1. Introduction
Epithelial ovarian cancer (EOC) is the most lethal
gynecologic malignancy worldwide. Epidemiology calculations of
lifetime risk for EOC are that 1 in 55 women is likely to develop
EOC during their lifetime (1).
Since EOC is more likely to be advanced stage with unfavorable
tumor biology, there are serious limitations to the surgical and
oncological treatment available. Therefore, it is crucial to
determine the earliest possible diagnosis. Early diagnosis and
surgical resection offer patients the best opportunity of excellent
long-term survival. On the other hand, patients who either present
with metastatic disease or develop distant relapse within 6 months
after surgery and chemotherapy have a poor prognosis. Among EOC,
clear cell carcinomas of the ovary (CCC) are frequently
characterized by chemoresistance and recurrence, resulting in a
poor prognosis (2). Since CCC are
resistant to conventional cytotoxic (platinum plus taxan-based)
chemotherapy, the prognosis is mostly poor (3). In the USA, the incidence of CCC is 5%
of all EOC, but the incidence in Japan is reported to be over 20%
of all EOC. Thus, Japanese oncologists have focused on
investigating the molecular pathogenesis and treatment strategies
of CCC.
At present, little is known about the molecular
genetic mechanisms that are involved in CCC tumorigenesis.
Accumulated somatic mutations found in a subset of cancer genes are
frequently noted. Such mutations include insertions/deletions
(indels) and base substitutions that act as the ‘driver mutations’
in oncogenesis. These mutations result in the activation of
proto-oncogenes (gain of function) or the inhibition of tumor
suppressor genes (loss of function) during the process of
carcinogenesis. Screening for regions with loss of heterozygosity
(LOH) in tumors is widely used to search for novel tumor suppressor
genes (4).
Endometriosis has been suggested to increase the
risk of developing EOC (5).
Compared to the serous surface epithelial subtype (serous
adenocarcinoma, SAC), which comprises the majority of EOC, some
types such as CCC and endometrioid adenocarcinoma (EAC) have been
associated with endometriosis (6).
CCC are most frequently associated with ovarian and/or pelvic
endometriosis in Japan (5). Even in
the case of endometriosis, LOH at 1, 5, 9, 10, 11, 17 and 22q is
especially common (7). In total,
approximately 30% of cases exhibited LOH at one or more of these
loci. These studies support the hypothesis that tumor suppressor
gene inactivation plays a role in the development of at least one
subset of cases (4,7). LOH studies have shown the involvement
of specific chromosomal regions (5, 6, 9, 10, 11, 17 and 22q) in
CCC (8). LOH of these chromosomes
is frequent not only in endometriosis but also in CCC, suggesting
that one or more common tumor suppressor genes are present in these
regions.
The purpose of this review is to summarize the
current knowledge on the molecular mechanisms involved in CCC
tumorigenesis and to provide the central role played by aberrant
chromatin remodeling. We specifically summarize what is currently
known about the salient features of members of the ARID protein
family and focus on recent developments in characterizing the
functional links of carcinogenesis between CCC and clear cell renal
cell carcinoma (ccRCC).
2. Materials and methods
The present article reviews the English-language
literature for chromatin remodeling studies on CCC. We searched
PubMed electronic databases over a 20-year period (1990–2010),
combining the keywords ‘ARID1A’, ‘SWI/SNF’, ‘chromatin remodeling’,
‘p53’, ‘pRb’, ‘cell cycle’, ‘check point’ with ‘endometriosis’,
‘ovarian cancer’, ‘endometriosis-associated ovarian cancer (EAOC)’,
‘clear cell carcinoma of the ovary’, or ‘clear cell renal cell
carcinoma’. Various recent studies are discussed in the context of
the pathogenesis of CCC. Additionally, references in each article
were searched to identify potentially missed studies for a 10-year
period. In the present review, we evaluate promising molecular
candidates for the development of CCC.
3. Article selection, data extraction and
assessment
Although the main focus of the present review was
the regulation of ARID1A mutations obtained from human cancer
samples, in vitro studies were included in the knowledge
base. Animal models performed to support human data were also
included. Initially, 37 potentially relevant studies were
identified by screening electronic databases. Additionally, 49
peer-reviewed journal articles were identified from references.
4. Chromatin remodeling complex in
cancers
Chromatin regulates transcriptional processes
through coordinated covalent modifications of DNA and its
associated nucleosomal histones (9). Such modifications include acetylation,
methylation and ubiquitination. Post-translational modifications of
histones play critical roles in chromosome dynamics. Epigenetic
mechanisms underlying the modification of chromatin structure rely
on the activity of complexes that control the accessibility of DNA
sequences to transcription factors, thereby determining its
different functional states (9).
Chromatin remodeling complexes are master regulators of
transcription factor action and enable gene transcription by aiding
in the coordination of the binding of transcription factors to
promoters and enhancers. These complexes are involved in various
processes that require alteration of chromatin structure including
DNA repair, DNA synthesis, mitosis and genomic stability (10). Mounting evidence shows that
alterations in the subunits of these complexes play a significant
role in human disease, including cancer (10). Core components of the chromatin
remodeling complexes are potent tumor suppressors that are
specifically inactivated in cancers. Alterations of the components
have been reported in a variety of cancer types, including central
nervous system, head and neck, leukemia, breast, lung,
neuroblastoma, renal, skin, gastric, colon, cervical and prostate
cancer (10). Results of various
studies have also shed light on the mechanistic basis of the action
by demonstrating that these components regulate the cell cycle to
prevent oncogenic transformation.
