Introduction
Osteosarcoma (OS) is the most frequent primary bone
malignancy and the eighth most common type of cancer among
children, comprising 2.4% of all malignancies in pediatric patients
and ∼35% of all types of bone cancer (1). OS is characterized by high local
aggression and a tendency to metastasize to the lungs and distant
bones. For patients with localized forms of OS, the recovery rate
is ∼65%. For those who present with metastases at the time of
diagnosis, the survival rate is 25% (2,3). Thus,
it is important to identify and confirm potential therapeutic
targets involved in OS progression.
Astrocyte elevated gene-1 (AEG-1), also known as
metadherin (MTDH), is a multifunctional oncogene. This gene is
overexpressed in a variety of types of human cancer, although it
was originally isolated as a novel HIV-1- and TNFα-induced
transcript from primary human fetal astrocytes (4,5). As a
downstream target of Ha-Ras, AEG-1 is important in regulating
tumorigenesis, invasion, metastasis and angiogenesis (6). In a study by Wang et al, AEG-1
was found to be overexpressed in OS tissues, and the overexpression
of AEG-1 strongly correlates with OS metastasis and poor survival
(7). The data suggest that AEG-1 is
important in OS progression via matrix metalloproteinase 2 (MMP-2),
and that AEG-1 may be a useful biomarker for the prediction of OS
progression and prognosis (7).
Endothelin-1 (ET-1) is expressed in a variety of
malignancies, and promotes tumor cell proliferation and survival
through the ET A receptor (ETAR) (8). ET-1 and ETAR are expressed in OS cells
and tissue (9,10). Felx et al revealed that ET-1
may promote OS cell invasion by inducing the synthesis of MMP-2
through ETAR, suggesting an important role of ET-1 in OS metastasis
(9). In vitro studies have
demonstrated that blocking ETAR leads to the inhibition of OS cell
invasion, suggesting that ETAR is a potential therapeutic target
for OS metastasis (9,10).
In the present study, we conducted the first
investigation into the interaction between AEG-1 and ET-1/ETAR
signaling in OS cells, and assessed how the functional interaction
may impact OS cell invasion and survival against chemotherapy
agents.
Materials and methods
Cells lines, plasmids and reagents
Saos-2 and MG-63 human OS cell lines were purchased
from the American Type Culture Collection (Rockville, MD, USA).
Human AEG-1 cDNA was subcloned into a pcDNA 3.1 expression vector.
AEG-1/MTDH (sc-77797-V) short hairpin RNA (shRNA) lentiviral
particles, control shRNA lentiviral particles-A (sc-108080), and
anti-ET-1 (sc-21625) and anti-MMP-2 (sc-10736) antibodies were
purchased from Santa Cruz Biotechnology, Inc. (Santa Cruz, CA,
USA). Anti-AEG-1 antibody (HPA010932) was purchased from Sigma (St.
Louis, MO, USA). All secondary antibodies were purchased from
Jackson ImmunoResearch Laboratories, Inc. (West Grove, PA, USA).
The ET-1 enzyme-linked immunosorbent assay (ELISA) kit was
purchased from R&D Systems (Minneapolis, MN, USA). The DeadEnd™
Fluorometric terminal deoxynucleotidyl transferase mediated
nick-end labeling (TUNEL) system was purchased from Promega
(Madison, WI, USA). Superfect™ transfection reagent was purchased
from Qiagen (Valencia, CA, USA). Puromycin, cisplatin, synthetic
ET-1, LY294002, BQ123 and reagent grade chemicals were purchased
from Sigma.
