Introduction
Treatment for aplastic anemia includes intensive
immunosuppressive therapy (IST) and allogeneic hematopoietic stem
cell transplantation (AHSCT). AHSCT is the treatment of choice for
young adults with severe aplastic anemia (SAA), with cure ranging
from 75 to 80% and overall survival at 6 years being more than 90%.
Cyclophosphamide (CY) plus antithymocyte globulin (ATG) is the most
commonly used regimen in AHSCT for SAA due to a low incidence of
graft rejection and chronic graft-versus-host disease (GVHD)
(1). However, 5–15% of SAA patients
receiving sibling AHSCT are likely to develop graft rejection,
particularly those patients that have been heavily transfused
(2).
Kaposi’s sarcoma (KS) was first described in 1872 by
Kaposi as a progressive sarcoma (3). It is a multicentric neoplasm of
lymphatic endothelium-derived cells infected with KS-associated
herpesvirus (KSHV). Four recognized clinical subsets can be
distinguished: the sporadic or classic subtype initially described
by Kaposi, the endemic subtype observed in sub-Saharan Africans,
the epidemic subtype in patients infected with human
immunodeficiency virus (HIV) and the iatrogenic subtype in patients
treated by immunosuppressive therapy particularly in organ
transplant recipients (4).
The majority of reported post-transplant KS cases
occurred in solid organ transplant recipients, and the likelihood
of KS developing following hematopoietic stem cell transplantation
(HSCT) is low. Only a few cases of KS following allogeneic stem
cell transplantation were previously reported (5–11). The
majority of these cases had presented with typical KS mucocutaneous
lesions. However, no study regarding KS with atypical presentations
mimicking those of post-transplant lymphoproliferative disorders
(PTLD) after AHSCT for SAA currently exists. This study examined a
patient with SAA who developed KS following matched sibling AHSCT
and succumbed to bone marrow failure due to graft rejection.
Case report
A 33-year-old Chinese male patient from South China
presenting with SAA received AHSCT from his HLA-identical brother
using a CY plus busulfan (BU) and ATG conditioning regimen. Prior
to the transplantation, the patient had been treated with
cyclosporine A (CsA) for 4 months, but without improvement and had
to receive red cell and platelet transfusions constantly. Both the
patient and the donor were serum-negative for hepatitis B, C, HIV,
cytomegalovirus (CMV) or Epstein-Barr virus (EBV). Following
transplantation, CsA was administered for GVHD prophylaxis. The
patient developed grade I acute GVHD with mucocutaneous changes and
diarrhea. His neutrophil count increased to
>1.5×109/l and his platelet count increased to
>50×109/l on days +10 and +14. Complete donor
engraftment was documented on day +30 by blood DNA-STR
amplification. A bone marrow examination showed hyperplasia with a
megakaryocyte count within normal range, but maturation hindrance
was noted. On day +40, the platelet count decreased and 15–30 mg
daily of prednisone was given and sustained to improve
megakaryocyte maturation. Two months after transplantation, the
patient had severe pancytopenia with fever, diarrhea and oral
mucositis. Although the symptoms disappeared following treatment
with antibiotics, the patient had to receive blood transfusion
every week. Three months after transplantation, he got varicella
and then fully recovered following treatment with oral acyclovir.
Another bone marrow examination at that time showed marrow
hypoplasia. The patient then developed fever and respiratory
infections twice and recovered. Blood transfusion was not required
until 5 months after transplantation. However, a second DNA-STR
analysis performed on day +112 revealed partial donor chimerism.
CsA was tapered and completely stopped within 6 months of
transplantation. On day +156, he was discharged. He was free of
infection and did not require blood transfusion.