5. Characteristics of SWI/SNF subunit
inactivation in CCC
SWI/SNF
The chromatin remodeling complexes include SWI/SNF,
ISWI (Imitation SWI), CHD/Mi-2 (chromodomain helicase DNA binding
protein), and INO80 (SNF2 family helicase). Of these complexes,
SWI/SNF is the most studied, consisting of an evolutionarily
conserved protein complex from yeast to humans. First, SWI/SNF is a
multimeric complex of proteins of variable composition, including
ATPase, core and accessory proteins. The functions and components
of the SWI/SNF complex have been thoroughly reviewed elsewhere
(10). This complex constitutes
SMARCs (SWI/SNF-related, matrix-associated, actin-dependent
regulators of chromatin; SMARCA/BRGI subunit or SMARCA2/BRM
subunit) and the BAF (BRM- or BRG1-associated factors) complex,
which comprises 10–12 protein subunits (10–12).
The role of this complex may be to protect cells against DNA damage
by ensuring DNA repair through cell cycle arrest and apoptosis. Key
observations link the SWI/SNF complex with cancer. The complex is
associated with multiple cancer-related pathways, and various
components of the SWI/SNF complex act as bona fide tumor
suppressors (13). Furthermore,
cancer-related proteins such as p21 (also known as p21WAF1/Cip1 and
CDKN1A, cyclin-dependent kinase inhibitor 1A), BRCA1 (breast cancer
1, early onset), LKB1 (also known as STK11, serine/threonine kinase
11), SMADs, FOS (FBJ murine osteosarcoma viral oncogene homolog),
MYC (v-myc myelocytomatosis viral oncogene homolog) and FANCA
(Fanconi anemia, complementation group A) have been associated with
certain components of the SWI/SNF complexes. A growing body of
evidence has demonstrated that these factors play a critical role
in several steps of carcinogenesis, particularly in alterations in
the cell cycle checkpoint machinery. p21WAF1/Cip1 mediates the
p53-dependent cell cycle G1 phase arrest in response to a variety
of stress stimuli (14). Epigenetic
inactivation of BRCA1 impairs the machinery involved in maintaining
genomic integrity and stability, and also acts as a bona
fide tumor suppressor (15).
LKB1/STK11 plays a role in apoptosis and cell cycle arrest, both of
which may require the tumor suppressor action of this kinase, whose
mutations occur in approximately 50% of lung cancers (16). LKB1 tumor suppressor enzyme is
vulnerable to inactivation by redox-active species. Other genes
such as SMADs, FOS, MYC and FANCA have been shown to play a role as
regulators of cell cycle progression, apoptosis and cellular
transformation (17–19).
BRG1 (Brahma/SWI2-related gene 1)
BRG1 (Brahma/SWI2-related gene 1) is a central
component of the SWI/SNF chromatin-remodeling complex that features
an ATPase activity (13). The role
of BRG1 is to arrest the cell cycle. Loss of BRG1 has been
associated with cancer development. BRG1 deficiency is associated
with a subset of lung, breast, prostate and pancreatic cancers
(13).
SNF5/INI1
The clearest functional link between the SWI/SNF
complex and cancer is evident from the subunit SNF5/INI1 (also
known as SMARCAB1 or BAF47). SNF5 is a core protein of the SWI/SNF
complex. SNF5 leads to a G1 cell cycle arrest associated with an
increase in p16INK4a (also known as CDKN2A, cyclin-dependent kinase
inhibitor 2A), E2F and cyclin D to prevent oncogenic transformation
(20). Importantly, the E2F
transcription factor plays a role in cell cycle control and is
intimately regulated by RB (retinoblastoma tumor suppressor gene).
Since SNF5 is a potent tumor suppressor, loss of SNF5 can lead to
aberrant cell cycle activity and subsequent tumor formation. In
particular, germ line or somatic mutations of the SNF5 gene were
detected in malignant rhabdoid tumors arising primarily in the
kidney and brain. This gene has been shown to bind the c-myc
proto-oncogene, as well as the BRCA1 and p53 tumor suppressors.
Furthermore, SNF5 cooperates with HDAC (histone deacetylase) and
loss of its function may thus inactivate HDAC. There is no evidence
that silencing of these components of the SWI/SNF complex occurs by
epigenetic means in EOC.
BAF
The mechanistic relationship between BAF complexes
and the chromatin architecture inhibits target genes (21). BAF250 [also known as ARID1
(adenine-thymine rich interactive domain 1)] is responsible for
directing the SWI/SNF complex to target promoters and regulates the
transcription of certain genes by altering the chromatin structure
around those genes (http://www.ncbi.nlm.nih.gov/gene/8289). ARID1 is also
involved in the modulation of hormone-responsive promoters. The
C-terminus of the protein is capable of stimulating glucocorticoid
receptor-dependent transcriptional activation. Additionally, ARID1
is an essential gene for FAS (TNF receptor superfamily, member
6)-mediated apoptosis (22). ARID1A
is one of the most frequently deleted genes across all cancer types
(23,24) and knockdown of this gene results in
a failure of cell cycle arrest (23,25).
This alteration strongly predominates in kidney, breast and lung
cancers in humans (13,26). All inactivating mutations of this
gene have been found in these cancers, suggesting that ARID1A plays
an essential role during cancer development as a tumor
suppressor.
