' contributions
SJF and CE designed the study, carried out the
experimental work. SJF, ECF and CE evaluated the data, confirmed
the authenticity of all the raw data and prepared the manuscript.
All authors read and approved the final version of the
manuscript.
Ethics approval and consent to
participate
Not applicable.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
De Blaineville HMD: Injection de matière
cerebrale dans des veines. Gaz Med Paris (Ser. 2). 1834.2524
|
|
2
|
Bächli E: History of tissue factor. Br J
Haematol. 110:248–255. 2000.PubMed/NCBI
|
|
3
|
Howell WH: The nature and action of the
thromboplastin (zymoplastic) substance of the tissues. Am J
Physiol. 1:31–59. 1912.
|
|
4
|
Zelaya H, Rothmeier AS and Ruf W: Tissue
factor at the crossroad of coagulation and cell signaling. J Thromb
Haemost. 16:1941–1952. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Carson SD and Konigsberg WH: Lipid
activation of coagulation factor III apoprotein (tissue
factor)-Reconstitution of the protein-membrane complex. Thromb
Haemost. 44:12–15. 1980. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Kurosawa S, Matsuda M and Aoki N: Urinary
procoagulant behaves as tissue factor by promoting factor
VIIa-catalyzed activation of factor X. Thromb Res. 33:595–606.
1984. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Sanders NL, Bajaj SP, Zivelin A and
Rapaport SI: Inhibition of tissue factor/factor VIIa activity in
plasma requires factor X and an additional plasma component. Blood.
66:204–212. 1985. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Hisada Y and Mackman N: Tissue factor and
cancer: Regulation, tumor growth, and metastasis. Semin Thromb
Hemost. 45:385–395. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
McVey JH: The role of the tissue factor
pathway in haemostasis and beyond. Curr Opin Hematol. 23:453–461.
2016. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Rondon AMR, Kroone C, Kapteijn MY,
Versteeg HH and Buijs JT: Role of tissue factor in tumor
progression and cancer-associated thrombosis. Semin Thromb Hemost.
45:396–412. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Cimmino G and Cirillo P: Tissue factor:
Newer concepts in thrombosis and its role beyond thrombosis and
hemostasis. Cardiovasc Diagn Ther. 8:581–593. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Li H, Yu Y, Gao L, Zheng P, Liu X and Chen
H: Tissue factor: A neglected role in cancer biology. J Thromb
Thrombolysis. 54:97–108. 2022. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Rao LV and Pendurthi UR: Tissue
factor-factor VIIa signaling. Arter. Thromb Vasc Biol. 25:47–56.
2005. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Rak J, Milsom C and Yu J: Tissue factor in
cancer. Curr Opin Hematol. 15:522–528. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Schaffner F and Ruf W: Tissue factor and
PAR2 signaling in the tumor microenvironment. Arterioscler Thromb
Vasc Biol. 29:1999–2004. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Nemerson Y and Repke D: Tissue factor
accelerates the activation of coagulation factor VII: The role of a
bifunctional coagulation cofactor. Thromb Res. 40:351–358. 1985.
View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Rao LV, Rapaport SI and Bajaj SP:
Activation of human factor VII in the initiation of tissue
factor-dependent coagulation. Blood. 68:685–691. 1986. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Madkhali Y, Featherby S, Collier ME,
Maraveyas A, Greenman J and Ettelaie C: The Ratio of Factor
VIIa:Tissue Factor content within microvesicles determines the
differential influence on endothelial cells. TH Open. 3:e132–e145.
2019. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Pradier A and Ettelaie C: The influence of
exogenous tissue factor on the regulators of proliferation and
apoptosis in endothelial cells. J Vasc Res. 45:19–32. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
20
|
ElKeeb AM, Collier ME, Maraveyas A and
Ettelaie C: Accumulation of tissue factor in endothelial cells
induces cell apoptosis, mediated through p38 and p53 activation.