However, on day +198 he was admitted again
complaining of high fever, cough, intermittent epigastric
discomfort and progressive emaciation. A physical examination
revealed a painful subcutaneous boil of pink color with a diameter
of 5 mm in the right temporal region without any other macula,
plaque or nodule changes in skin. Severe pancytopenia and platelet
transfusion refractoriness with platelet counts of
<20×109/l even after frequent platelet transfusion
were noted. A chest X-ray revealed right pneumonia. The sputum and
blood cultures were positive for different gram negative bacteria
which responded to treatment with antibiotics. Both abdominal B
ultrasound and computer tomography revealed hepatosplenomegaly and
multiple hepatic, splenic hilar and intrabdominal lymphadenopathy.
Bone marrow smear and biopsy examinations were typical of SAA. The
patient remained serum-negative for HIV, hepatitis B virus,
hepatitis C virus, CMV or syphilis. Treatment with combined
antibiotics and anti-fungal medicine was ineffective. One month
later, multiple superficial lymphadenopathy was noted and tender
swelling in his right canthal area gradually developed and
exacerbated. Within a few days, the right periorbital and right
cheek area was badly swollen and right exophthalmos was present
resulting in local exudation and ulceration. Blood samples sent for
CMV antigen and EBV polymerase chain reaction (PCR) analysis were
both negative. Blood DNA-STR analysis on day +190 confirmed
complete graft rejection. With the patient’s consent and blood
transfusion support, excisional biopsy of a cervical lymph node was
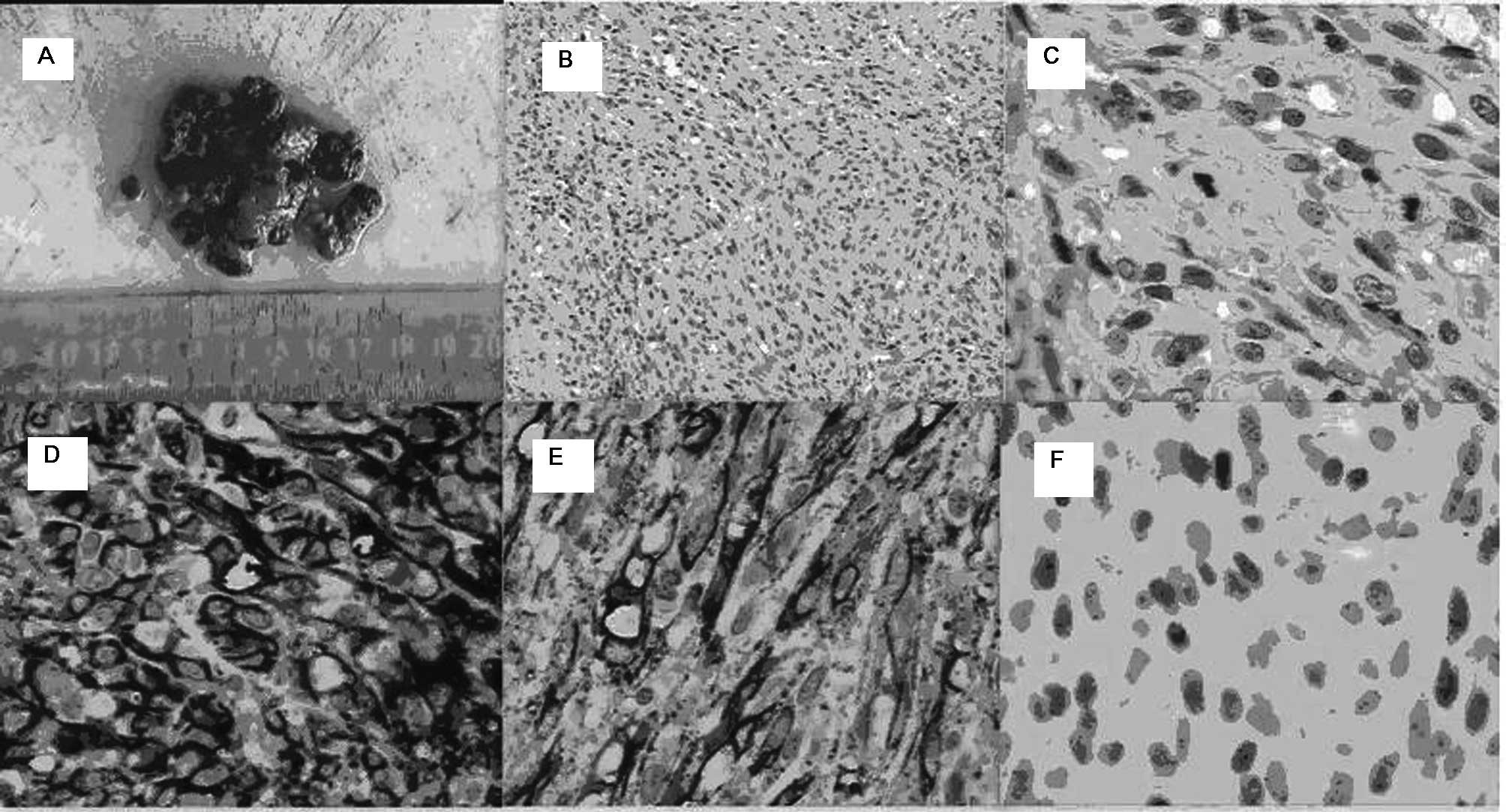
performed and KS was confirmed by positive morphologic,
immunohistochemistry examination results (Fig. 1). The patient then refused further
treatment and was discharged against advice. He succumbed to
intracranial bleeding as a result of thrombocytopenia on day +230
at home.
Discussion
The patient described in this study received
HLA-matched sibling AHSCT with ATG-containing conditioning regimen
after 4 months of CsA treatment for SAA. The DNA-STR results on day
+30 showed complete donor engraftment. However, repeat DNA-STR
analysis performed 4 months after transplantation confirmed a
mosaic phenotype which, together with marrow hypoplasia, were
indicative of partial graft rejection. Early CsA tapering appeared