More recent data from two groups have provided
information about the profiles of CCC tumors with ARID1A
inactivation (27,28). Inactivating mutations of ARID1A were
identified in sporadic CCC samples and a variety of CCC cancer cell
lines. ARID1A is considered to be the most commonly altered gene in
EAOC, particularly in CCC, while alterations are rare in SAC. The
mutation spectrum was enriched for C to T transitions at 5′-CG base
pairs. The mutations in ARID1A result in a stop codon or an
out-of-frame insertion or deletion. Atypical endometriotic lesions
adjacent to the tumor (and not distant lesions) also exhibited the
ARID1A mutations (27). Taken
together, the data show that CCC may arise from atypical
endometriosis with the mutation of ARID1A. Even in cases where
genetic mutations of ARID1A have not been identified, other
aberrations affecting its activity have been noted, such as
abnormal promoter hyper-methylation of ARID1A or aberrations of the
downstream target genes, leading to low or normal protein levels,
respectively. No conclusive evidence currently exists to support
the existence of a role for ARID1A in tumor suppression in CCC.
Identification of ARID-containing
proteins and ARID1A-interacting components
A number of distinct human ARID proteins have been
identified, including p270 (also known as ARID1A), KIAA1235
(ARID1B), RBP1 (retinol binding protein 1), RBP1L1 (ARID4B), RBP2
(retinol binding protein 2), SMCY/SMCX [KDM5D, lysine (K)-specific
demethylase 5D], Plu-1 (KDM5B), jumonji (JARID2), Bright (DRIL1)
(ARID3A), Bdp (DRIL-2) (ARID3B), MRF1 (ARID5A) and MRF2 (ARID5B)
(29). Proteins of the ARID family
are required for embryonic development and patterning, and maintain
the expression pattern of homeotic genes at the chromatin level. As
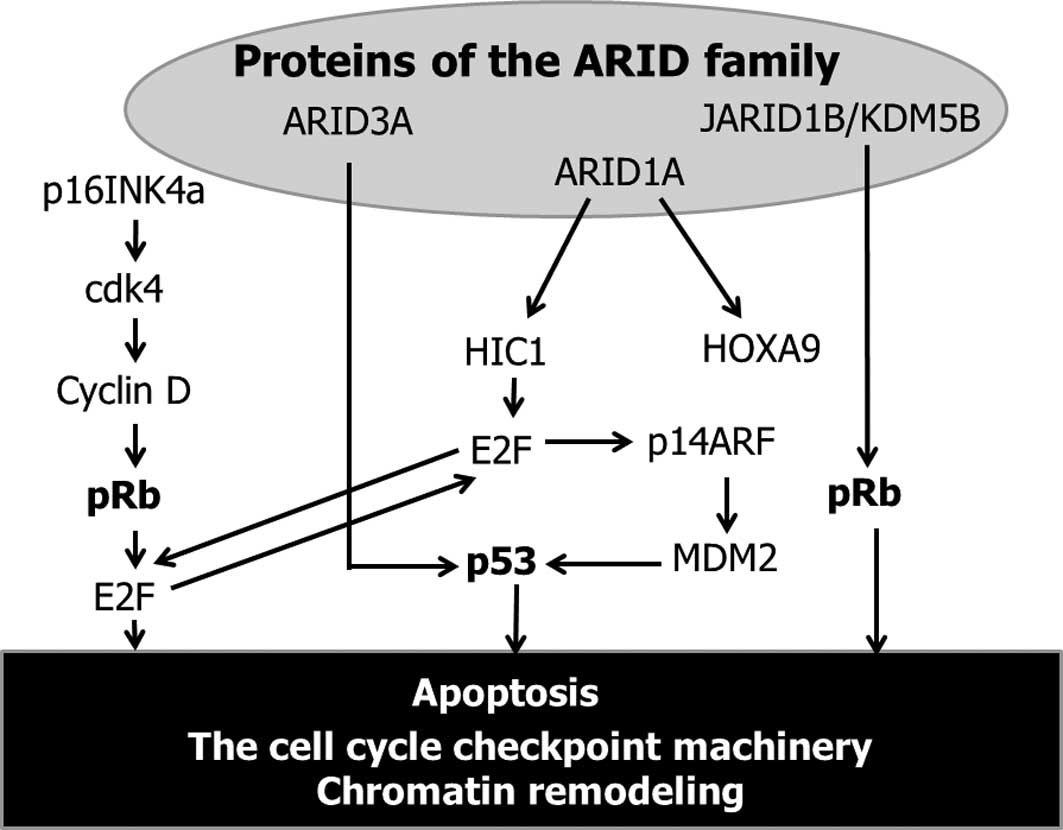
shown in Fig. 1, various BAF250a
(ARID1A)-interacting proteins (transcription factor) exist,
including HOXA9 and HIC1. ARID1A is a positive regulator of HOXA9
(30). HOXA9 expression is
spatially and temporally regulated during hematopoiesis and
embryonic development. Reduced HOXA9 transcript levels are
reportedly associated with breast cancer aggression, metastasis and
patient mortality (31). ARID1A
also directly interacts with HIC1 (hypermethylated in cancer 1), a
tumor suppressor gene, which is epigenetically inactivated in
numerous human cancers (32,33).
This gene encodes a transcriptional repressor involved in
regulatory loops modulating p53-dependent and E2F-dependent cell
survival and damage/stress responses through the recruitment of
ARID1A. E2F is a critical regulator of genes required for apoptosis
by up- regulating p53 and enhancing the p53-mediated activation of
downstream pro-apoptotic genes (34). ARID3A is known as E2FBP1 (E2F
binding protein-1; a protein that interacts with E2F) (35). ARID3A also interacts with p53 and is
involved in the p53 regulatory pathway, suggesting that ARID3A
plays a role in growth suppression mediated by p53 (35). In addition, p14ARF inhibits
formation of the MDM2-p53 complex and subsequently prevents
MDM2-induced p53 degradation (36).
p14ARF stops cell growth at the G1/S and G2/M phases (36). Loss of the p53 signaling pathway
occurs in human cancer either by p53 gene mutation (a direct
mechanism) or by loss of cell signaling upstream and downstream of
p53 (an indirect mechanism) in the remaining cancers expressing
wild-type p53 gene (37).