Thromb Haemost. 114:364–378. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Ethaeb AM, Mohammad MA, Madkhali Y,
Featherby S, Maraveyas A, Greenman J and Ettelaie C: Accumulation
of tissue factor in endothelial cells promotes cellular apoptosis
through over-activation of Src1 and involves β1-integrin
signalling. Apoptosis. 25:29–41. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Madkhali Y, Rondon AMR, Featherby S,
Maraveyas A, Greenman J and Ettelaie C: Factor VIIa regulates the
level of cell-surface tissue factor through separate but
cooperative mechanisms. Cancers (Basel). 13:37182021. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Huang SZ, Wei MN, Huang JR, Zhang ZJ,
Zhang WJ, Jiang QW, Yang Y, Wang HY, Jin HL, Wang K, et al:
Targeting TF-AKT/ERK-EGFR pathway suppresses the growth of
hepatocellular carcinoma. Front Oncol. 9:1502019. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Kakkar AK, Lemoine NR, Scully MF, Tebbutt
S and Williamson RC: Tissue factor expression correlates with
histological grade in human pancreatic cancer. Br J Surg.
82:1101–1104. 1995. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Ettelaie C, Collier MEW, Featherby S,
Benelhaj NE, Greenman J and Maraveyas A: Analysis of the potential
of cancer cell lines to release tissue factor-containing
microvesicles: Correlation with tissue factor and PAR2 expression.
Thromb J. 14:22016. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Nakayama K and Nakayama K: Cip/Kip
cyclin-dependent kinase inhibitors: Brakes of the cell cycle engine
during development. Bioessays. 20:1020–1029. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Pavletich NP: Mechanisms of
cyclin-dependent kinase regulation: Structures of Cdks, their
cyclin activators, and Cip and INK4 inhibitors. J Mol Biol.
287:821–828. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Harper JW: Cyclin dependent kinase
inhibitors. Cancer Surv. 29:91–107. 1997.PubMed/NCBI
|
|
29
|
Vidal A and Koff A: Cell-cycle inhibitors:
Three families united by a common cause. Gene. 247:1–15. 2000.
View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Cánepa ET, Scassa ME, Ceruti JM, Marazita
MC, Carcagno AL, Sirkin PF and Ogara MF: INK4 proteins, a family of
mammalian CDK inhibitors with novel biological functions. IUBMB
Life. 59:419–426. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Blagosklonny MV and Pardee AB: The
restriction point of the cell cycle. Cell Cycle. 1:103–110. 2002.
View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Abbas T and Dutta A: p21 in cancer:
Intricate networks and multiple activities. Nat Rev Cancer.
9:400–414. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Kreis NN, Louwen F and Yuan J: The
multifaceted p21 (Cip1/Waf1/CDKN1A) in cell differentiation,
migration and cancer therapy. Cancers (Basel). 11:12202019.
View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Cheng M, Olivier P, Diehl JA, Fero M,
Roussel MF, Roberts JM and Sherr CJ: The p21(Cip1) and p27(Kip1)
CDK ‘inhibitors’ are essential activators of cyclin D-dependent
kinases in murine fibroblasts. EMBO J. 18:1571–1583. 1999.
View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Gartel AL: Is p21 an oncogene? Mol Cancer
Ther. 5:1385–1386. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Qureshi AW, Altamimy R, El Habhab A, El
Itawi H, Farooq MA, Zobairi F, Hasan H, Amoura L, Kassem M, Auger
C, et al: Ageing enhances the shedding of splenocyte microvesicles
with endothelial pro-senescent effect that is prevented by a
short-term intake of omega-3 PUFA EPA:DHA 6:1. Biochem Pharmacol.
173:1137342020. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Abbas M, Jesel L, Auger C, Amoura L,
Messas N, Manin G, Rumig C, León-González AJ, Ribeiro TP, Silva GC,
et al: Endothelial microparticles from acute coronary syndrome
patients induce premature coronary artery endothelial cell aging
and thrombogenicity: Role of the Ang II/AT1 Receptor/NADPH
Oxidase-mediated activation of MAPKs and PI3-Kinase pathways.