Real-time quantitative reverse
transcription (RT)-PCR
RNA was prepared from brain tissue samples using the
TRIzol reagent followed by purification with the TURBO DNA-free
system (Ambion; Austin, TX, USA). SuperScript II reverse
transcriptase (Invitrogen; Carlsbad, CA, USA) was used to
synthesize cDNA. Real-time quantitative PCR was performed in the
LightCycler thermal cycler system (Roche Diagnostics; Indianapolis,
IN, USA) using the SYBR-Green I kit (Roche Diagnostics) as per the
manufacturer’s instructions. Results were normalized against those
of the housekeeping gene glyceraldehyde-3-phosphate dehydrogenase
(GAPDH) in the same sample. The primer sequences used were
as follows: Forward: 5′-TCCTCTGCTGGTTCCTGACT-3′ and reverse:
5′-CAGAAACTCCACCCCTGTGT-3′ for human ET-1; forward:
5′-GACTCATGACCACAGTCCATGC-3′ and reverse:
5′-AGAGGCAGGGATGATGTTCTG-3′ for human GADPH. Each experiment
was repeated twice and performed in triplicate.
Transfection and lentiviral
transduction
The AEG-1 expression construct was transfected into
Saos-2 cells using the Superfect transfection reagent according to
the manufacturer’s instructions. Pools of stable transductants were
generated via selection with puromycin (5 μg/ml) according
to the manufacturer’s protocol. The AEG-1/MTDH shRNA lentiviral
particles contained expression constructs encoding target-specific
19–25 nt, as well as hairpin, shRNA designed to specifically
knockdown AEG-1 gene expression. The control shRNA lentiviral
particles contained a scrambled shRNA sequence that is not capable
of initiating the degradation any cellular mRNA, and were used as
negative controls for AEG-1/MTDH shRNA lentiviral particles.
Lentiviral transduction was performed in Saos-2 and MG-63 cells.
Pools of stable transductants were generated via selection with
puromycin (5 μg/ml) according to the manufacturer’s protocol
(Santa Cruz Biotechnology, Inc.).
In vitro cell invasion assay
Transwell® cell invasion assays (Corning
Life Sciences; Lowell, MA, USA) were performed as previously
described (23). Briefly, Transwell cell-culture chambers (pore
size, 8 μm; BD Biosciences; Bedford, MA, USA) for 24-well
plates were coated with 50 μl Matrigel (10 mg/ml; BD
Biosciences) diluted 1:3 in Roswell Park Memorial Institute
(RPMI)-1640 medium. Saos-2 and MG-63 cells were seeded in the upper
chamber at 5×105 cells/well in RPMI-1640 serum-free
medium. Complete medium (600 ml) was added to the lower chamber.
Cells were treated with ET-1 (10 or 100 pM) and/or LY294002 (50
μM) or BQ123 (5 μM) and allowed to migrate for 24 h
followed by fixation and staining with crystal violet. Migrated
cells were counted in 10 random fields per chamber under a
microscope. Each experiment was repeated three times and conducted
in triplicate.
Immunoassays
Secreted ET-1 levels in cell culture supernatants
were determined using an ET-1 ELISA kit. In brief, cells were grown
to confluence in 10-cm dishes in RPMI-1640 medium supplemented with
10% fetal bovine serum (FBS) in a humidified atmosphere of 95% air
and 5% CO2 at 37°C.. The medium was then replaced with
serum-free medium and cells were further incubated for 16 h. Cell
culture supernatants were collected for ELISA according to the
manufacturer’s instructions (R&D Systems). ELISA-detected ET-1
concentrations were normalized against the cell number (per
106 cells) and are shown as the fold change relative to
that of the normal control cells (designated as 1). Each ELISA
experiment was repeated three times and performed in duplicate. For
western blot analyses, protein was extracted by a lysis buffer
containing 150 mM NaCl, 2% Triton X-100, 0.1% SDS, 50 mM Tris (pH
8.0) and 10% protease inhibitor cocktail (Sigma), and stored at
−20°C. Equal amounts of protein (25 μg) for each sample were
loaded into pre-cast 7.5% Mini Protean TGX gels (BioRad; Hercules,
CA, USA) and separated by electrophoresis for 50 min at 200 V. The
separated proteins were transferred onto a polyvinylidene fluoride
(PVDF) transfer membrane (Amersham Biosciences/GE Healthcare;
Piscataway, NJ, USA) for 55 min at 100 V. Membranes were incubated
for 1 h with a 1:500 dilution of anti-AEG-1, anti-MMP-2 or
anti-ET-1 antibody, and then washed and revealed using secondary
antibodies with horseradish peroxidase conjugate (1:5000; 1 h).