Certain ARID family proteins are linked genetically
with E2F-mediated transcriptional factor, which is regulated in
part by pRb, an active retinoblastoma tumor suppressor gene,
resulting in cell cycle arrest (38). The p16INK4a-cdk4-cyclin D-pRb
pathway is modulated by ARID-dependent E2F signaling, resulting in
cell cycle regulation (38). pRb
phosphorylation leads to cell release from G1 arrest and to
promotion entry into the S-phase (36). In addition, JARID1B/KDM5B, a member
of the ARID family, exerts cell cycle control via maintenance of
pRb (39). These data allow us to
speculate that proteins of the ARID family directly or indirectly
regulate the cell cycle, particularly alterations in the cell cycle
checkpoint machinery. In contrast to various other human tumor
types, p53 and pRb mutations are only rarely detected in CCC.
ARID1A mutations may be involved in the carcinogenesis of CCC
through inactivation, but not mutation, of the cell cycle
regulatory proteins such as p53 and pRb.
6. A marked resemblance between CCC and
ccRCC
Clear cell cancers possess a marked similarity in
gene expression profiles between CCC and ccRCC (40), suggesting that CCC and ccRCC are
also pathogenetically similar. ccRCC is characterized by the
presence of inactivating mutations in the VHL (von Hippel-Lindau)
gene in the majority of cases (41). The VHL protein acts as the substrate
recognition module of an E3 ubiquitin ligase complex by binding the
substrate and adapters (42,43).
This protein directly interacts with Elongin C, whereas Elongin B
connects VHL-Elongin C to cullin2-ROC1 (Rbx1) (Fig. 2A). These substrates include
hypoxia-inducible factors (HIF-1α and -2α). VHL forms a complex
that retains the ability to ubiquitinate HIFs, resulting in
degradation by targeting the hydroxylated HIF-α subunit for
ubiquitination and proteasomal degradation. VHL protein acts as a
master regulator of HIF-activity loss of the VHL protein due to
germline or somatic mutations disrupting the formation of this
complex, thereby reducing the ability of the VHC protein to
ubiquitinate HIFs, resulting in accumulation of HIFs to high levels
(42). Overexpression of HIF genes
stimulates the expression of a number of significant genes, such as
vascular endothelial growth factor (VEGF), platelet-derived growth
factor (PDGF) and transforming growth factor α (TGFα) (44).
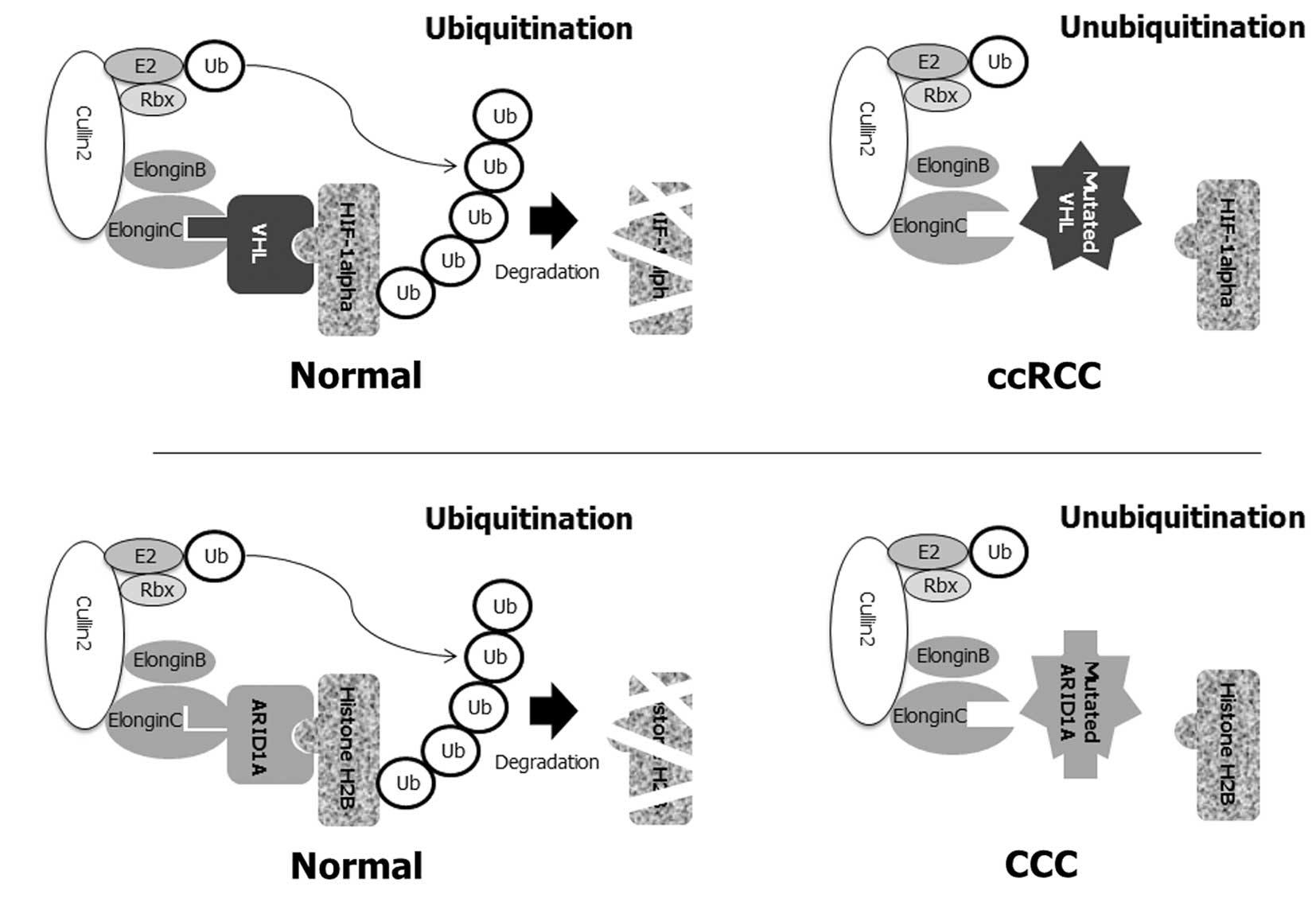 | Figure 2Putative molecular pathogenesis of
CCC with ARID1A mutations. (A) The majority of ccRCC are caused by
the mutation of the von Hippel-Lindau (VHL) tumor suppressor gene
(54). VHL protein is part of an
ubiquitin ligase complex that comprises elongin B, elongin C,
cullin 2 and Rbx1, which are involved in ubiquitin-mediated
destruction. This complex targets a hypoxia-inducible factor (HIF)
transcription factor. In the absence of VHL, HIF-responsive genes
and its downstream targets are activated. In ccRCC, when VHL
protein either cannot function due to a mutation or is abnormally
low/absent in the cell, HIF-1α cannot be bound to the ubiquitin
ligase, cannot be degraded, and thus is constitutively present at a
higher level in the nucleus. High levels of HIF-1α in turn lead to
the overexpression of VEGF and other angiogenesis factors in
carcinogenesis. (B) The molecular pathogenesis of ccRCC with VHL
mutations resembles that noted in CCC patients with ARID1A
mutations. ARID1A protein is also part of an ubiquitin ligase
complex for ubiquitin-mediated destruction of histone H2B. Similar
to the VHL gene, once histone H2B binds to ARID1A, an ubiquitin