Circulation. 135:280–296. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Hempel D, Sierko E and Wojtukiewicz MZ:
Protease-activated receptors-biology and role in cancer. Postepy
Hig Med Dosw (Online). 70:775–786. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Ruf W: Roles of factor Xa beyond
coagulation. J Thromb Thrombolysis. 52:391–396. 2021. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Koizume S and Miyagi Y: Tissue factor in
cancer-associated thromboembolism: Possible mechanisms and clinical
applications. Br J Cancer. 127:2099–2107. 2022. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Wojtukiewicz MZ, Hempel D, Sierko E,
Tucker SC and Honn KV: Protease-activated receptors (PARs)-biology
and role in cancer invasion and metastasis. Cancer Metastasis Rev.
34:775–796. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Dorfleutner A, Hintermann E, Tarui T,
Takada Y and Ruf W: Cross-talk of integrin alpha3beta1 and tissue
factor in cell migration. Mol Biol Cell. 15:4416–4425. 2004.
View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Kocatürk B and Versteeg HH: Tissue
factor-integrin interactions in cancer and thrombosis: Every Jack
has his Jill. J Thromb Haemost. 11 (Suppl 1):S285–S293. 2013.
View Article : Google Scholar
|
|
44
|
Collier ME and Ettelaie C: Induction of
endothelial cell proliferation by recombinant and
microparticle-tissue factor involves beta1-integrin and
extracellular signal regulated kinase activation. Arterioscler
Thromb Vasc Biol. 30:1810–1817. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Mirza H, Yatsula V and Bahou WF: The
proteinase activated receptor-2 (PAR-2) mediates mitogenic
responses in human vascular endothelial cells-Molecular
characterization and evidence for functional coupling to the
thrombin receptor. J Clin Invest. 97:1705–1714. 1996. View Article : Google Scholar : PubMed/NCBI
|
|
46
|
Xie L, Duan Z, Liu C, Zheng Y and Zhou J:
Protease-activated receptor 2 agonist increases cell proliferation
and invasion of human pancreatic cancer cells. Exp Ther Med.
9:239–244. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
47
|
Hu L, Xia L, Zhou H, Wu B, Mu Y, Wu Y and
Yan J: TF/FVIIa/PAR2 promotes cell proliferation and migration via
PKCα and ERK-dependent c-Jun/AP-1 pathway in colon cancer cell line
SW620. Tumour Biol. 34:2573–2581. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
48
|
R&D Biosystems Quantikine ELISA Human
Coagulation Factor III/Tissue Factor protocol, . chrome-extension://efaidnbmnnnibpcajpcglclefindmkaj/https://www.bio-techne.com/datasheet-pdf?src=rnd&pdf=dcf300.pdf2016.
|
|
49
|
Khorana AA, Francis CW, Menzies KE, Wang
JG, Hyrien O, Hathcock J, Mackman N and Taubman MB: Plasma tissue
factor may be predictive of venous thromboembolism in pancreatic
cancer. J Thromb Haemost. 6:1983–1985. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
50
|
Versteeg HH, Schaffner F, Kerver M,
Petersen HH, Ahamed J, Felding-Habermann B, Takada Y, Mueller BM
and Ruf W: Inhibition of tissue factor signaling suppresses tumor
growth. Blood. 111:190–199. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
51
|
Carson SD, Ross SE, Bach R and Guha A: An
inhibitory monoclonal antibody against human tissue factor. Blood.
70:490–493. 1987. View Article : Google Scholar : PubMed/NCBI
|
|
52
|
Ettelaie C, Collier ME, Mei MP, Xiao YP
and Maraveyas A: Enhanced binding of tissue factor-microparticles
to collagen-IV and fibronectin leads to increased tissue factor
activity in vitro. Thromb Haemost. 109:61–71. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
53
|
Livak KJ and Schmittgen TD: Analysis of
relative gene expression data using real-time quantitative PCR and
the 2(−Delta Delta C(T)) method. Methods. 25:402–408. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
54
|
Zheng N, Fraenkel E, Pabo CO and Pavletich
NP: Structural basis of DNA recognition by the heterodimeric cell
cycle transcription factor E2F-DP. Genes Dev. 13:666–674. 1999.
