Introduction
Chronic obstructive pulmonary disease (COPD), a
common chronic respiratory lung ailment, is characterized by
long-term airflow limitation that is not fully reversible. It is
estimated that COPD will become the third leading cause of
mortality by 2030 in the world (1). A total of 212.3 million patients with
COPD have been reported in 2019 worldwide; this is estimated to
increase to 600 million prevalent cases of COPD by 2050 worldwide
(2,3). It is widely accepted that persistent
exposure to cigarette smoke (CS) is by far the primary etiological
factor for COPD and has been documented in >80% of COPD cases
(4). As a serious health burden,
COPD has an important impact on the quality of life of patients,
and causes a huge socioeconomic and medical burden (5). Therefore, it is imperative to
identify COPD biomarkers and elucidate the molecular mechanisms of
pathogenesis involved in COPD.
Accumulated evidence indicates that multiple
biological functions are implicated in the pathogenesis of COPD
(6–8). Among them, autophagy exerts a crucial
effect on the development of COPD (9). Autophagy is a highly conserved
homeostatic process, during which autophagosomes devour and degrade
damaged or aged organelles for further processing and recycling to
maintain cellular homeostasis (10). Notably, autophagy eliminates the
damaged mitochondria, a process known as mitophagy, which is the
main part of the mitochondrial quality control system (11). Dysregulation of mitophagy
contributes to the development of diverse lung disorders, including
asthma, acute lung injury, bronchopulmonary dysplasia and COPD
(12). Substantial evidence
suggests that CS impairs mitophagy, resulting in the accumulation
of damaged mitochondria (13,14).
By regulating mitophagy in cigarette smoke extract (CSE)-induced
airway epithelial cells, MAPK15-Unc-51-like kinase signaling
participates in the development of COPD (15). Nevertheless, the genes related to
mitophagy in COPD have not been fully clarified and further studies
are required for their identification. The exploration of the
potential mitophagy-related genes during COPD development will
provide the possibility of identifying potential biomarkers for the
treatment of this condition.
In the present study, the mitophagy-associated
differentially expressed genes (DEGs) were analyzed in the Gene Set
Enrichment (GSE) 76925 dataset in COPD and control samples. The
expression patterns of these DEGs were analyzed by cluster
analysis. Subsequently, Gene Ontology (GO) and Kyoto Encyclopedia
of Genes and Genomes (KEGG) enrichment analyses were performed for
the identifications of DEGs and their biological functions and
interaction networks were assessed. In addition, least absolute
shrinkage and selection operator (LASSO) regression analysis was
used to screen signature genes and analyze their relationship with
immune cells. Finally, biological experiments were conducted to
investigate the involvement of signature genes in the progression
of COPD.
Materials and methods
Acquisition of datasets and
mitophagy-related genes
The COPD datasets were downloaded from the Gene
Expression Omnibus (GEO) datasets (http://www.ncbi.nlm.nih.gov/geo/) on December 5, 2023.
The GSE76925 dataset which included 111 COPD samples and 40 control
samples was downloaded from the GPL10558 platform and was used as
the training set. In addition, a study of 18 patients with COPD and
36 control samples (GSE8545) downloaded from the GPL570 platform
was selected as the validation set. The ‘limma’ R package
(https://bioconductor.org/packages/release/bioc/html/limma.html)
was employed to pre-process the raw microarray data for quality
control. A total of 29 mitophagy-related genes sets
(REACTOME_MITOPHAGY) were downloaded from the Molecular Signatures
Database (MSigDB; http://www.gsea-msigdb.org/gsea/index.jsp).
Identification of differentially
expressed mitophagy-related genes (DEMRGs)
The DEMRGs identified in COPD and control samples
were analyzed with the application of the Wilcoxon test. Plot box
plots were made using the ‘ggpubr’ package (https://rpkgs.datanovia.com/ggpubr/).
GO and KEGG enrichment analysis
The GO and KEGG enrichment analyses were used to
investigate the biological functions and signaling pathways for
DEMRGs using ‘ClusterProfiler’ R package (https://yulab-smu.top/biomedical-knowledge-mining-book/).
The results are visualized using the ‘ggplot2’ R package
(https://ggplot2.tidyverse.org/).
Clustering and differential
analyses
To analyze the differential expression
characteristics of DEMRGs in patients with COPD,
‘ConsensusClusterPlus’ (https://www.bioconductor.org/packages/release/bioc/html/ConsensusClusterPlus.html)
was used in the R package to perform hierarchical clustering
according to the expression of DEMRGs. The parameter was set to 50
replicates (rep=50) with a resampling rate of 80% (pItem=0.8). The
DEGs between clusters were analyzed by the ‘limma’ R package, using
|log2FC| >1 and the adjusted P<0.05 was used as cutoff value.
The Volcano plot of DEGs was generated using the ‘ggrepel’ package
(https://ggrepel.slowkow.com/).
Screening for signature genes using
LASSO regression analysis
LASSO regression analysis was conducted to screen
genes with COPD diagnostic significance with the application of
‘glmnet’ R package (https://glmnet.stanford.edu/). The genes and their
coefficients were determined by the optimal penalty parameter λ
with a minimum 10-fold cross validation. Receiver operating
characteristic (ROC) curve was employed to calculate the area under
the curve (AUC) to accurately evaluate the validity of the
signature genes as diagnostic markers. ROC curves were obtained
using the R package ‘pROC’ (https://xrobin.github.io/pROC/).
Validation of the signature genes in
the test set
The diagnostic performance was validated in
validation sets (GSE8545) by calculating the AUC of the ROC curve.
The ROC curves were plotted by means of the R software package
‘pROC’.
Analysis of immune infiltration
The immune cell infiltration in the
microenvironment, involving 28 immune cells, was analyzed by
single-sample gene set enrichment analysis to obtain a score file
representing the immune cell composition in each individual sample
in the GSE76925 dataset. The immune cell scores of the COPD and
control groups were statistically analyzed by the t-test. The
results were visually represented by box plots. The association
between signature genes and immune cells was analyzed with the
application of the correlation test function in the R software
(v.4.3.3; http://cran.r-project.org/).
Subsequently, the results were visualized with the ‘ggplot’ R
package. A correlation analysis was used to measure the
relationship between the dysregulated immune cells and a signature
gene, which was presented graphically.
Culture of 16HBE cells
The normal human bronchial epithelial cell line
16HBE was accessed from Procell Life Science & Technology Co.,
Ltd. The cells were nurtured in a mixture of RPMI-1640 medium
(HyClone; Cytiva) containing 10% fetal bovine serum (HyClone;
Cytiva), 100 U/ml penicillin and 0.1 mg/ml streptomycin (Beyotime
Institute of Biotechnology) in an incubator at 37°C in the presence
of 5% CO2.
Preparation of CSE and treatment
CSE was prepared as previously reported (16). The smoke of 2 3R4F reference
cigarettes was bubbled through 10 ml RPMI-1640 medium. The CSE
solution was titrated to pH 7.4 and sterilized by filtration
through a 0.22-µm pore filter, which was regarded as 100%
concentration of CSE. The solution was diluted to the required
concentrations (0.5, 1, 2 and 4%) with RPMI-1640 medium to treat
16HBE cells for 24 h; the treatment aimed to simulate the lung cell
destruction condition observed in COPD.
Cell viability assay
16HBE cells were replanted in 96-well plates
(2×104 cells/well). The following day, the cells were
cultured in fresh medium containing different concentrations of CSE
(0.5, 1, 2 and 4%) for an additional 24 h. Subsequently, 10 µl Cell
Counting Kit-8 (CCK-8) working liquid (cat. no. C0037; Beyotime
Institute of Biotechnology) was mixed with each well and the
samples were incubated for 2 h. The absorbance values at 450 nm
were detected with a microplate reader (Bio-Rad Laboratories,
Inc.).
Gene interference
Experiments with knockdown of expression of
mitochondrial transcription termination factor 3 (MTERF3) were
performed. Specifically, 16HBE cells were transfected with small
interfering RNA (siRNA) targeting MTERF3 and the sequences were as
follows: siRNA-MTERF3-1 (50 nM), sense,
5′-UGGAUUUGUCCAAGAUAGAAAAA-3′, anti-sense,
5′-UUUUUCUAUCUUGGACAAAUCCA-3′; siRNA-MTERF3-2 (50 nM), sense,
5′-AGGCUGUUUAAGGUCAAAGAAAG-3′, anti-sense,
5′-CUUUCUUUGACCUUAAACAGCCU-3′) or 50 nM scrambled negative control
(siRNA-NC, sense, 5′-ACCACAAGAUGAAGAGCACCAAU-3′, anti-sense,
5′-AUUGGUGCUCUUCAUCUUGUGGU-3′) customized by Shanghai GeneChem Co.,
Ltd. The cells were transfected using Lipofectamine™ 3000
(Invitrogen; Thermo Fisher Scientific, Inc.) for 48 h at 37°C
following the operating instructions. The cells were cultured for
48 h at 37°C following transfection and immunoblotting was adopted
for the detection of the transfection efficiency.