Peroxidase activity was revealed using a GE Healthcare ECL kit.
Proteins were quantified prior to being loaded onto the gel, and
equal loading of protein was verified by Ponceau coloration.
Measurement of apoptosis by TUNEL
assay
The TUNEL assay was performed using the DeadEnd
Fluorometric TUNEL system according the manufacturer’s
instructions. Cells were treated with cisplatin (10 nM) in the
presence or absence of ET-1 (10 or 100 pM) and/or LY294002 (50
μM) or BQ123 (5 μM) for ≤8 h. Apoptotic cells
exhibited a green nuclear fluorescence that was detected using a
standard fluorescein filter. Cells stained with
4′,6-diamidino-2-phenylindole (DAPI) exhibited a blue nuclear
fluorescence. Slides were observed under fluorescence microscopy
with the relative number of apoptotic cells determined by counting
the number of TUNEL-positive cells in five random fields
(magnification, ×100) for each sample.
Statistical analysis
Statistical analyses were performed with the
Statistical Package for the Social Sciences (SPSS) software for
Windows, version 10.0. Data values were expressed as the mean ± SD.
Comparisons of means among multiple groups were performed with
one-way ANOVA followed by post hoc pairwise comparisons
using the least significant difference method. The significance
level of this study was set at a two- tailed α=0.05.
Results
Effect of overexpression and knockdown of
AEG-1 on ET-1 expression in OS cells
Saos-2 cells were found to exhibit a relatively low
constitutive AEG-1 expression compared with MG-63 cells (Fig. 1). Thus, to investigate the
interaction between ET-1 and AEG-1 in OS cells, Saos-2 cells were
stably transfected with an AEG-1 expression vector to induce AEG-1
overexpression, while MG-63 cells were stably transfected with
AEG-1-shRNA to knock down AEG-1. Compared with the controls, AEG-1
was overexpressed by >3-fold in Saos-2 cells, and the endogenous
AEG-1 level was knocked down by >70% in MG-63 cells. ET-1 was
detected at a lower constitutive level in Saos-2 cells compared
with that of MG-63 cells. In Saos-2 cells, overexpression of AEG-1
increased the ET-1 level by >2-fold compared with the controls.
This effect was eradicated by the addition of the selective
phosphatidylinositol 3-kinase (PI3K) inhibitor, LY294002. In MG-63
cells, knockdown of AEG-1 decreased the level of ET-1 by >2-fold
compared with the controls, while treatment with LY294002
demonstrated no more significant effects. Similar results were
observed at the secreted ET-1 level in the two cell lines,
suggesting that AEG-1 signaling regulates ET-1 expression in a
PI3K-dependent manner in OS cells.
Real-time RT-PCR revealed that overexpression of
AEG-1 in Saos-2 cells increased the ET-1 mRNA level by >4-fold
compared with the controls. This effect was eradicated by adding
LY294002 (Fig. 2A). By contrast,
knockdown of AEG-1 in MG-63 cells decreased the ET-1 mRNA level by
∼3-fold compared with the controls, while treatment with LY294002
demonstrated no significant further effects (Fig. 2B). The results indicate that AEG-1
signaling regulates ET-1 expression at the transcriptional level in
a PI3K-dependent manner in OS cells.