to have a positive effect, since the blood cell count began to rise
and the patient did not require blood transfusion for over 2 months
after that. However, graft rejection developed, which was confirmed
by the DNA-STR results performed on day +190. It is well-known that
AHSCT is curative in SAA. Nevertheless, graft rejection, infections
and GVHD have limited the effectiveness thereof. Graft rejection
decreased with the introduction of ATG into conditioning regimen,
but it still occurs, particularly in highly transfused patients.
The number of prior transfusions is associated with rejection and
survival after AHSCT due to the sensitization by the
histocompatibility antigens infused with blood products (12). Marsh suggested that SAA patients
with eligible sibling bone marrow transplant (BMT) donors should
not receive IST, but should receive an early transplant before
patients become sensitized to HLA and non-HLA antigens from
multiple transfusions (2). Multiple
transfusions prior to AHSCT may be the main precipitating factors
underlying the graft failure in this patient.
As the patient’s hematopoietic function got worse,
another attack of mixed infections were observed, which was
preceded by KSHV infection resulting in KS and followed by
secondary sepsis. The KS lesion firstly presented as an atypical
skin boil in the right temporal area together with fever, multiple
deep lymphadenopathy and hepatosplenomegaly resembling
presentations of PTLD. PTLDs are a heterogeneous group of
lymphoproliferative disorders associated with immunosuppression in
recipients of solid organ or allogeneic stem cell transplantation.
The clinical presentations of PTLD may vary from symptomless or
non-specific early symptoms, such as fever, malaise and weight loss
in certain patients to typical involvement of lymph nodes and
extranodal involvement in other patients. Most cases of PTLD are
associated with EBV infection from B-lymphocytes (13). In our case, both the EBV PCR
analysis of the patient’s blood sample and Epstein-Barr encoded RNA
(EBER) in situ hybridization test on his pathologic tissue
were negative. Although 20–30% of PTLDs were reportedly
EBV-negative and a case of HHV-8-positive PTLD has been reported
(14), the typical pathological
alterations of the lymph node biopsy in this case exclude the
diagnosis of PTLD.