Flow cytometric analysis
The induction of apoptosis of 16HBE cells was tested
by using an Annexin V-fluorescein isothiocyanate (FITC)/propidium
iodide (PI) Kit (cat. no. C1062L; Beyotime Institute of
Biotechnology). 16HBE cells were seeded in a 6-well plate at a
density of 1×105/well. Following transfection and
treatment with CSE, 16HBE cells were resuspended in binding buffer
and stained with Annexin V-FITC and PI for 15 min in dark at 37°C.
The cells were quantified with flow cytometry (BD FACSCalibur; BD
Biosciences). A total of 10,000 events were collected per sample
and the apoptotic rate of the cells was analyzed using FlowJo 10.8
software (FlowJo; BD Biosciences).
Analysis of mitochondrial reactive
oxygen species (mitoROS)
The generation of mitoROS was assessed with the
application of Mito SOX Red (cat. no. M36008; Invitrogen; Thermo
Fisher Scientific, Inc.) assay. Specifically, 16HBE cells were
seeded in 96 well plates (5×104 cells/ml). Following the
indicated treatment, the cells were treated with 5 mM Mito SOX Red
probe for 15 min at 37°C. Subsequently, the cells were incubated
with PBS and the fluorescence images were acquired using a
fluorescence microscope (Olympus Corporation).
Assessment of oxidative stress
markers
The levels of malonaldehyde (MDA; cat. no. A003-4-1)
and superoxide dismutase (SOD; cat. no. A001-3-2) in the
supernatant of cultured 16HBE cells were detected using the
commercially available kits (Nianjing Jiancheng Bioengineering
Institute). The absorbance value of the samples, at 530 nm for MDA
and 450 nm for SOD, was detected using a microplate reader (Bio-Rad
Laboratories, Inc.).
Detection of mitochondrial membrane
potential (MMP)
MMP was evaluated using the
5,5′,6,6′-tetrachloro1,1′,3,3′-tetramethylbenzimidazolylcarbocyanine
iodide (JC-1) dye (cat. no. C2005; Beyotime Institute of
Biotechnology). Following the indicated treatment, 16HBE cells were
stained at 37°C for 20 min with the JC-1 dye. Fluorescence images
were obtained using a fluorescence microscope (Olympus
Corporation). The red fluorescence represents the JC-1 aggregates,
while the green fluorescence represents JC-1 monomers.
Immunoblotting
The cells were lysed with radioimmunoprecipitation
assay buffer (Beyotime Institute of Biotechnology) to extract the
total proteins. The total protein concentration was assessed with
the application of the Bradford assay. Each quantity of boiled
protein (40 µg) was added to 10% sodium dodecyl
sulfate-polyacrylamide gel electrophoresis (SDS-PAGE) for
separation; the proteins were subsequently transferred to the
nitrocellulose films. Following 1 h blocking with 5% bovine serum
albumin (Beyotime Institute of Biotechnology) at room temperature,
these membranes were incubated with primary antibodies overnight at
4°C. Subsequently, an anti-rabbit IgG HRP-linked secondary antibody
(1:10,000; cat. no. 7074P2; Cell Signaling Technology, Inc.) was
incubated with the transfer membranes. The detection was performed
using the enhanced chemiluminescence (MilliporeSigma). GAPDH acted
as a reference gene. The intensities of the proteins were assessed
quantitatively using ImageJ Software (version 1.8.0; National
Institutes of Health). The primary antibodies used were the
following: Anti-MTERF3 (1:1,000; cat. no. ab230232) and
anti-putative kinase 1 (PINK1; 1:1,000; cat. no. ab216144)
antibodies were acquired from Abcam. Anti-B cell lymphoma-2 (Bcl-2;
1:1,000; cat. no. 3498T), anti-Bcl-2-associated X protein (Bax;
1:1,000; cat. no. 2772T), anti-cleaved casapse3 (1:1,000; cat. no.
9664T), anti-caspase3 (1:1,000; cat. no. 14220T), anti-LC3II/LC3I
(1:1,000; cat. no. 12741T), anti-Beclin-1 (1:1,000; cat. no.
3495T), anti-p62 (1:1,000; cat. no. 5114T), anti-Parkin (1:1,000;
cat. no. 2132S), anti-COX IV (1:1,000; cat. no. 4850T) and
anti-GAPDH (1:1,000; cat. no. 2118T) antibodies were provided by
Cell Signaling Technology, Inc.
Statistical analysis
The statistical analysis was performed using R
software (v.4.3.3). Wilcoxon rank-sum test or unpaired Student's
t-test was used to compare the significance differences between the
two groups of samples. Adjusted P<0.05 was used as a threshold
for significant difference. Results are shown as the mean ±
standard deviation from three independent experiments. GraphPad 8.0
software (GraphPad; Dotmatics) was used for statistical analysis.
The different groups were compared by one-way analysis of variance
with post-hoc Tukey's analysis. P<0.05 was considered to
indicate a statistically significant difference.
Results
Identification and functional
enrichment analysis of DEMRGs
Dysregulation of mitophagy is involved in the
development of COPD (17,18). To determine the mitophagy-related
core gene profiles of patients with COPD, the collated datasets
were downloaded from the GEO database. The information included in
the study was listed in Table I.
The GSE76925 dataset (COPD, 111; control, 40) was used to screen
the DEGs. A total of 29 mitophagy-related genes were obtained from
the MSigDB database. The expression levels of mitophagy-related
genes in the 111 COPD and 40 control samples were analyzed. As
displayed in Fig. 1A, among the 29
mitophagy-related genes, 14 genes demonstrated significant
differences in their expression levels between the COPD and control
samples (P<0.05); these were defined as DEMRGs. Subsequent GO
analysis suggested that these DEMRGs were mainly involved in the
biological processes of mitophagy, mitochondrial breakdown and
selective autophagy (Fig. 1B). In
addition, the results of the KEGG analysis, which indicated that
DEMRGs were mainly enriched in mitophagy, the NOD receptor and the
NF-κB signaling pathways were identified (Fig. 1C).