Effect of overexpression and knockdown of
AEG-1 on OS cell invasion and MMP-2 expression
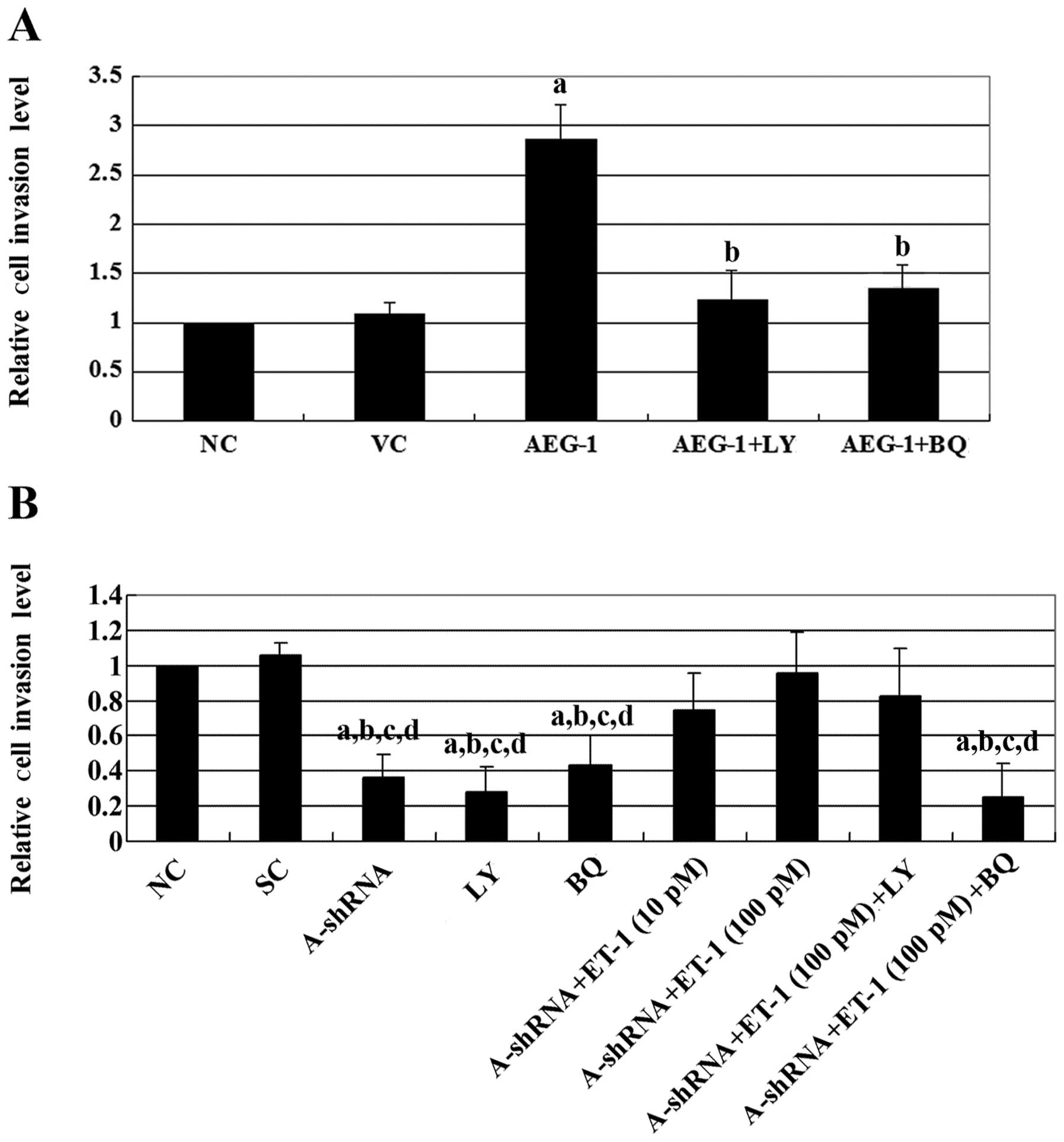
Both AEG-1 and ET-1 have been demonstrated to
promote OS cell invasion through MMP-2 (7,9). To
investigate the effect of the interaction between AEG-1 and
ET-1/ETAR signaling on OS invasion, we performed in vitro
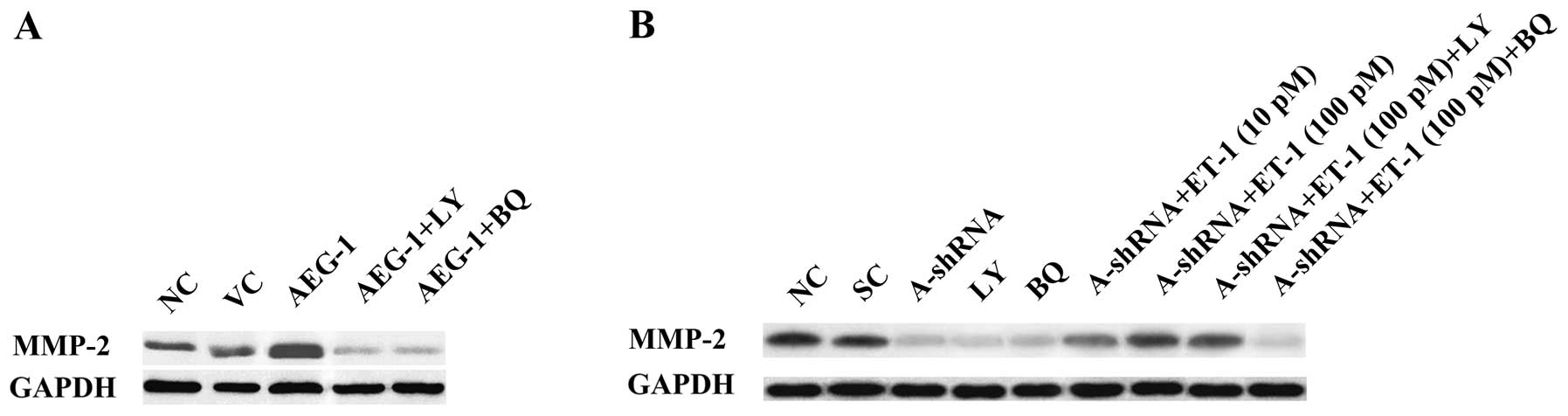
cell invasion assays and examined the MMP-2 expression level in the
two cell lines. Overexpression of AEG-1 in Saos-2 cells increased
cell invasion by ∼2.5-fold compared with the controls (Fig. 3). This effect was eradicated by the
addition of either LY294002 or the ETAR inhibitor, BQ123. By
contrast, knockdown of AEG-1 in MG-63 cells decreased cell invasion
by >2-fold compared with the controls. Treatment with exogenous
ET-1 increased cell invasion in a dose-dependent manner in cells in