ligase complex binds H2B, leading to ubiquitination of H2B, and
marking it for degradation by the proteasomal machinery of the
cell. The right panels of the figure show the disruption of this
normal regulatory process when ARID1A function is aberrant by
somatic mutations. In the absence of functional ARID1A, the
ubiquitin ligase complex cannot bind H2B, resulting in the
accumulation of H2B in the nucleus. Inactivation of the ARID1A
tumor-suppressor protein and subsequent loss of function in the
ARID1A complex result in dysfunction in the ubiquitination of
histone H2B, which is crucial in the aberration of chromatin
remodeling and cell cycle checkpoint machinery, and subsequent
evasion of apoptosis. |
As described above, the majority of CCC possess
somatic inactivating mutations in the ARID1A gene (27,28).
As with VHL, ARID1A exists in complex with a series of other
proteins, including elongin B, elongin C, cullin2 and Rbx1, to form
an ubiquitin ligase complex (30)
(Fig. 2B). Loss of the ARID1A tumor
suppressor, which occurs with VHL mutations in ccRCC, promotes CCC
tumorigenesis primarily through loss of the assembly of the
Cullin-Elongin-Rbx1-E2 complex and subsequent ARID1A-mediated
histone H2B regulation. The binding of H2B to ARID1 and to the
ubiquitin ligase complex causes H2B to be ubiquitinated and
degraded by the proteasomal complex (30). Mutated ARID1A may not bind to H2B,
and consequently is not degraded. Loss of the ARID1A protein may
cause a failure to regulate the H2B assembly, resulting in
accumulation of H2B to high levels. Results using siRNA-knockdown
approach indicate that ARID1A is required for cell-cycle arrest
(26). Ubiquitination of H2B was
associated with transcriptional activation (45). For example, ubiquitinated H2B has
been identified in the yeast Saccharomyces cerevisiae, and
mutation of the conserved ubiquitination site is shown to confer
defects in mitotic cell growth and meiosis. H2B ubiquitination is a
prerequisite for a second modification on a different histone.
Methylation of H3 at lysine residues depends on the ubiquitination
of histone H2B (46). Reduced
ubiquitination of H2B reduces H3 lysine-79 di-methylation and
correlates with a decreased gene expression. The addition of
ubiquitin to chromatin components such as H2B affects various
DNA-based processes, such as cell cycle progression, DNA damage
repair and gene silencing (47).
These results indicate the role of ARID1A mutations in components
of the chromatin modification machinery in CCC.
Notably, mutations of the following three genes
encoding enzymes involved in histone modification were found in
ccRCC cases: UTX (KDM6A, a histone H3 lysine 27 demethy lase),
SETD2 (a histone H3 lysine 36 methyltransferase), and JARID1C
(KDM5C, a histone H3 lysine 4 demethylase) (48). KDM6A catalyzes the demethylation of
tri/dimethylated histone H3 (48).
This gene regulates cell proliferation and acts as a tumor
suppressor via pRb-dependent pathways (49). SETD2 is a methyltransferase specific
to histone H3 lysine-36, and methylation of this residue is
associated with active chromatin. SETD2 behaves in the same manner
as a potential tumor suppressor gene in breast cancer (50) and ccRCC (51). KDM5C encodes a protein with one ARID
domain and is involved in the regulation of transcription and
chromatin remodeling. These results emphasize the role of mutations
in other components of the chromatin modification machinery in
ccRCC (48).
Finally, the somatic mutation spectrum of the ARID1A
gene in CCC tissue samples was calculated using the data from two
recently published articles (27,28).