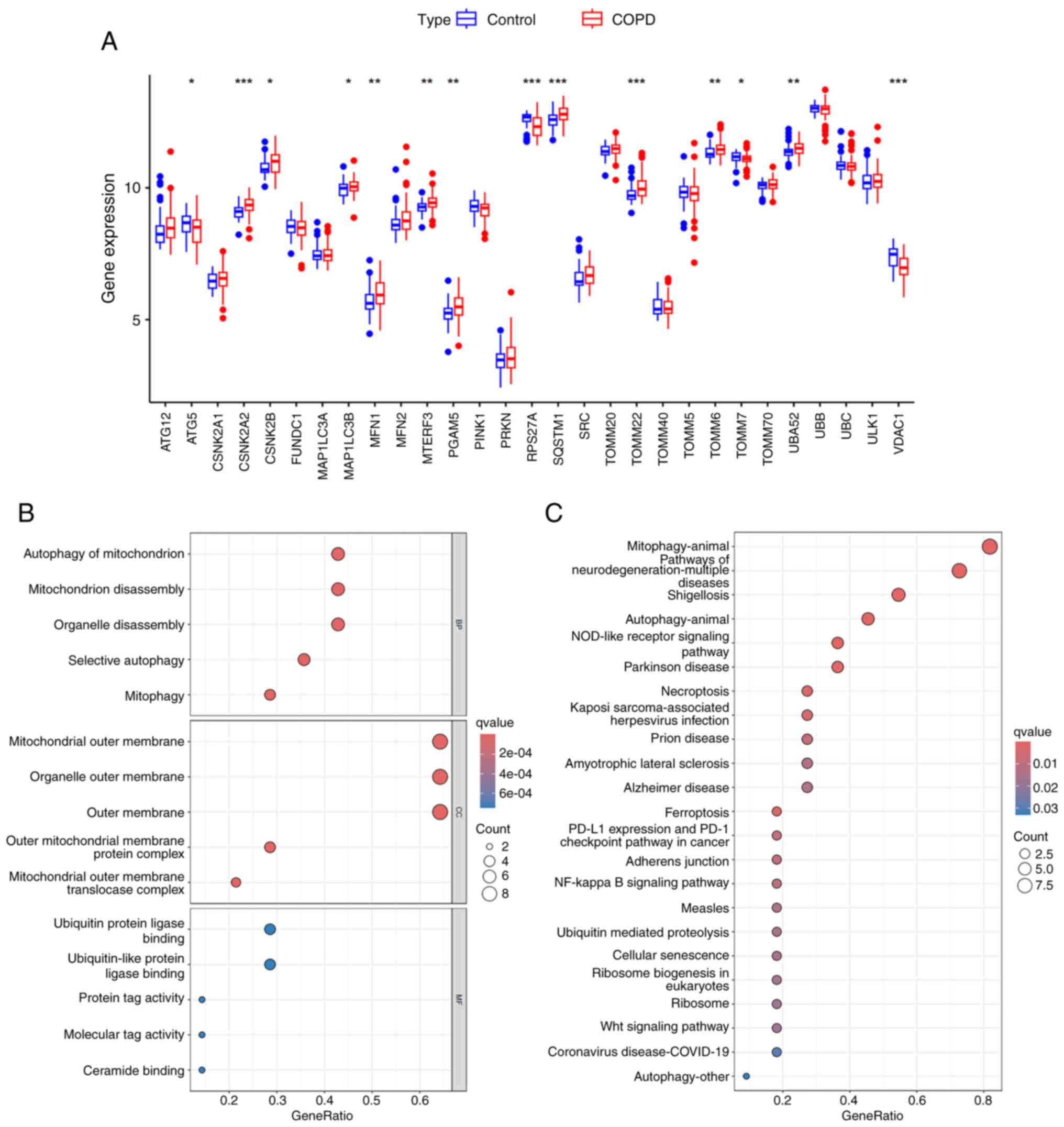 | Figure 1.Identification and functional
enrichment analysis of DEMRGs. (A) Plot box plots indicated the
DEMRGs between COPD and control samples in the GSE76925 dataset
(COPD, 111; control, 40). *P<0.05, **P<0.01 and
***P<0.001. (B) GO enrichment analysis of DEMRGs, including BP,
CC and MF. (C) KEGG enrichment analysis of DEMRGs. The size of dots
indicates the gene number and the shade of the color indicates the
scale of -log10 (P-value). DEMRGs, differentially expressed
mitophagy-related genes; COPD, chronic obstructive pulmonary
disease; GO, Gene Ontology; BP, biological processes; CC, cellular
components; MF, molecular functions; KEGG, Kyoto Encyclopedia of
Genes and Genomes. |
 | Table I.Characteristics of the datasets used
in the present study. |
Table I.
Characteristics of the datasets used
in the present study.
| Dataset | Platform | Samples | Total | Normal | COPD |
|---|
| GSE76925 | GPL10558 | Lung tissue | 141 | 40 | 111 |
| GSE8545 | GPL570 | Airway epithelial
cells | 54 | 36 | 18 |
The expression pattern of DEMRGs
To explore the expression levels of DEMRGs in
patients with COPD, hierarchical clustering analysis was performed
in 111 COPD and 40 control samples according to the expression
levels of these DEMRGs. All samples were divided into two groups
(cluster1, n=87; cluster2, n=64; Fig.
2A-C). The DEGs between cluster 1 and 2 were analyzed and 1,207
DEGs were screened, of which 73 genes were upregulated and 1,134
genes were downregulated (Fig.
2D). These DEGs were mainly involved in biological processes,
such as transcription and translation (Fig. 2E). In addition, the expression
levels of these DEMRGs between cluster 1 and 2 were also explored.
The results indicated that except for microtubule-associated
protein 1 light chain-3β (MAP1LC3B), the expression levels of other
genes were significantly different (Fig. 2F). To assess the correlation of the
expression levels of DEMRGs, correlation analysis was conducted. As
depicted in Fig. 2G, the
expression of ribosomal protein S27A was positively correlated with
the expression of voltage-dependent anion channel 1 (VDAC1),
whereas it was negatively correlated with the expression levels of
other genes. Moreover, strong interactions were noted between
mitofusin-1 and phosphoglycerate mutase family member 5 (PGAM5) as
well as with translocase of outer mitochondrial membrane 22 and
TOMM6. Analysis of immune infiltration between cluster 1 and 2 is
shown in Fig. 2H.
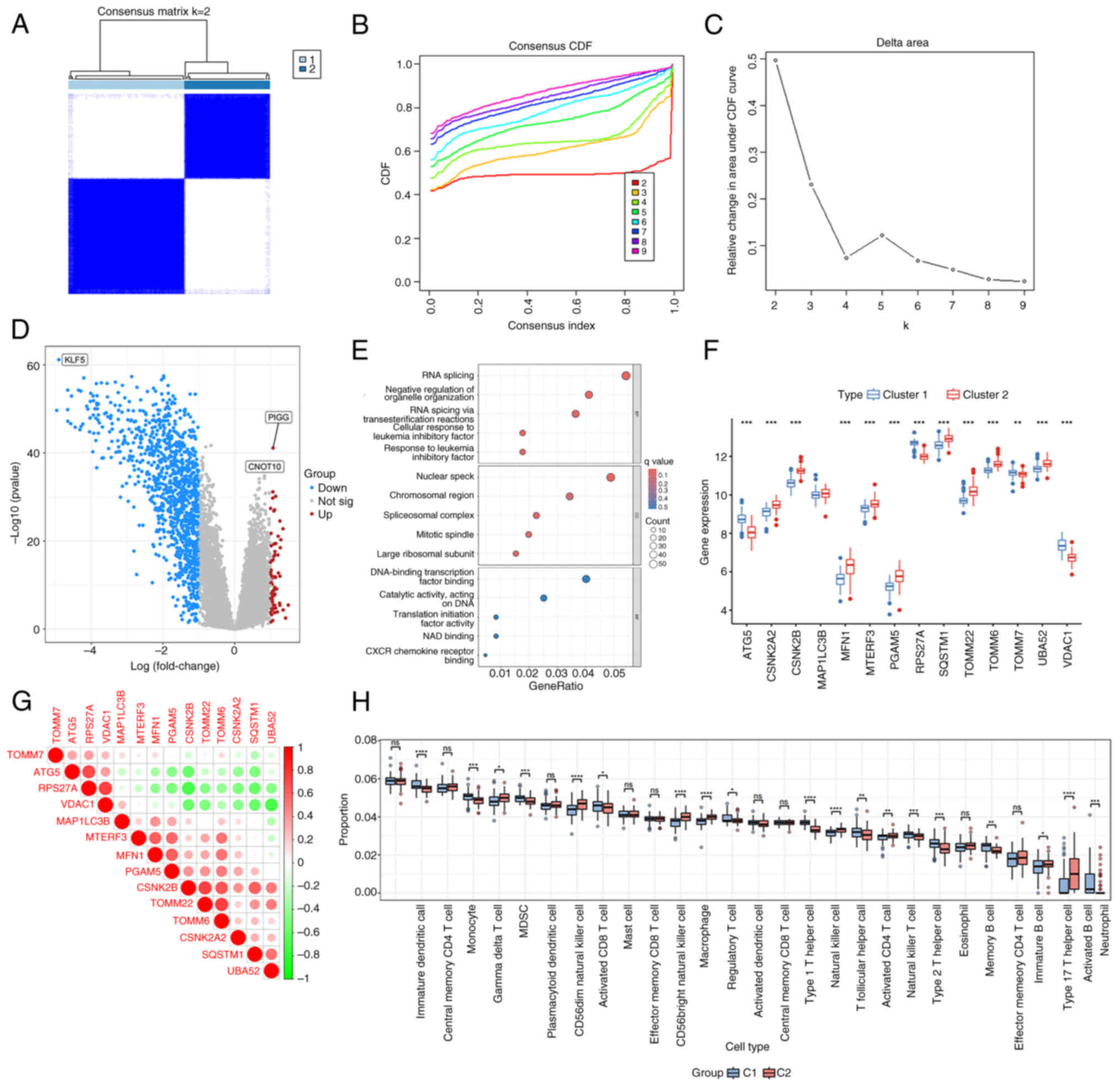 | Figure 2.The expression pattern of DEMRGs.
(A-C) Hierarchical cluster. (D) Volcano plot of 1,207 DEGs
including 14 DEMRGs. The red and blue dots represent the
upregulated and downregulated genes, respectively. (E) GO
enrichment analysis of DEGs, including BP, CC and MF. (F) Plot box
plots indicated the expression levels of DEMRGs between cluster 1
and 2. (G) Correlation analysis of 14 DEMRGs. (H) The proportion of
28 types of immune cells was analyzed in cluster 1 and 2.