which AEG-1 had been knocked down. This effect was blocked by
BQ123, but not by LY294002. Similar results were observed with
MMP-2 expression (Fig. 4). The
results suggest that AEG-1 promotes OS cell invasion primarily
through ET-1/ETAR, which functions downstream of PI3K and regulates
MMP-2 expression.
Effect of overexpression and knockdown of
AEG-1 on OS cell survival against cisplatin-induced apoptosis
Both AEG-1 and ET-1/ETAR signaling have been
demonstrated to promote tumor cell survival and chemoresistance
(11,12). To investigate the effect of the
interaction between AEG-1 and ET-1/ETAR signaling on OS cell
survival, we examined the rate of cell apoptosis in the two cell
lines that had been treated with 10 nM cisplatin, an
apoptosis-inducing chemotherapeutic agent commonly used to treat
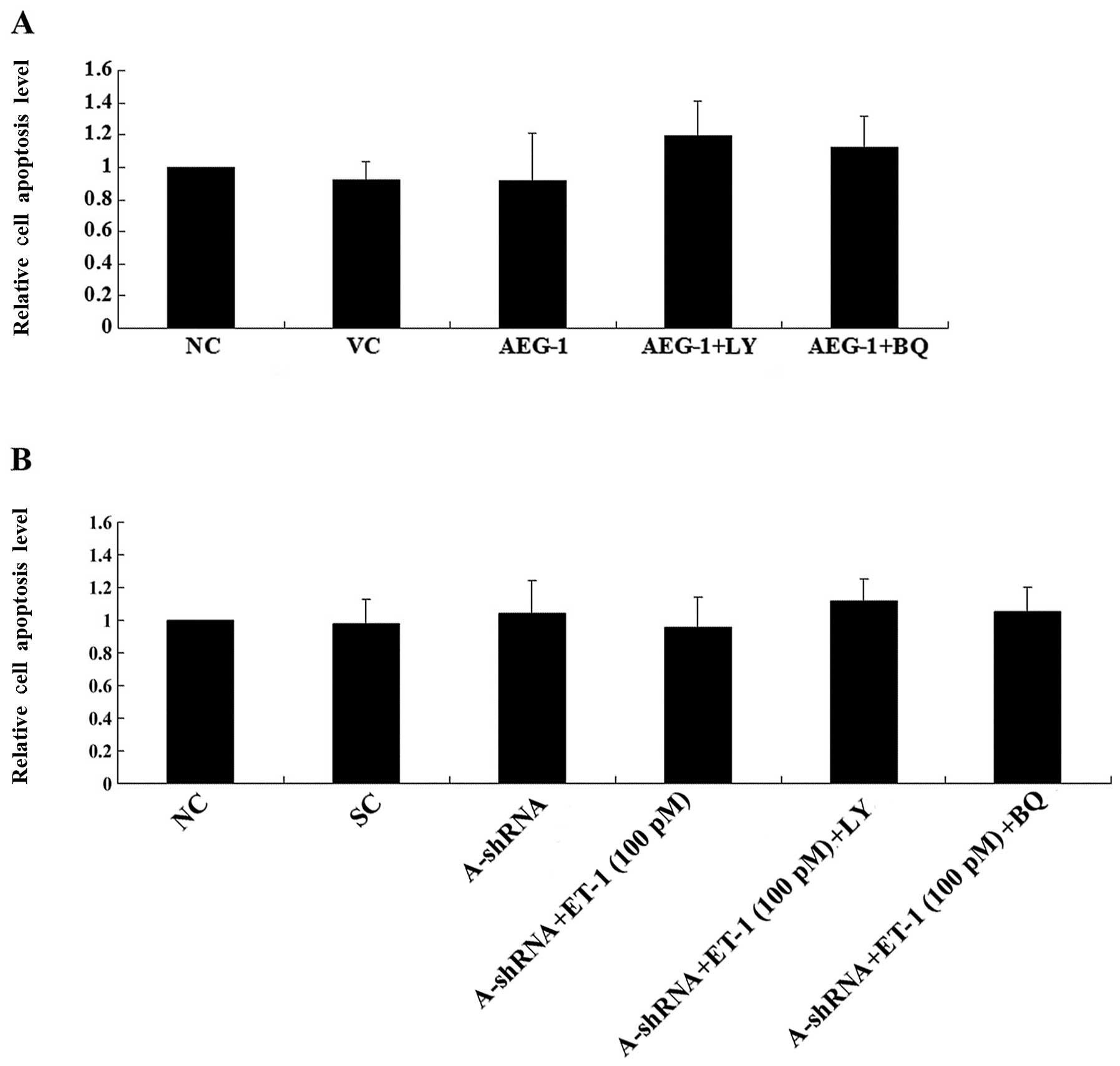
OS. Overexpression or knockdown of AEG-1 in the presence or absence
of ET-1 (100 pM) and/or LY294002 (50 μM) or BQ123 (5
μM) for ≤8 h did not significantly alter the rate of cell
apoptosis in normal culture conditions (Fig. 5). In Saos-2 cells treated with
cisplatin, over-expression of AEG-1 significantly decreased the
rate of cell apoptosis compared with that of the controls, and was
reversed by LY294002 or BQ123 (Fig.
6A). In MG-63 cells, knockdown of AEG-1 significantly increased
cell apoptosis in the presence of cisplatin, and was reversed by
treatment with exogenous ET-1. The rescue effect of ET-1 was
completely blocked by BQ123 and partially blocked by LY294002
(Fig. 6B). Taken together, the
results suggest that AEG-1 promotes OS cell survival against
cisplatin partially, but significantly, through ET-1/ETAR, which
functions downstream of PI3K.