We calculated that the majority of these mutations were C to T
mutations (68%) (Table I). The
mutation spectrum was enriched for transitions (75%), indicating
that C to T transition mutations were the major type of base
substitution. Notably, the mutation spectrum in ccRCC was dominated
by C to T/G to A transitions, but not transversion (48). The mutation spectrum in CCC was
dominated by C to T transition as has been noted in ccRCC. These
data allow us to hypothesize that CCC and ccRCC arise from a common
pathogenesis. The data on the ccRCC gene mutations may provide
insights into pathogenesis as well as the opportunity to gain a
better understanding of the role of genetic and epigenetic subtypes
of CCC. VHL inactivation alone induces senescence, suggesting a
requirement for additional mutations to further drive ccRCC
development in VHL mutant cases (48). No data exist regarding whether
ARID1A inactivation alone induces CCC phenotype. Therefore, the
role of ARID1A and the manner in which its inactivation affects
cancer development should be investigated.
 | Table IBase substitution mutations of the
ARID1A gene observed in CCC tissue samples. |
Table I
Base substitution mutations of the
ARID1A gene observed in CCC tissue samples.
| Base substitution
mutations | No. | (%) |
|---|
| Transition
total | 21 | (75.0) |
| C to T | 19 | (67.9) |
| A to G | 1 | (3.6) |
| G to A | 1 | (3.6) |
| Transversion
total | 7 | (25.0) |
| C to A | 1 | (3.6) |
| C to G | 1 | (3.6) |
| A to C | 1 | (3.6) |
| G to T | 2 | (7.1) |
| T to A | 1 | (3.6) |
| T to G | 1 | (3.6) |
7. Conclusions
Although the broad principles of the biology of CCC
are not fully understood, this tumor type is often associated with
endometriosis. The somatic mutations of CCC have been investigated.
Although p53, KRAS and PTEN genes are frequently mutated in other
types of human cancer, contribution of these genes to neoplastic
transformation of CCC is limited (27,52).
One of the novel mutated genes involved in CCC was found to be
ARID1A, a chromatin remodeling modifier (27,28).
This review focuses on the potential role of histone modifiers such
as ARID1A in CCC carcinogenesis. The mutation spectrum of ARID1A in
CCC was dominated by C to T transitions, leading to nonsense
mutations (27,28). Since ARID1A mutation and subsequent
loss or dysfunction of BAF250a protein is observed in preneoplastic
lesions (atypical endometriosis), it has been reported that this
mutation is an early event in the transformation of endometriosis
into CCC (27). The biological role
of ARID1A is to ubiquitinate and rapidly degrade histone H2B
through the proteosomal complex (30). This process includes the pathway
from aberrations in ARID1A to dysregulated (unubiquitinated)
histone H2B, leading to downstream changes in transcription genes.
Post-translational modifications of the histone H2B are centrally
involved in the regulation of all DNA-templated processes,
including gene transcription, DNA replication, recombination, and
repair, thus regulating a wide range of cellular processes and
functions.
Histone modifications and other epigenetic
mechanisms work together in maintaining gene activity states.
Epigenetic information in chromatin includes covalent
modifications, such as acetylation, methylation, phosphorylation
and ubiquitination, of histones. Therefore, ARID1A, a
chromatin-modifying factor, plays an essential role in DNA
processing pathways that dictate cellular functions. ARID1A may
contribute to cell cycle arrest and induced apoptosis, possibly
through p53- and/or pRb-dependent signaling cascades, suggesting a
role of ARID1A in the cell cycle checkpoint machinery (Fig. 2). Loss or dysfunction of the ARID1A
gene by mutations may lead to aberrant chromatin remodeling,
alterations in the cell cycle checkpoint machinery and subsequent
apoptosis evasion. ARID1A acts as a tumor suppressor gene, that
stimulates cell signaling, leading to cell cycle arrest and cell
death in the event of DNA damage. Therefore, the dysfunction of
ARID1A may lead to susceptibility to CCC carcinogenesis through a
defect in the repair or replication of damaged DNA. Loss of
expression and dysfunction of the ARID family may markedly alter in
carcinogenesis. However, the functions and regulation of histone
H2B ubiquitination and deubiquitination by ARID1A have yet to be
fully understood. This is a significant consideration for various
tumors showing the downregulation of ARID1A expression since
acquired resistance to the activation of apoptosis in tumor cells
is a serious limitation of current anticancer therapies. Thus
investigating the manner in which ARID1A levels in CCC affect the
prognosis of the disease upon treatment with various DNA-damaging
agents is crucial.
Although the broad principles of the biology of CCC
are not fully understood, significant advances have been made.
Recent biochemical studies based on genome-wide expression analysis
technology have noted a specific expression of a transcription
factor, hepatocyte nuclear factor-1β (HNF-1β), in CCC (8,53).
HNF-1β overexpression is common in CCC. By comparing CCC to the
contiguous atypical endometriotic lesions, the same overexpression
of HNF-1β may be present in the putative precursor lesions
(atypical endometriosis) and in a variety cancers. The distant
endometriotic lesions have also shown a moderate expression of
HNF-1β. In the case of EAC, however, HNF-1β expression was not
present in the atypical endometriosis nor in the tumors. A
correlation between chromatin remodeling gene ARID1A and the
CCC-specific gene HNF-1β has yet to be elucidated.
Although ARID1A mutation may be significant factor
for CCC carcinogenesis, various questions have yet to be answered.
Investigation into whether i) histone modification is the most
critical event for oncogenesis; ii) other, as yet understudied,
genes are also involved; and iii) these events are more critical
for CCC and not in another organ system, is required to determine
new treatment modalities for CCC.