*P<0.05, **P<0.01, ***P<0.001 and ****P<0.0001. DEMRGs,
differentially expressed mitophagy-related genes; DEGs,
differentially expressed genes; GO, Gene Ontology; BP, biological
processes; CC, cellular components; MF, molecular functions; ns,
not significant. |
Selection and validation of COPD
signature genes
To screen signature genes with COPD diagnostic
significance, LASSO regression analysis was used in the present
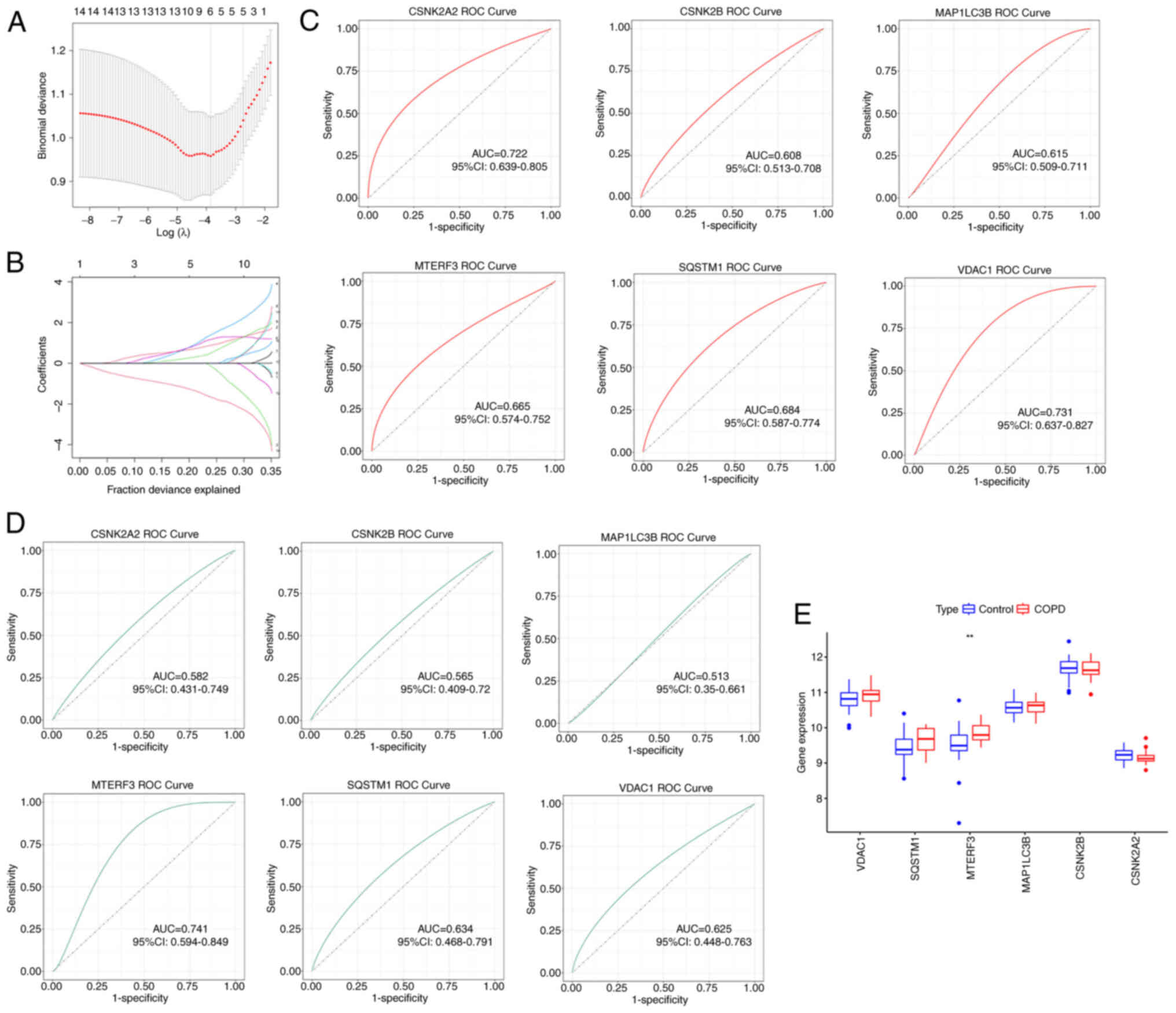
study. As shown in Fig. 3A and B,
the optimal performance was achieved when 6 genes (CSNK2A2, CSNK2B,
MAP1LC3B, MTERF3, SQSTM1 and VDAC1) were included in the LASSO
regression analysis. Therefore, these 6 genes were identified as
diagnostic markers for COPD. Subsequently, ROC curve was employed
to calculate the AUC to accurately evaluate the validity of the
signature genes as diagnostic markers. In the GSE76925 dataset, the
AUC values of the ROC curves for the 6 signature genes are
demonstrated in Fig. 3C. It was
noted that CSNK2A2 (AUC: 0.722), MTERF3 (AUC: 0.665), SQSTM1 (AUC:
0.684) and VDAC1 (AUC: 0.731) exhibited the highest AUC values,
suggesting that these 4 genes exhibited an optimal diagnostic value
for distinguishing patients with COPD from control subjects.
Subsequently, the 6 signature genes were validated in COPD using
the GSE8545 dataset. As displayed in Fig. 3D, the AUC value of MTERF3 (AUC:
0.741) was the highest than that of other genes. In addition, only
MTERF3 expression was significantly different between COPD and
control samples (Fig. 3E).
Therefore, the molecular mechanism of MTERF3 in COPD was further
explored in subsequent experiments.