Discussion
As a multifunctional oncoprotein, AEG-1 has been
demonstrated to enhance the aggressiveness of multiple types of
human cancer, including OS (7,13,14).
The ET-1/ETAR signaling pathway is a potential therapeutic target
for the control of OS metastasis (9,10). In
the present study, we explored the functional interaction between
AEG-1 and ET-1/ETAR signaling in OS cells, and assessed its impact
on OS cell invasion and survival.
We showed that Saos-2 cells exhibited a relatively
low constitutive expression of AEG-1, while AEG-1 was amply
expressed in MG-63 cells. Thus, overexpression and knockdown of
AEG-1 were respectively performed in the two cell lines in order to
utilise opposite approaches to the same study objective. Our
results showed that AEG-1 regulated ET-1 expression at the
transcriptional level in a PI3K-dependent manner in OS cells, which
is concordant with the results of previous studies. A number of
studies have demonstrated that AEG-1 triggers PI3K/Akt signaling in
cancer cells (13,14). Additionally, Zhang et al
found that AEG-1 regulated nuclear β-catenin accumulation in
colorectal cell lines (15), while
Sun et al demonstrated that nuclear β-catenin signaling
regulated ET-1 transcription in a PI3K-dependent manner in prostate
cancer cells (16). In the present
study, we provide the first evidence that AEG-1 regulates ET-1
expression in a PI3K-dependent manner in OS cells. Further studies
are required to address whether AEG-1 regulates ET-1 expression
through nuclear β-catenin signaling.
Both AEG-1 and ET-1 have been demonstrated to
promote OS cell invasion through MMP-2 (7,9). The
present in vitro cell invasion assay results suggest that
ET-1/ETAR signaling functions downstream of PI3K and primarily
mediates the effect of AEG-1 on OS cell invasion. This is due to
the fact that exogenous ET-1 was capable of restoring cell invasion
and MMP-2 expression levels in MG-63 cells, in which AEG-1 had been
knocked down, in the presence of a selective PI3K inhibitor
(LY294002), but not in the presence of a selective ETAR inhibitor
(BQ123). However, exogenous ET-1 only partially rescued cell
survival against cisplatin-induced apoptosis in the presence of
LY294002, in cells in which AEG-1 had been knocked down, suggesting
that unlike that of cell invasion, alternative signaling pathways
downstream of PI3K (other than ET-1/ETAR signaling) are involved in
AEG-1-induced chemoresistance in OS cells.
Cisplatin elicits DNA repair mechanisms by
crosslinking DNA, which in turn activates apoptosis when repair is
not possible (17). It remains
unclear whether the functional interaction between AEG-1 and
ET-1/ETAR signaling is capable of impacting OS cell survival
against other types of chemotherapy agents. Further studies with
additional types of chemotherapy agents and OS cell lines are
required.
In conclusion, we have demonstrated that AEG-1
regulates ET-1 expression at the transcription level in a
PI3K-dependent manner in OS cells. Downstream of PI3K, ET-1/ETAR
signaling primarily mediates the promoting effect of AEG-1 on OS
cell invasion, likely through the upregulation of MMP-2 expression,
while ET-1/ETAR signaling partially, but significantly, mediates
the AEG-1-induced chemoresistance in OS cells. This study provides
the first evidence of a functional link between AEG-1 and ET-1/ETAR
signaling in OS cells, which adds novel insights into the molecular
mechanism of OS metastasis and chemoresistance.
References
|
1.
|
Ottaviani G and Jaffe N: The epidemiology
of osteosarcoma. Cancer Treat Res. 152:3–13. 2010. View Article : Google Scholar
|
|
2.
|
Gorlick R, Anderson P, Andrulis I, et al:
Biology of childhood osteogenic sarcoma and potential targets for
therapeutic development: meeting summary. Clin Cancer Res.