View Article : Google Scholar : PubMed/NCBI
|
|
55
|
Adrian R, Black AR and Azizkhan-Clifford
J: Regulation of E2F: A family of transcription factors involved in
proliferation control. Genes. 237:281–302. 1999.
|
|
56
|
Aksoy O, Chicas A, Zeng T, Zhao Z,
McCurrach M, Wang X and Lowe SW: The atypical E2F family member
E2F7 couples the p53 and RB pathways during cellular senescence.
Genes Dev. 26:1546–1557. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
57
|
Rabinovich A, Jin VX, Rabinovich R, Xu X
and Farnham PJ: E2F in vivo binding specificity: Comparison of
consensus versus nonconsensus binding sites. Genome Res.
18:1763–1777. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
58
|
Ertosun MG, Hapil FZ and Osman Nidai O:
E2F1 transcription factor and its impact on growth factor and
cytokine signaling. Cytokine Growth Factor Rev. 31:17–25. 2016.
View Article : Google Scholar : PubMed/NCBI
|
|
59
|
Collier MEW, Akinmolayan A and Goodall AH:
Comparison of tissue factor expression and activity in foetal and
adult endothelial cells. Blood Coagul Fibrinolysis. 28:452–459.
2017. View Article : Google Scholar : PubMed/NCBI
|
|
60
|
Noble JR, Zhong ZH, Neumann AA, Melki JR,
Clark SJ and Reddel RR: Alterations in the p16(INK4a) and p53 tumor
suppressor genes of hTERT-immortalized human fibroblasts. Oncogene.
23:3116–3121. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
61
|
Chapman EJ, Hurst CD, Pitt E, Chambers P,
Aveyard JS and Knowles MA: Expression of hTERT immortalises normal
human urothelial cells without inactivation of the p16/Rb pathway.
Oncogene. 25:5037–5045. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
62
|
Collier ME and Ettelaie C: Regulation of
the incorporation of tissue factor into microparticles by serine
phosphorylation of the cytoplasmic domain of tissue factor. J Biol
Chem. 286:11977–11984. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
63
|
Georgiadou D, Sergentanis TN, Sakellariou
S, Filippakis GM, Zagouri F, Vlachodimitropoulos D, Psaltopoulou T,
Lazaris AC, Patsouris E and Zografos GC: Cyclin D1, p16(INK) (4A)
and p27(Kip1) in pancreatic adenocarcinoma: Assessing prognostic
implications through quantitative image analysis. APMIS.
122:1230–1239. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
64
|
Tsiambas E, Karameris A, Gourgiotis S,
Salemis N, Athanassiou AE, Karakitsos P, Papalois A, Merikas E,
Kosmidis P and Patsouris E: Simultaneous deregulation of p16 and
cyclin D1 genes in pancreatic ductal adenocarcinoma: A combined
immunohistochemistry and image analysis study based on tissue
microarrays. J BUON. 12:261–267. 2007.PubMed/NCBI
|
|
65
|
Kreisel F, Kulkarni S, Kerns RT, Hassan A,
Deshmukh H, Nagarajan R, Frater JL and Cashen A: High resolution
array comparative genomic hybridization identifies copy number
alterations in diffuse large B-cell lymphoma that predict response
to immuno-chemotherapy. Cancer Genet. 204:129–137. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
66
|
Collier MEW, Li C and Ettelaie C: The
Influence of Tissue factor on the methylation of oestrogen receptor
alpha gene during breast cancer. Thrombosis Res. 120:PO–77. 2007.
View Article : Google Scholar
|
|
67
|
Zhang Y, Hyle J, Wright S, Shao Y, Zhao X,
Zhang H and Li C: A cis-element within the ARF locus mediates
repression of p16INK4A expression via long-range chromatin
interactions. Proc Natl Acad Sci USA. 116:26644–22652. 2019.
View Article : Google Scholar : PubMed/NCBI
|
|
68
|
Kobayashi T, Wang J, Al-Ahmadie H and
Abate-Shen C: ARF regulates the stability of p16 protein via
REGγ-dependent proteasome degradation. Mol Cancer Res. 11:828–833.
2013. View Article : Google Scholar : PubMed/NCBI
|