Immune cell infiltration in COPD and
control samples
An increasing number of immune cell types have been
associated with risk of COPD and prognosis of affected individuals
(19,20). The degree of the infiltration of 28
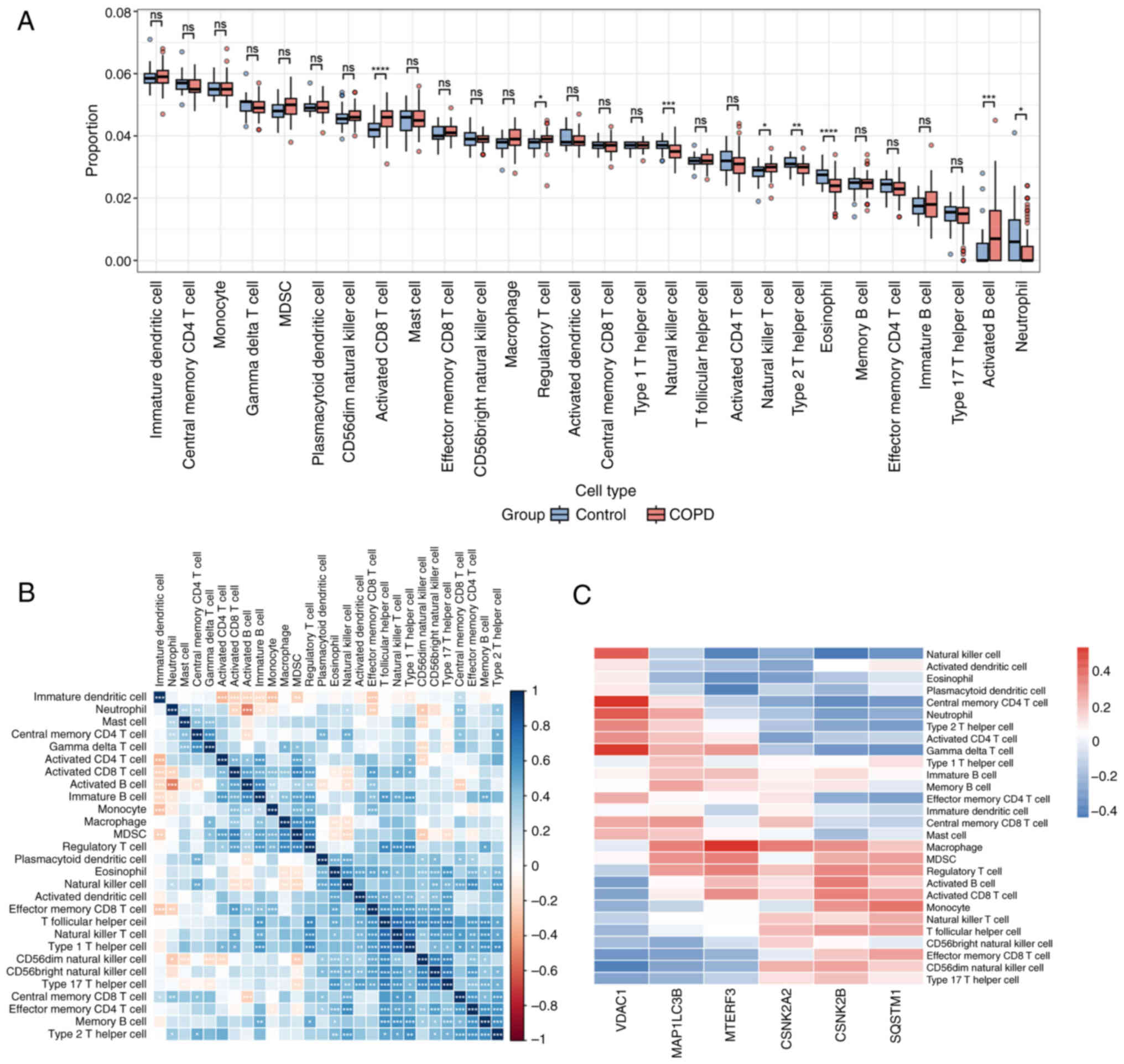
immune cells in COPD and control samples was analyzed. As displayed
in Fig. 4A, the infiltrating
abundance of the majority of immune cells was higher in COPD
samples than that observed in control samples. As revealed in
Fig. 4B, immature B cells and
effector memory cluster of differentiation (CD) 8 T cells were
highly correlated with the majority of the immune cells. Moreover,
the correlation between the expression of the 6 signature genes and
immune cells was examined and it was found that MTERF3 exhibited
optimal correlation with macrophages, myeloid-derived suppressor
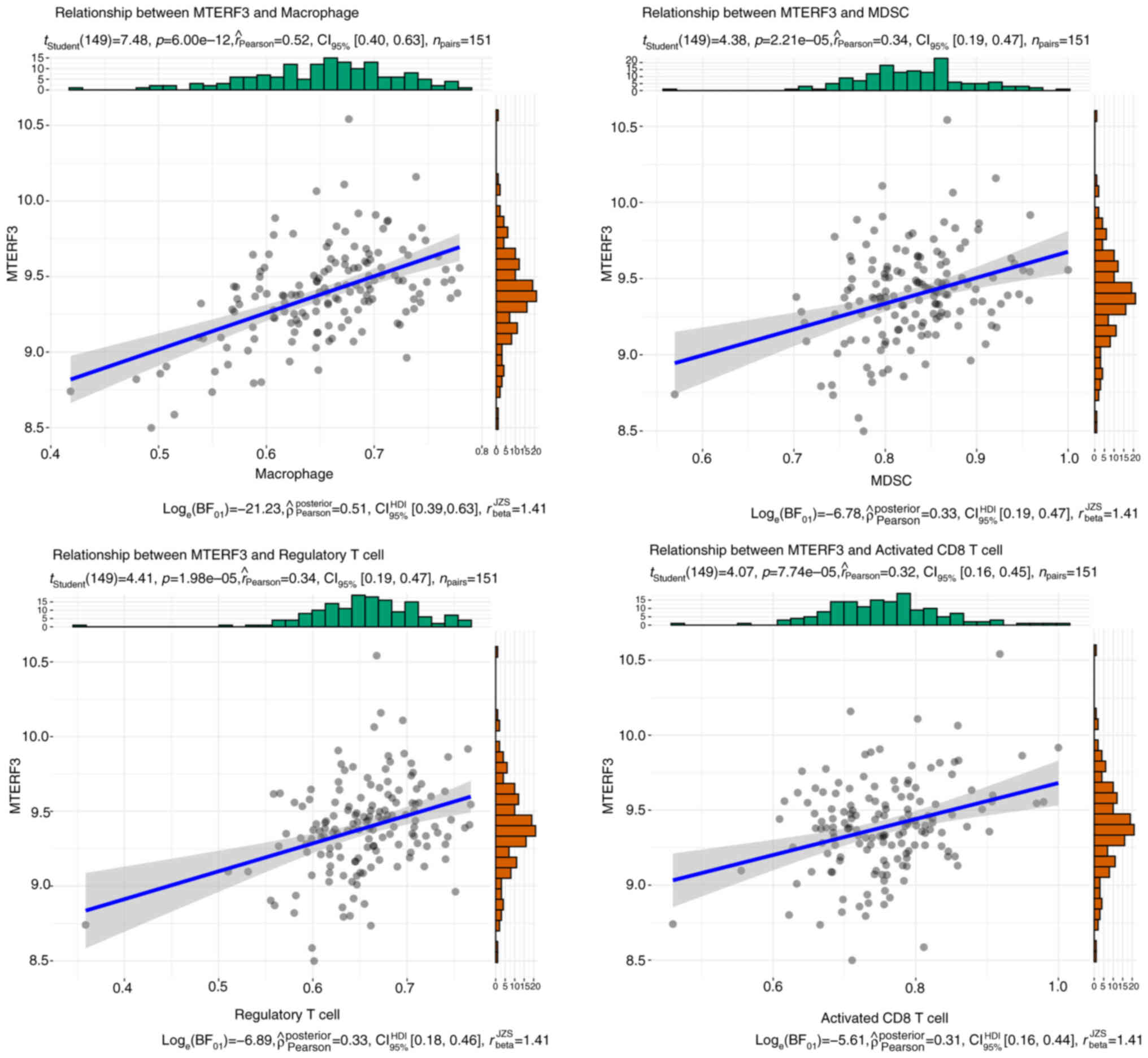
cells (MDSC), regulatory T cell and activated CD8 T cells (Fig. 4C). It is noteworthy that MTERF3
exhibited the highest correlation with macrophages (Fig. 5).
MTERF3 expression is upregulated in
CSE-treated 16HBE cells and MTERF3 deficiency suppresses the
induction of apoptosis of 16HBE cells exposed to CSE
Based on the aforementioned results, the signature
gene MTERF3 was experimentally validated in the following study. As
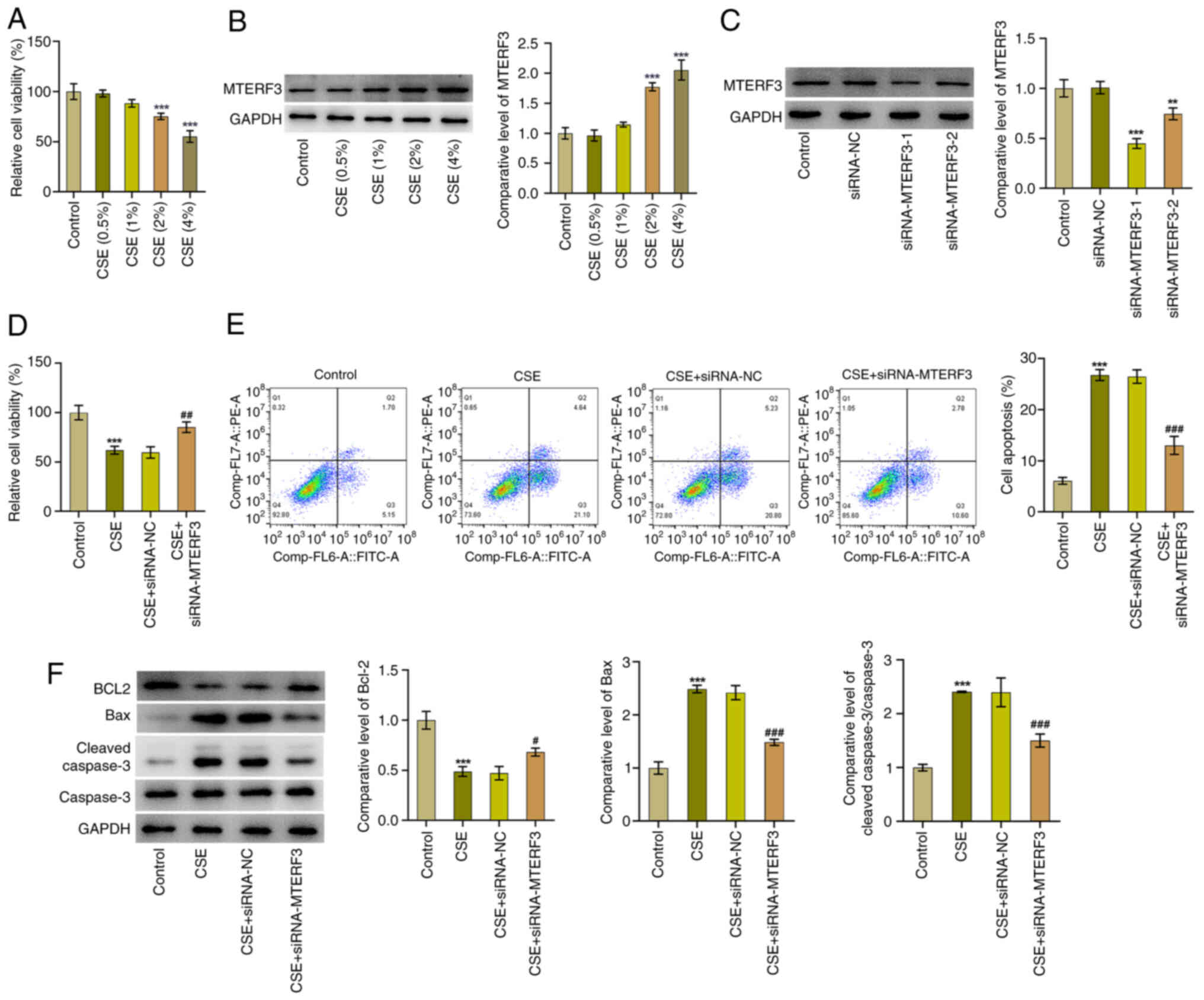
shown in Fig. 6A, the viability of
16HBE cells was gradually decreased with the increase of CSE
concentration compared with the control group. Immunoblotting
revealed that CSE induction led to the upregulated MTERF3
expression; the highest MTERF3 expression level was observed in the
CSE (4%) group (Fig. 6B).