9:5442–5453. 2003.PubMed/NCBI
|
|
3.
|
Wittig JC, Bickels J, Priebat D, et al:
Osteosarcoma: a multi-disciplinary approach to diagnosis and
treatment. Am Fam Physician. 65:1123–1132. 2002.
|
|
4.
|
Su ZZ, Chen Y, Kang DC, Chao W, Simm M,
Volsky DJ and Fisher PB: Customized rapid subtraction hybridization
(RaSH) gene microarrays identify overlapping expression changes in
human fetal astrocytes resulting from human immunodeficiency
virus-1 infection or tumor necrosis factor-alpha treatment. Gene.
30:67–78. 2003.
|
|
5.
|
Liao WT, Guo L, Zhong Y, Wu YH, Li J and
Song LB: Astrocyte elevated gene-1 (AEG-1) is a marker for
aggressive salivary gland carcinoma. J Transl Med. 9:2052011.
View Article : Google Scholar : PubMed/NCBI
|
|
6.
|
Lee SG, Su ZZ, Emdad L, Sarkar D and
Fisher PB: Astrocyte elevated gene-1 (AEG-1) is a target gene of
oncogenic Ha-ras requiring phosphatidylinositol 3-kinase and c-Myc.
Proc Natl Acad Sci U S A. 103:17390–17395. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
7.
|
Wang F, Ke ZF, Sun SJ, et al: Oncogenic
roles of astrocyte elevated gene-1 (AEG-1) in osteosarcoma
progression and prognosis. Cancer Biol Ther. 12:539–548. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
8.
|
Nelson J, Bagnato A, Battistini B and
Nisen P: The endothelin axis: emerging role in cancer. Nat Rev
Cancer. 3:110–116. 2003. View
Article : Google Scholar : PubMed/NCBI
|
|
9.
|
Felx M, Guyot MC, Isler M, et al:
Endothelin-1 (ET-1) promotes MMP-2 and MMP-9 induction involving
the transcription factor NF-κB in human osteosarcoma. Clin Sci
(Lond). 110:645–654. 2006.PubMed/NCBI
|
|
10.
|
Zhao Y, Liao Q, Zhu Y and Long H:
Endothelin-1 promotes osteosarcoma cell invasion and survival
against cisplatin-induced apoptosis. Clin Orthop Relat Res.
469:3190–3199. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
11.
|
Yoo BK, Chen D, Su ZZ, et al: Molecular
mechanism of chemoresistance by astrocyte elevated gene-1. Cancer
Res. 70:3249–3258. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
12.
|
Rosanò L, Cianfrocca R, Spinella F, et al:
Acquisition of chemoresistance and EMT phenotype is linked with
activation of the endothelin A receptor pathway in ovarian
carcinoma cells. Clin Cancer Res. 17:2350–2360. 2011.PubMed/NCBI
|
|
13.
|
Ying Z, Li J and Li M: Astrocyte elevated
gene 1: biological functions and molecular mechanism in cancer and
beyond. Cell Biosci. 1:362011. View Article : Google Scholar : PubMed/NCBI
|
|
14.
|
Yoo BK, Emdad L, Lee SG, et al: Astrocyte
elevated gene-1 (AEG-1): a multifunctional regulator of normal and
abnormal physiology. Pharmacol Ther. 130:1–8. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
15.
|
Zhang F, Yang Q, Meng F, Shi H, Li H,
Liang Y and Han A: Astrocyte elevated gene-1 interacts with
β-catenin and increases migration and invasion of colorectal
carcinoma. Mol Carcinog. Mar 16–2012.(Epub ahead of print).
View Article : Google Scholar
|
|
16.
|
Sun P, Xiong H, Kim TH, Ren B and Zhang Z:
Positive inter-regulation between β-catenin/T cell factor-4
signaling and endothelin-1 signaling potentiates proliferation and
survival of prostate cancer cells. Mol Pharmacol. 69:520–531.
2006.
|
|
17.
|
Rosenberg B, VanCamp L, Trosko JE, et al:
Platinum compounds: a new class of potent antitumour agents.
Nature. 222:385–386. 1969. View
Article : Google Scholar : PubMed/NCBI
|