Clinical features of KS include mucocutaneous and
visceral involvement. Mucocutaneous lesions have been reported in
more than 90% of post-transplantation KS cases usually starting as
macules that progress and coalesce to form large plaques or nodular
and fungiform tumors. These lesions are mainly localized on the
lower limbs, but are also frequently observed on the trunk and
upper limbs, face, genitalia and oropharyngeal mucosa.
Additionally, KS frequently involves lymph nodes and visceral
organs, notably the respiratory and gastrointestinal tracts. Other
unusual locations of KS involvement include the musculoskeletal
system, nervous system, larynx, eye, salivary glands, endocrine
glands, heart, thoracic duct, urinary system, breast and wounds
(15,16). The development of KS following AHSCT
is rare. A review of the current literature shows that only a few
KS cases have been reported following AHSCT in patients with sickle
cell disease or leukemia. Development of KS following AHSCT for SAA
has never been reported. All of the reported KS cases had presented
with typical mucosal or skin lesions more than 6 months after AHSCT
(5–11). The atypical and complex clinical
characteristics of KS in this case made it difficult to make an
earlier diagnosis.
KS occurs in patients infected with human
herpesvirus type 8 (HHV-8), also known as KSHV, and the level of
immunosuppression is the main factor for the development and
progression of the disease. HHV-8 belongs to a family of
double-stranded DNA viruses involving herpesviruses that are able
to escape from complete clearance by the human immune system. The
ability of these viruses to become latent is due to their delicate
interference with the immune system. Consequently, some of these
viruses are regarded as tumor viruses. Herpesviridae comprises
three main subfamilies: α- , β- and γ-herpesviruses.
α-herpesviruses consist of human herpesvirus-1 (HHV-1), HHV-2
(genital herpes virus) and HHV-3 (varicella-zoster virus; VZV).
β-herpesviruses comprise HHV-5 (CMV), HHV-6 and HHV-7. The
subfamily of γ-herpesviruses comprises HHV-4 (EBV) and HHV-8 (KSHV)
(17). Of note is that during the
post-transplant period, the patient in this study got varicella
caused by HHV-3 infection during the 2nd post-transplant month.
Thus, this patient had infections by two subtypes of herpesvirus at
different post-transplant periods. The most frequently reported
herpesvirus infections following AHSCT are CMV or EBV (18). Mixed infections of two or three
herpesviruses have been reported, mostly CMV and EBV infections.
Other herpesviruses include successive EBV/HHV-7, HHV-6/CMV and
CMV/HHV-8 infections (10,19,20).
Successive VZV and HHV-8 infections in post-transplantation
patients have never been reported and may be an indication of
sustained immunosuppression in the patient.
The origin of KSHV infection in this patient is
unknown as no examination was performed for HHV-8 infection in
either the donor or the recipient prior to transplantation. KSHV
infection in an immunocompetent host is usually asymptomatic.
Therefore, we cannot exclude latent KSHV infection in the donor or
the recipient prior to transplantation although neither the donor
nor the recipient had presented with any clinical manifestations of
KS. In the case reported in this study, KSHV infection may have
been transmitted from the donor’s latent infection, the result of
reactivation of the recipient’s previous infection or through blood
transfusions. Studies on viral serology suggest that
post-transplant KS was primarily due to HHV-8 reactivation in
endemic areas and to primary infection in non-endemic areas
(10). KS is rare in the majority
of Chinese regions, with the exception of Xinjiang. The
seroprevalence of KSHV in the general population is 9.5–12.3% and
that in volunteer blood donors ranged from 5.65 to 16.2% (21,22).