Acknowledgements
This review was supported by KAKENHI (Japan Society
for the Promotion of Science (JSPS) Grant-in-Aid). We thank all the
study participants for their time and efforts. We thank Mr. Mikiko
Kita for editorial assistance.
References
|
1
|
Piver MS: Prophylactic oophorectomy:
reducing the U.S. death rate from epithelial ovarian cancer. A
continuing debate. Oncologist. 1:326–330. 1996.PubMed/NCBI
|
|
2
|
Kennedy AW, Biscotti CV, Hart WR and
Webster KD: Ovarian clear cell adenocarcinoma. Gynecol Oncol.
32:342–349. 1998. View Article : Google Scholar
|
|
3
|
Ryu SY, Park SI, Nam BH, et al: Prognostic
significance of histological grade in clear-cell carcinoma of the
ovary: a retrospective study of Korean Gynecologic Oncology Group.
Ann Oncol. 20:1032–1036. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Viganó P, Somigliana E, Chiodo I, Abbiati
A and Vercellini P: Molecular mechanisms and biological
plausibility underlying the malignant transformation of
endometriosis: a critical analysis. Hum Reprod Update. 12:77–89.
2006.PubMed/NCBI
|
|
5
|
Kobayashi H, Sumimoto K, Moniwa N, et al:
Risk of developing ovarian cancer among women with ovarian
endometrioma: a cohort study in Shizuoka, Japan. Int J Gynecol
Cancer. 17:37–43. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Bell DA: Origins and molecular pathology
of ovarian cancer. Mod Pathol. 18(Suppl 2): S19–S32. 2005.
View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Jiang X, Hitchcock A, Bryan EJ, et al:
Microsatellite analysis of endometriosis reveals loss of
heterozygosity at candidate ovarian tumor suppressor gene loci.
Cancer Res. 56:3534–3539. 1996.PubMed/NCBI
|
|
8
|
Kobayashi H, Kajiwara H, Kanayama S, et
al: Molecular pathogenesis of endometriosis-associated clear cell
carcinoma of the ovary (Review). Oncol Rep. 22:233–240.
2009.PubMed/NCBI
|
|
9
|
Yoo AS and Crabtree GR: ATP-dependent
chromatin remodeling in neural development. Curr Opin Neurobiol.
19:120–126. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Weissman B and Knudsen KE: Hijacking the
chromatin remodeling machinery: impact of SWI/SNF perturbations in
cancer. Cancer Res. 69:8223–8230. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Osipovich OA, Subrahmanyam R, Pierce S,
Sen R and Oltz EM: Cutting edge: SWI/SNF mediates antisense Igh
transcription and locus-wide accessibility in B cell precursors. J
Immunol. 183:1509–1513. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Kundu S and Peterson CL: Role of chromatin
states in transcriptional memory. Biochim Biophys Acta.
1790:445–455. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Rodriguez-Nieto S and Sanchez-Cespedes M:
BRG1 and LKB1: tales of two tumor suppressor genes on chromosome
19p and lung cancer. Carcinogenesis. 30:547–554. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Abbas T and Dutta A: p21 in cancer:
intricate networks and multiple activities. Nat Rev Cancer.
9:400–414. 2009. View
Article : Google Scholar : PubMed/NCBI
|
|
15
|
Toyota M and Suzuki H: Epigenetic drivers
of genetic alterations. Adv Genet. 70:309–323. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Fan D, Ma C and Zhang H: The molecular
mechanisms that underlie the tumor suppressor function of LKB1.
Acta Biochim Biophys Sin (Shanghai). 41:97–107. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Hess J, Angel P and Schorpp-Kistner M:
AP-1 subunits: quarrel and harmony among siblings. J Cell Sci.
117:5965–5973. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Hydbring P and Larsson LG: Cdk2: a key
regulator of the senescence control function of Myc. Aging.
2:244–250. 2010.PubMed/NCBI
|
|
19
|
Bogliolo M, Cabré O, Callén E, et al: The
Fanconi anaemia genome stability and tumour suppressor network.
Mutagenesis. 17:529–538. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Sansam CG and Roberts CW: Epigenetics and
cancer: altered chromatin remodeling via Snf5 loss leads to
aberrant cell cycle regulation. Cell Cycle. 5:621–624. 2006.
View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Watanabe H, Mizutani T, Haraguchi T, et
al: SWI/SNF complex is essential for NRSF-mediated suppression of
neuronal genes in human nonsmall cell lung carcinoma cell lines.
Oncogene. 25:470–479. 2006.PubMed/NCBI
|
|
22
|
Luo B, Cheung HW, Subramanian A, et al:
Highly parallel identification of essential genes in cancer cells.
Proc Natl Acad Sci USA. 105:20380–20385. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Crow E, Du Z and Li L: New insights into
prion biology from the novel [SWI+] system. Prion.
2:141–144. 2008.
|
|
24
|
Mertens F, Johansson B, Höglund M and
Mitelman F: Chromosomal imbalance maps of malignant solid tumors: a
cytogenetic survey of 3185 neoplasms. Cancer Res. 57:2765–2780.
1997.PubMed/NCBI
|
|
25
|
Nagl NG Jr, Patsialou A, Haines DS, et al:
The p270 (ARID1A/SMARCF1) subunit of mammalian SWI/SNF-related
complexes is essential for normal cell cycle arrest. Cancer Res.