Therefore, CSE (4%) was selected for subsequent studies. It was
shown (Fig. 6C) that MTERF3
expression was significantly downregulated in the siRNA-MTERF3-1
and siRNA-MTERF3-2 groups. 16HBE cells transfected with
siRNA-MTERF3-1 were used to conduct subsequent experiments due to
the lower MTERF3 expression. CCK-8 assay revealed that MTERF3
deficiency significantly elevated the viability of 16HBE cells
treated by CSE compared with the CSE + siRNA-NC group (Fig. 6D). In addition, the apoptotic rate
of 16HBE cells triggered by CSE was significantly decreased
following knockdown of MTERF3 expression, as accompanied by the
increased BCL2 expression and the decreased Bax and cleaved caspase
3 expressions (Fig. 6E and F).
Knockdown of MTERF3 expression
promotes mitophagy in CSE-treated 16HBE cells
The effect of MTERF3 knockdown on mitophagy in
CSE-treated 16HBE cells was explored in the following experiments.
As shown in Fig. 7A and B, the
increased MDA content and decreased SOD activity in the CSE group
were significantly attenuated following MTERF3 loss-of-function in
16HBE cells. Of note, Mito SOX Red staining indicated that
CSE-induced mitoROS accumulation in 16HBE cells was alleviated
following siRNA-MTERF3 transfection (Fig. 7C). In addition, the decreased MMP
expression caused by CSE in 16HBE cells was elevated following
knockdown of MTERF3 expression (Fig.
7D). Moreover, CSE treatment prominently downregulated
LC3II/LC3I, Beclin-1, PINK1 and Parkin (mitochondrion) expression
levels, while it upregulated p62 and Parkin (cytoplasm) expression
levels; this effect was reversed by MTERF3 depletion (Fig. 7E).
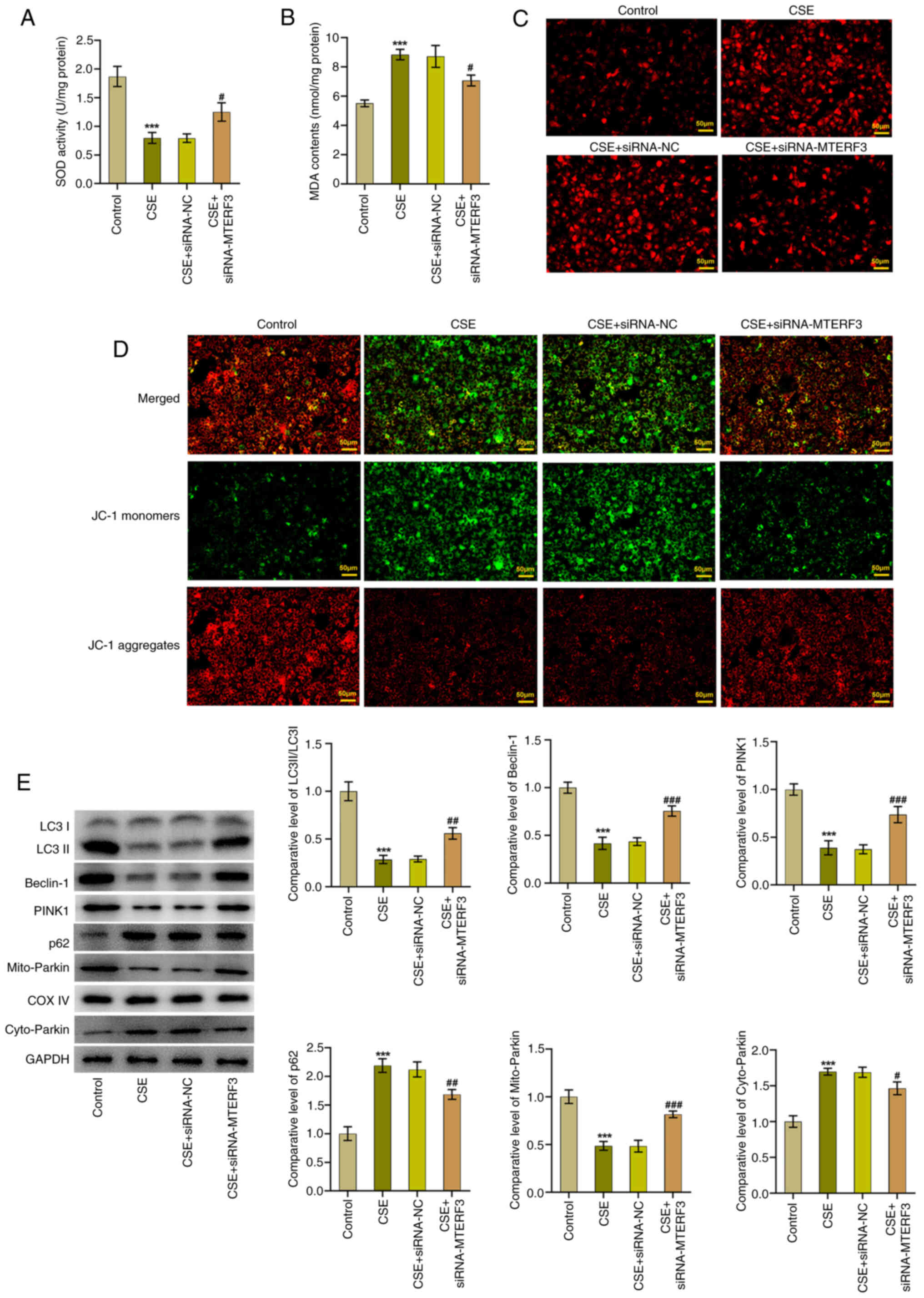 | Figure 7.Knockdown of MTERF3 expression
promotes mitophagy in CSE-treated 16HBE cells. (A) MDA content and
(B) SOD activity were detected using the corresponding kits. (C)
The generation of mitoROS was assessed with the application of Mito
SOX Red staining. (D) MMP was evaluated using JC-1 staining. (E)
Immunoblotting was employed to assess the expression of
mitophagy-related proteins. ***P<0.001 vs. control group;
#P<0.05, ##P<0.01 and
###P<0.001 vs. CSE + siRNA-NC group. MTERF3,
mitochondrial transcription termination factor 3; CSE, cigarette
smoke extract; MDA, malondialdehyde; SOD, superoxide dismutase;
MMP, matrix metalloproteinase; JC-1,
5,5′,6,6′-tetrachloro1,1′,3,3′-tetramethylbenzimidazolylcarbocyanine
iodide; siRNA, small interfering RNA; NC, negative control. |
Discussion
As a heterogeneous lung disease, COPD is a global
health burden (21). Based on
publicly available databases, disease signature genes were screened
for COPD. In vitro cell experiments were conducted using a
CSE-induced bronchial epithelial cell model for validation and
exploration. In the present study, 14 DEMRGs were identified.
Following validation using the test dataset and LASSO regression,
one DEMRG (MTERF3) was acquired. The in vitro cellular
experiments further supported the regulation of MTERF3 on
mitophagy.