Rosenzwajg et al studied the seropositivity of antibodies to
HHV-8 latent nuclear antigen in 200 allogeneic BMT recipients and
their donors. These authors did not find any association between
the presence of antibodies prior to or after transplantation to
chronic GVHD or to overall BM transplantation survival. However,
their study suggests that blood transfusions increase the risk of
HHV-8 infection following BMT (23). It was estimated that KS develops in
0.1–5% of transplant recipients (24). Risk for KS development following
organ transplantation is 500 times higher than that in the general
population and increases with immunosuppressive therapy. Compared
to more frequently reported KS cases following solid organ
transplantation, only a few cases of KS following AHSCT have been
reported thus far. It has been suggested that the intense cytotoxic
conditioning regimens in AHSCT destroy host lymphoid tissues and
potential HHV-8 harbouring cells, whereas the immunosuppressive
regimens used in solid organ transplants do not eradicate such
cells (10). This may partially
explain the reason that KS is rare following AHSCT. The above
results indicate that the patient in this study got KSHV infection
through blood transfusions as compared to donor-derived infection
or reactivation of KSHV infection.
The main treatment of post-transplantation KS
involves tapering down immunosuppressive regimens to the lowest
possible level. In this case, CsA was discontinued 6 months after
transplantation. However, a small dose of prednisone was sustained
from 1 month after transplantation to the last month. Moreover, the
intractable and progressive graft rejection led to the recurrence
of aplastic anemia resulting in refractory neutropenia and
secondary infections in the patient. This event combined with
sustained immnuosuppression therapy caused severe immunosuppression
and rapid progression of KS in the patient. Other treatment choices
for KS include cryotherapy, surgical removal or intralesional
chemotherapy for localized mucocutaneous lesions and chemotherapies
comprising vinblastine, bleomycin, liposomal anthracycline, taxanes
or thalidomide for advanced cases with visceral lesions (3). The patient in this study received none
of the therapies due to severe pancytopenia, complicating
infections and economical reasons. As in the majority of cases with
KS, the patient did not succumb to KS, but to marrow failure caused
by the recurrence of SAA.
In conclusion, this report examined a patient with
KS following AHSCT for SAA presenting with atypical clinical
features resembling those of PTLD. Sustained immunosuppression and
graft rejection were the main risk factors of KS infection in this
case. In post-transplantation patients with PTLD-like
presentations, differentiation from KS should be included.
Acknowledgements
The authors would like to thank Professor Qifa Liu
from the Department of Hematology of Nanfang hospital for his help
in the diagnosis and treatment of the case.
References
|
1
|
Ommati LV, Rodrigues CA, Silva AR, Silva
LP, Chaufaille ML and Oliveira JS: A retrospective comparison of
cyclophosphamide plus antithymocyte globulin with cyclophosphamide
plus busulfan as the conditioning regimen for severe aplastic
anemia. Braz J Med Biol Res. 42:244–250. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Marsh J: Making therapeutic decisions in
adults with aplastic anemia. Hematology Am Soc Hematol Educ
Program. 78–85. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Ahmadpoor P: Human herpesvirus-8 and
Kaposi sarcoma after kidney transplantation: mechanisms of tumor
genesis. Iran J Kidney Dis. 3:121–126. 2009.PubMed/NCBI
|
|
4
|
Abbaszadeh S and Taheri S: Kaposi’s
sarcoma after renal transplantation. Saudi J Kidney Dis Transpl.
20:775–758. 2009.
|
|
5
|
Helg C, Adatto M, Salomon D, et al:
Kaposi’s sarcoma following allogeneic bone marrow transplantation.
Bone Marrow Transplant. 14:999–1001. 1994.
|
|
6
|
Gluckman E, Parquet N, Scieux C, et al:
KS-associated herpesvirus-like DNA sequences after allogeneic
bone-marrow transplantation. Lancet. 346:1558–1559. 1995.
View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Erer B, Angelucci E, Muretto P, et al:
Kaposi’s sarcoma after allogeneic bone marrow transplantation. Bone
Marrow Transplant. 19:629–631. 1997.
|
|
8
|
De Medeiros BC, Rezuke WN, Ricci A Jr, et
al: Kaposi’s sarcoma following allogeneic hematopoietic stem cell
transplantation for chronic myelogenous leukemia. Acta Haematol.