65:9236–9244. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Wang X, Nagl NG Jr, Flowers S, et al:
Expression of p270 (ARID1A), a component of human SWI/SNF
complexes, in human tumors. Int J Cancer. 112:6362004. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Wiegand KC, Shah SP, Al-Agha OM, et al:
ARID1A mutations in endometriosis-associated ovarian carcinomas. N
Engl J Med. 363:1532–1543. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Jones S, Wang TL, Shih IeM, et al:
Frequent mutations of chromatin remodeling gene ARID1A in ovarian
clear cell carcinoma. Science. 330:228–231. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Wilsker D, Patsialou A, Dallas PB and
Moran E: ARID proteins: a diverse family of DNA binding proteins
implicated in the control of cell growth, differentiation, and
development. Cell Growth Differ. 13:95–106. 2002.PubMed/NCBI
|
|
30
|
Li XS, Trojer P, Matsumura T, Treisman JE
and Tanese N: Mammalian SWI/SNF – a subunit BAF250/ARID1 is an E3
ubiquitin ligase that targets histone H2B. Mol Cell Biol.
30:1673–1688. 2010.
|
|
31
|
Gilbert PM, Mouw JK, Unger MA, et al:
HOXA9 regulates BRCA1 expression to modulate human breast tumor
phenotype. J Clin Invest. 120:1535–1550. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Dehennaut V and Leprince D: Implication of
HIC1 (hypermethylated in cancer 1) in the DNA damage response. Bull
Cancer. 96:E66–E72. 2009.PubMed/NCBI
|
|
33
|
Van Rechem C, Boulay G and Leprince D:
HIC1 interacts with a specific subunit of SWI/SNF complexes,
ARID1A/BAF250A. Biochem Biophys Res Commun. 385:586–590.
2009.PubMed/NCBI
|
|
34
|
Iaquinta PJ and Lees JA: Life and death
decisions by the E2F transcription factors. Curr Opin Cell Biol.
19:649–657. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Ma K, Araki K, Ichwan SJ, et al:
E2FBP1/DRIL1, an AT-rich interaction domain-family transcription
factor, is regulated by p53. Mol Cancer Res. 1:438–444.
2003.PubMed/NCBI
|
|
36
|
Kim YT and Zhao M: Aberrant cell cycle
regulation in cervical carcinoma. Yonsei Med J. 46:597–613. 2005.
View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Bourdon JC: p53 and its isoforms in
cancer. Br J Cancer. 97:277–282. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Staehling-Hampton K, Ciampa PJ, Brook A
and Dyson N: A genetic screen for modifiers of E2F in
Drosophila melanogaster. Genetics. 153:275–287.
1999.PubMed/NCBI
|
|
39
|
Roesch A, Mueller AM, Stempfl T, et al:
RBP2-H1/JARID1B is a transcriptional regulator with a tumor
suppressive potential in melanoma cells. Int J Cancer.
122:1047–1057. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Zorn KK, Bonome T, Gangi L, et al: Gene
expression profiles of serous, endometrioid, and clear cell
subtypes of ovarian and endometrial cancer. Clin Cancer Res.
11:6422–6430. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Gnarra JR, Tory K, Weng Y, et al:
Mutations of the VHL tumour suppressor gene in renal carcinoma. Nat
Genet. 7:85–90. 1994. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Hacker KE, Lee CM and Rathmell WK: VHL
type 2B mutations retain VBC complex form and function. PLoS One.
3:E38012008. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Vodermaier HC: APC/C and SCF: controlling
each other and the cell cycle. Curr Biol. 14:R787–R796. 2004.
View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Clark PE: The role of VHL in clear-cell
renal cell carcinoma and its relation to targeted therapy. Kidney
Int. 76:939–945. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Beisel C, Imhof A, Greene J, Kremmer E and
Sauer F: Histone methylation by the Drosophila epigenetic
transcriptional regulator Ash1. Nature. 419:857–862.
2002.PubMed/NCBI
|
|
46
|
Ezhkova E and Tansey WP: Proteasomal
ATPases link ubiquitylation of histone H2B to methylation of
histone H3. Mol Cell. 13:435–442. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
47
|
Atanassov BS, Koutelou E and Dent SY: The
role of deubiquitinating enzymes in chromatin regulation. FEBS
Lett. Oct 26–2010.(Epub ahead of print).
|
|
48
|
Dalgliesh GL, Furge K, Greenman C, et al:
Systematic sequencing of renal carcinoma reveals inactivation of
histone modifying genes. Nature. 463:360–363. 2010. View Article : Google Scholar
|
|
49
|
Wang JK, Tsai MC, Poulin G, et al: The
histone demethylase UTX enables RB-dependent cell fate control.
Genes Dev. 24:327–332. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
50
|
Newbold RF and Mokbel K: Evidence for a
tumour suppressor function of SETD2 in human breast cancer: a new
hypothesis. Anticancer Res. 30:3309–3311. 2010.PubMed/NCBI
|
|
51
|
Duns G, van den Berg E, van Duivenbode I,
et al: Histone methyltransferase gene SETD2 is a novel tumor
suppressor gene in clear cell renal cell carcinoma. Cancer Res.
70:4287–4291. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
52
|
Kurman RJ and Shih IeM: Pathogenesis of
ovarian cancer: lessons from morphology and molecular biology and
their clinical implications. Int J Gynecol Pathol. 27:151–160.
2008.PubMed/NCBI
|
|
53
|
Kajihara H, Yamada Y, Kanayama S, et al:
Clear cell carcinoma of the ovary: Potential pathogenic mechanisms
(Review). Oncol Rep. 23:1193–1203. 2010.PubMed/NCBI
|
|
54
|
Sufan RI, Jewett MA and Ohh M: The role of
von Hippel-Lindau tumor suppressor protein and hypoxia in renal
clear cell carcinoma. Am J Physiol Renal Physiol. 287:F1–F6. 2004.
View Article : Google Scholar : PubMed/NCBI
|