Among them, autophagy exerts a crucial effect on the
development and prognosis of COPD (22). Autophagy is a highly conserved
homeostatic process, during which autophagosomes devour and degrade
damaged or aged organelles for further processing and recycling to
maintain cellular homeostasis (10). Notably, autophagy eliminates the
damaged mitochondria, a process known as mitophagy, which is the
main part of the mitochondrial quality control system (11). Dysregulation of mitophagy
contributes to the development of diverse lung disorders, including
asthma, acute lung injury, bronchopulmonary dysplasia and COPD
(12).
A previous study has identified 40 potential
autophagy-related genes of COPD through bioinformatics analysis,
but no basic cell experiments were performed for verification
(22). Mitophagy is the selective
autophagy subtype specific for mitochondria, which exerts crucial
effects on cell survival by maintaining energy homeostasis via its
own catabolic activity (23,24).
Impaired mitophagy, resulting in damaged or dysfunctional
accumulation of mitochondria, has been demonstrated to be related
to diverse pathological conditions; insufficient mitophagy
participates in the pathogenesis of COPD (14,25).
By enhancing mitophagy, Bufei Yishen formula III attenuates COPD
rat pulmonary dysfunction and CSE-induced airway epithelial cell
damage (26). PINK1 and Parkin,
two key markers related to mitophagy, exhibit downregulated
expression levels in lung tissues of COPD mice exposed to CSE
(27). Recently, Wang et al
(28) demonstrated that
pre-treatment with a mitophagy activator alleviated CSE-induced
oxidative stress, improved mitochondrial dysfunction, downregulated
p62 levels and elevated LC3B-II/I ratio in alveolar and airway
epithelial cells. In the present study, CSE treatment impaired
mitophagy in human bronchial epithelial cells, as demonstrated by
downregulation of LC3II/LC3I, Beclin-1, PINK1 and Parkin
(mitochondrion) expressions, upregulation of p62 and Parkin
(cytoplasm) expressions as well as enhanced MitoSOX fluorescence
intensity and reduced MMP levels.
In the present study, MTERF3 displayed significant
variability in expression in both the training and validation sets,
demonstrating high specificity and diagnostic performance. MTERF3
is the most highly conserved member of the MTERF protein family
(29). As reported, MTERF3
functions as a negative regulator of mitochondrial DNA
transcription (30). Accumulating
research has confirmed that MTERF3 is implicated in diseases
associated with mitochondrial dysfunction (31,32).
A recent study on Parkinson's disease has shown that MTERF3
facilitates 1-methyl-4-phenylpyridinium ion-induced mitochondrial
dysfunction in SH-SY5Y cells (33). In addition, MTERF3 expression is
upregulated in lung adenocarcinoma and interference with MTERF3
obviously restrains the proliferation and migration of lung cancer
cells while contributing to mitophagy and the production of MitoSOX
(34). The present study was the
first to explore the expression and role of MTERF3 in CSE-treated
16HBE cells. The results demonstrated that MTERF3 expression was
elevated in CSE-treated 16HBE cells and MTERF3 depletion suppressed
the induction of apoptosis and promoted mitophagy in CSE-treated
16HBE cells.
Accumulating evidence has shown that immune cells
are linked to the development of COPD (35,36).
Significantly higher type 1 T helper cells but lower levels of type
2 T helper cells have been found in the peripheral blood from
patients with COPD (37). A
previous study has also reported the elevated alveolar macrophage
alveolar space in patients with COPD (38). A negative association has been
shown between the number of neutrophils and the degree of alveolar
destruction in smokers, in contrast to alveolar macrophages
(39). As reported, the percentage
of natural killer cells notably decreased in patients with COPD
compared with healthy non-smokers (40). The increases in B cell counts lung
tissues of patients with COPD is correlated directly with COPD
severity (41). In patients with
COPD, long-term CS increases regulatory T cells in the airways
(42). Specifically, Sales et
al (43) demonstrated that the
role of regulatory T cells in regulating immune response may vary
in different lung regions of patients with COPD. Emerging evidence
has supported the notion that CD8 T cells are notably increased in
lung tissues of patients with mild-moderate COPD (38). Consistent with the aforementioned
studies, the current study revealed that the infiltrating abundance
of the majority of immune cells, such as activated CD8 T cells,
regulatory T cells and activated B cells, was higher in COPD
samples than that noted in control samples. Additionally,
significantly reduced proportion of natural killer cells and
neutrophils were found in in COPD samples. These results suggested
that these immune cells were potentially associated with the
pathogenesis of COPD. Moreover, it was found that MTERF3 exhibited
optimal correlation with macrophages, MDSCs, regulatory T cells and
activated CD8 T cells. The present study suggested that MTERF3
exhibited optimal correlation with multiple immune cells. The exact
role of immune cells and MTERF3 requires further investigations
using basic research experiments in the future.
However, certain limitations should be noted in the
current study. First of all, the bioinformatics results were
acquired from lung tissues of patients with COPD. Nevertheless, the
verification was conducted in small airway epithelium of included
individuals. Secondly, there was no information about the inclusion
and exclusion criteria used in selecting the COPD and control
samples on GEO datasets. Additionally, the exact role of immune
cells and their association with signature genes require further
investigations using basic research experiments. Lastly, the
inclusion of additional validation cohorts is crucial to ensure the
sensitivity and reliability of the diagnostic performance noted in
the present study.
In summary, the present study identified a key
signature gene (MTERF3) related to mitophagy in COPD via
bioinformatic analysis. In vitro experiments demonstrated
that MTERF3 expression was upregulated in CSE-treated bronchial
epithelial cells and that MTERF3 deficiency promoted mitophagy.
These findings expand our understanding on COPD and offer novel
perspectives for the diagnosis and treatment of this condition.
Acknowledgements
Not applicable.
Funding
Funding: No funding was received.
Availability of data and materials
The data generated in the present study may be
requested from the corresponding author.
Authors' contributions
JC and GS contributed to the conception and design
of this study. JC and XZ analyzed the data and drafted the
manuscript. GS contributed to make the figures and revised the
manuscript. JC, GS and XZ participated in the experiments. All
authors read and approved the final version of the manuscript. JC
and GS confirm the authenticity of all the raw data.
Ethics approval and consent to
participate
Not applicable.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
López-Campos JL, Tan W and Soriano JB:
Global burden of COPD. Respirology. 21:14–23. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Safiri S, Carson-Chahhoud K, Noori M,
Nejadghaderi SA, Sullman MJM, Ahmadian Heris J, Ansarin K,
Mansournia MA, Collins GS, Kolahi AA and Kaufman JS: Burden of
chronic obstructive pulmonary disease and its attributable risk
factors in 204 countries and territories, 1990-2019: Results from
the Global Burden of Disease Study 2019. BMJ. 378:e0696792022.
View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Boers E, Barrett M, Su JG, Benjafield AV,
Sinha S, Kaye L, Zar HJ, Vuong V, Tellez D, Gondalia R, et al:
Global burden of chronic obstructive pulmonary disease through
2050. JAMA Netw Open. 6:e23465982023. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Hikichi M, Mizumura K, Maruoka S and Gon
Y: Pathogenesis of chronic obstructive pulmonary disease (COPD)
induced by cigarette smoke. J Thorac Dis. 11 (Suppl
17):S2129–S2140. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
GBD 2019 Diseases Injuries Collaborators,
. Global burden of 369 diseases and injuries in 204 countries and
territories, 1990-2019: A systematic analysis for the Global burden
of disease study 2019. Lancet. 396:1204–1222. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Mei J, Zhang Y, Lu S and Wang J: Long
non-coding RNA NNT-AS1 regulates proliferation, apoptosis,
inflammation and airway remodeling of chronic obstructive pulmonary
disease via targeting miR-582-5p/FBXO11 axis. Biomed Pharmacother.
129:1103262020. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Lee KY, Ho SC, Sun WL, Feng PH, Lin CW,
Chen KY, Chuang HC, Tseng CH, Chen TT and Wu SM: Lnc-IL7R
alleviates PM(2.5)-mediated cellular senescence and apoptosis
through EZH2 recruitment in chronic obstructive pulmonary disease.
Cell Biol Toxicol. 38:1097–1120. 2022. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Barnes PJ, Baker J and Donnelly LE:
Autophagy in asthma and chronic obstructive pulmonary disease. Clin
Sci (Lond). 136:733–746. 2022. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Lv X, Li K and Hu Z: Chronic obstructive
pulmonary disease and autophagy. Adv Exp Med Biol. 1207:559–567.