104:115–118. 2000.
|
|
9
|
Tamariz-Martel R, Maldonado MS, Carrillo
R, Crespo D, Pérez-Caballero C and Muñoz A: Kaposi’s sarcoma after
allogeneic bone marrow transplantation in a child. Haematologica.
85:884–885. 2000.
|
|
10
|
Bruno B, Sorasio R, Barozzi P, et al:
Kaposi’s sarcoma triggered by endogenous HHV-8 reactivation after
non-myeloablative allogeneic haematopoietic transplantation. Eur J
Haematol. 76:342–347. 2006.
|
|
11
|
Avital I, Moreira AL, Klimstra DS, et al:
Donor-derived human bone marrow cells contribute to solid organ
cancers developing after bone marrow transplantation. Stem Cells.
25:2903–2909. 2007. View Article : Google Scholar
|
|
12
|
Dulley FL, Vigorito AC, Aranha FJ, et al:
Addition of low-dose busulfan to cyclophosphamide in aplastic
anemia patients prior to allogeneic bone marrow transplantation to
reduce rejection. Bone Marrow Transplant. 33:9–13. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Choi JH, Park BB, Suh C, Won JH, Lee WS
and Shin HJ: Clinical characteristics of monomorphic
post-transplant lymphoproliferative disorders. J Korean Med Sci.
25:523–526. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Sathy SJ, Martinu T, Youens K, et al:
Symptomatic pulmonary allograft Kaposi’s sarcoma in two lung
transplant recipients. Am J Transplant. 8:1951–1956.
2008.PubMed/NCBI
|
|
15
|
Lebbé C, Legendre C and Francès C: Kaposi
sarcoma in transplantation. Transplant Rev. 22:252–261. 2008.
|
|
16
|
Pantanowitz L and Dezube BJ: Kaposi
sarcoma in unusual locations. BMC Cancer. 8:1902008. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Ahmadpoor P: Human herpesvirus-8 and
Kaposi sarcoma after kidney transplantation: mechanisms of tumor
genesis. Iran J Kidney Dis. 3:121–126. 2009.PubMed/NCBI
|
|
18
|
Razonable RR and Eid AJ: Viral infections
in transplant recipients. Minerva Med. 100:479–501. 2009.PubMed/NCBI
|
|
19
|
Zawilińska B, Kosz-Vnenchak M,
Piatkowska-Jakubas B, Kopeć J, Daszkiewicz E and Skotnicki AB:
Herpesviruses mixed infections in allogeneic steam cell recipients
(allo-HSCT). Przegl Epidemiol. 62:39–46. 2008.PubMed/NCBI
|
|
20
|
Wang LR, Dong LJ, Zhang MJ and Lu DP:
Correlations of human herpesvirus 6B and CMV infection with acute
GVHD in recipients of allogeneic haematopoietic stem cell
transplantation. Bone Marrow Transplant. 42:673–677. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Zhu B, Xie YR and Chen YG: Kaposi’s
sarcoma-associated herpesvirus infection in portion population of
China and the possible mode of transmission. Zhonghua Liu Xing Bing
Xue Za Zhi. 30:528–529. 2009.
|
|
22
|
Mei Q, Ming ZW, Ping YX, et al: HHV-8
seroprevalence in blood donors and HIV-positive individuals in
Shandong area, China. J Infect. 55:89–90. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Rosenzwajg M, Fery N, Bons V, Damaj G,
Gluckman E and Gluckman J: Human herpes virus 8 (HHV8) serology in
allogeneic bone marrow transplant recipients. Bone Marrow
Transplant. 24:351–354. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Stoebner PE, Fabre C, El Kabbaj N, Bismuth
M, Pageaux GP and Meunier L: Koebnerizing Kaposi’s sarcoma mimics a
laparotomic hypertrophic scar in a liver transplant recipient.
Liver Transpl. 15:994–996. 2009.PubMed/NCBI
|