2020. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Mizushima N and Komatsu M: Autophagy:
Renovation of cells and tissues. Cell. 147:728–741. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Lu Y, Li Z, Zhang S, Zhang T, Liu Y and
Zhang L: Cellular mitophagy: Mechanism, roles in diseases and small
molecule pharmacological regulation. Theranostics. 13:736–766.
2023. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Sharma A, Ahmad S, Ahmad T, Ali S and Syed
MA: Mitochondrial dynamics and mitophagy in lung disorders. Life
Sci. 284:1198762021. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Ito S, Araya J, Kurita Y, Kobayashi K,
Takasaka N, Yoshida M, Hara H, Minagawa S, Wakui H, Fujii S, et al:
PARK2-mediated mitophagy is involved in regulation of HBEC
senescence in COPD pathogenesis. Autophagy. 11:547–559. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Ahmad T, Sundar IK, Lerner CA, Gerloff J,
Tormos AM, Yao H and Rahman I: Impaired mitophagy leads to
cigarette smoke stress-induced cellular senescence: Implications
for chronic obstructive pulmonary disease. FASEB J. 29:2912–2929.
2015. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Zhang M, Fang L, Zhou L, Molino A,
Valentino MR, Yang S, Zhang J, Li Y and Roth M: MAPK15-ULK1
signaling regulates mitophagy of airway epithelial cell in chronic
obstructive pulmonary disease. Free Radic Biol Med. 172:541–549.
2021. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Zhang MY, Jiang YX, Yang YC, Liu JY, Huo
C, Ji XL and Qu YQ: Cigarette smoke extract induces pyroptosis in
human bronchial epithelial cells through the ROS/NLRP3/caspase-1
pathway. Life Sci. 269:1190902021. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Liu YB, Hong JR, Jiang N, Jin L, Zhong WJ,
Zhang CY, Yang HH, Duan JX and Zhou Y: The role of mitochondrial
quality control in chronic obstructive pulmonary disease. Lab
Invest. 104:1003072024. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Tulen CBM, Wang Y, Beentjes D, Jessen PJJ,
Ninaber DK, Reynaert NL, van Schooten FJ, Opperhuizen A, Hiemstra
PS and Remels AHV: Dysregulated mitochondrial metabolism upon
cigarette smoke exposure in various human bronchial epithelial cell
models. Dis Model Mech. 15:dmm0492472022. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Trivedi A, Khan MA, Bade G and Talwar A:
Orchestration of neutrophil extracellular traps (Nets), a unique
innate immune function during chronic obstructive pulmonary disease
(COPD) development. Biomedicines. 9:532021. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Schivo M, Albertson TE, Haczku A, Kenyon
NJ, Zeki AA, Kuhn BT, Louie S and Avdalovic MV: Paradigms in
chronic obstructive pulmonary disease: Phenotypes, immunobiology,
and therapy with a focus on vascular disease. J Investig Med.
65:953–963. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Mathioudakis AG, Janssens W, Sivapalan P,
Singanayagam A, Dransfield MT, Jensen JS and Vestbo J: Acute
exacerbations of chronic obstructive pulmonary disease: In search
of diagnostic biomarkers and treatable traits. Thorax. 75:520–527.
2020. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Sun S, Shen Y, Wang J, Li J, Cao J and
Zhang J: Identification and validation of autophagy-related genes
in chronic obstructive pulmonary disease. Int J Chron Obstruct
Pulmon Dis. 16:67–78. 2021. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Ni HM, Williams JA and Ding WX:
Mitochondrial dynamics and mitochondrial quality control. Redox
Biol. 4:6–13. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Palikaras K, Lionaki E and Tavernarakis N:
Mechanisms of mitophagy in cellular homeostasis, physiology and
pathology. Nat Cell Biol. 20:1013–1022. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Wang S, Long H, Hou L, Feng B, Ma Z, Wu Y,
Zeng Y, Cai J, Zhang DW and Zhao G: The mitophagy pathway and its
implications in human diseases. Signal Transduct Target Ther.
8:3042023. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Li MY, Qin YQ, Tian YG, Li KC, Oliver BG,
Liu XF, Zhao P and Li JS: Effective-component compatibility of
Bufei Yishen formula III ameliorated COPD by improving airway
epithelial cell senescence by promoting mitophagy via the
NRF2/PINK1 pathway. BMC Pulm Med. 22:4342022. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Wang Q, Unwalla H and Rahman I:
Dysregulation of mitochondrial complexes and dynamics by chronic
cigarette smoke exposure Utilizing MitoQC reporter mice.
Mitochondrion. 63:43–50. 2022. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Wang S, Song X, Wei L, Liu Q, Li C and
Wang J: Role of mitophagy in cigarette smoke-induced lung
epithelial cell injury in vitro. Curr Mol Med. 23:1130–1140. 2023.
View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Roberti M, Polosa PL, Bruni F, Deceglie S,
Gadaleta MN and Cantatore P: MTERF factors: A multifunction protein
family. Biomol Concepts. 1:215–224. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Park CB, Asin-Cayuela J, Cámara Y, Shi Y,
Pellegrini M, Gaspari M, Wibom R, Hultenby K, Erdjument-Bromage H,
Tempst P, et al: MTERF3 is a negative regulator of mammalian mtDNA
transcription. Cell. 130:273–285. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Zheng Z, Zhao Y, Yu H, Wang T, Li J, Xu L,
Ding C, He L, Wu L and Dong Z: Suppressing MTERF3 inhibits
proliferation of human hepatocellular carcinoma via ROS-mediated
p38 MAPK activation. Commun Biol. 7:182024. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Andersson DC, Fauconnier J, Park CB, Zhang
SJ, Thireau J, Ivarsson N, Larsson NG and Westerblad H: Enhanced
cardiomyocyte Ca(2+) cycling precedes terminal AV-block in
mitochondrial cardiomyopathy Mterf3 KO mice. Antioxid Redox Signal.
15:2455–2464. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Zhu S, Xu N, Han Y, Ye X, Yang L, Zuo J
and Liu W: MTERF3 contributes to MPP+-induced mitochondrial
dysfunction in SH-SY5Y cells. Acta Biochim Biophys Sin (Shanghai).
54:1113–1121. 2022. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Wang J, Liu K and Li J, Zhang H, Gong X,
Song X, Wei M, Hu Y and Li J: Constructing and evaluating a
mitophagy-related gene prognostic model: Implications for immune
landscape and tumor biology in lung adenocarcinoma. Biomolecules.
14:2282024. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Ma R, Su H, Jiao K and Liu J: Role of Th17
cells, Treg cells, and Th17/Treg imbalance in immune homeostasis
disorders in patients with chronic obstructive pulmonary disease.
Immun Inflamm Dis. 11:e7842023. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Kim RY and Oliver B: Innate immune
reprogramming in chronic obstructive pulmonary disease: New
mechanisms for old questions. Am J Respir Cell Mol Biol.
68:470–471. 2023. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Chen G, Mu Q and Meng ZJ: Cigarette
smoking contributes to Th1/Th2 cell dysfunction via the cytokine
milieu in chronic obstructive pulmonary disease. Int J Chron
Obstruct Pulmon Dis. 18:2027–2038. 2023. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Villaseñor-Altamirano AB, Jain D, Jeong Y,
Menon JA, Kamiya M, Haider H, Manandhar R, Sheikh MDA, Athar H,
Merriam LT, et al: Activation of CD8(+) T cells in chronic
obstructive pulmonary disease lung. Am J Respir Crit Care Med.
208:1177–1195. 2023. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Kotlyarov S: Involvement of the innate
immune system in the pathogenesis of chronic obstructive pulmonary
disease. Int J Mol Sci. 23:9852022. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Urbanowicz RA, Lamb JR, Todd I, Corne JM
and Fairclough LC: Altered effector function of peripheral
cytotoxic cells in COPD. Respir Res. 10:532009. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Polverino F, Seys LJ, Bracke KR and Owen
CA: B cells in chronic obstructive pulmonary disease: moving to
center stage. Am J Physiol Lung Cell Mol Physiol. 311:L687–l695.
2016. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Smyth LJ, Starkey C, Vestbo J and Singh D:
CD4-regulatory cells in COPD patients. Chest. 132:156–163. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Sales DS, Ito JT, Zanchetta IA, Annoni R,
Aun MV, Ferraz LFS, Cervilha DAB, Negri E, Mauad T, Martins MA and
Lopes FDTQS: Regulatory T-Cell distribution within lung
compartments in COPD. COPD. 14:533–542. 2017. View Article : Google Scholar : PubMed/NCBI
|