Introduction
Hepatocellular carcinoma (HCC) is a leading cause of
cancer-related mortality worldwide and the prognosis for HCC
patients is poor. Vitamin K2 (VK2) belongs to the vitamin K family
and has been used clinically as an activator of homeostasis
(1) and an inhibitor of
osteoporosis (2). In addition, VK2
is able to exert cell growth inhibitory effects in various human
cancer cells such as SMMC-7721 HCC cells. In particular, induction
of apoptosis by VK2 is associated with p53 in SMMC-7721 HCC cells
(3). p53 is a multi-faceted tumor
suppressor gene capable of inducing cell cycle arrest and DNA
repair, irreversible growth arrest, terminal differentiation or
apoptosis (4,5). Furthermore, one important pathway
through which p53 induces apoptosis may involve the transcriptional
activation of proapoptotic target genes, including proapoptotic
members of the BCL-2 family of proteins such as Bax and Bid,
leading to intrinsic/mitochondrial apoptosis via the activation of
caspase cascades (6).
BCL-2 is an antiapoptotic protein that is frequently
overexpressed in numerous tumors. Modulation of multiple
antiapoptotic signaling pathways involving BCL-2, which are
associated with growth factor-stimulated signal transduction in
cell survival, is essential for enhancement of the cytotoxic effect
of anticancer drugs (7). Several
exceptions to the hypothesis that BCL-2 inhibits and p53 promotes
apoptosis have been encountered (8). Furthermore, BCL-2 is an antagonist to
Bax and inhibits mitochondrial membrane disruption, a mechanism
that likely accounts for drug resistance in BCL-2-overexpressing
tumors (9).
Therefore, the aim of this study was to enhance the
anticancer efficacy of VK2 through suppression of BCL-2 expression,
which is known as a ‘double-targeted’ therapeutic approach.
Suppression of BCL-2 expression by antisense and RNAi compounds
inhibited tumor growth and enhanced apoptosis in a variety of tumor
models (10,11). Silencing BCL-2 reportedly results in
improvement of the antitumor effect of the chemotherapeutic agent
5-FU (12). In the present study,
we examined the anticancer effect of VK2 by silencing BCL-2
expression with BCL-2 RNAi.
Materials and methods
RNA interference
siRNA duplexes were produced by Shanghai Genepharma
Co. Inc. (Shanghai, China) against human BCL-2 (5′-AAG GUG UCU UCC
AGA UCC UGA-3′). Scrambled fluorescent-labeled siRNA (5′-AAA UGU
GUG UAC GUC UCC UCC-3′) (siNC) was also designed and used as the
negative control in this study.
Cell culture
SMMC-7721 cells were cultured at 37°C under 5%
CO2 in RPMI-1640 supplemented with 10% fetal bovine
serum (FBS). Cells were treated with VK2 and 2% ethanol where
appropriate.
Combination treatment of tumor cells with
BCL-2 siRNA and VK2
SMMC-7721 cells at 70–90% confluence in 6-well
plates were transfected with 100 pmol BCL-2 siRNA using a
Lipofectamine 2000 reagent following the manufacturer’s
instructions (Invitrogen, Carlsbad, CA, USA). After 12 h, cells
were treated with VK2 at a multiplicity of concentrations. Control
groups included cells that were transfected with siNC.
RNA extraction and reverse
transcription-PCR analysis
Total RNA was isolated from treated cells using
TRIzol reagent (Invitrogen) according to the manufacturer’s
instructions. Total RNA (1 μg) was reverse-transcribed into cDNA
using M-MLV reverse transcriptase (Invitrogen). The cDNA was used
to amplify the BCL-2 fragments. For normalization of RNA, the
housekeeping gene GAPDH was also amplified from each sample. The
primer sequences used were: BCL-2 forward:
5′-ATGTGTGTGGAGAGCGTCAA-3′ and reverse:
5′-CAGGAGAAATCAAACAGAGGC-3′, 173 bp; and GAPDH forward:
5′-GGATTTGGTGGTATTGGG-3′ and reverse: 5′-GGAAGATGGTGATGGGATT-3′,
205 bp.
Quantitative real-time RT-PCR amplification was
carried out using Real-Time MIX (SYBR Premix Ex Taq™, Takara Bio,
Inc., Shiga, Japan). Specifically, total RNA was extracted by
TRIzol reagent (Invitrogen) as described and cDNA was synthesized
with RNA reverse transcriptase. The CT (threshold cycle) value of
BCL-2 and GAPDH was quantitated by Q-PCR in triplicate using an ABI
Prism 7500 HT sequence detector (AB Applied Biosciences, Foster
City, CA, USA) according to the manufacturer’s instructions, and
was normalized over the CT of the GAPDH control.
Western blot analysis
Cells were harvested at the indicated times, and
proteins were separated by sodium dodecyl sulfate polyacrylamide
gel electrophoresis in 10% SDS-polyacrylamide Tris-glycine gels for
protein expression. The immunoblotting was performed with BCL-2
(Cell Signaling Technology, Inc., Danvers, MA, USA), p53 (Cell
Signaling), p21 (Proteintech, Chicago, IL, USA) and mouse β-actin
(Sigma, St. Louis, MO, USA), followed by detection with a
horseradish peroxidase-conjugated secondary antibody.
MTT assay
The 3-(4,5-dimethylthiazol-2-yl)-2,
5-diphenyl-2H-tetrazolium bromide (MTT) assay was performed to
assess the effect of VK2 in combination with RNA interference on
cell proliferation. SMMC-7721 cells were seeded in 96-well plates
at a concentration of 1×105 cells/well. At the end of
the incubation period, 20 μl of 5 mg/ml MTT (Sigma) in PBS was
added to each well. Each experiment was repeated three times.
Absorbance was measured following incubation for a further 4 h at
37°C with a solution of MTT (0.2 mg/ml) that contained 12.5 μM
dimethyl sulfoxide (DMSO). The absorbance was measured on a
spectrophotometer microplate reader at a wavelength of 490 nm.
Cell cycle
Cells were seeded at 1×106 cells per well
in flat-bottomed 6-well plates. Cells were harvested at 6, 12, 24
and 36 h following treatment with 40 μM VK2 and 18, 24, 36 and 48 h
following transfection with siBCL-2. Cells were washed twice with
PBS and stained with 10 μg/ml PI. Cell cycle distribution was
determined by flow cytometry. Cell cycle analysis was performed
using FACS Calibur (Becton-Dickinson and company, Franklin Lakes,
NJ, USA). Software were obtained from CELL Quest software.
Statistical analysis
All experiments were performed in triplicate, and
the data were expressed as mean ± SD. The comparative Ct method was
applied in the quantitative real-time RT-PCR assay according to the
delta-delta Ct method. The data were analyzed with Student’s t-test
or by one-way analysis of variance, and results were considered
statistically significant at p≤0.05.
Results
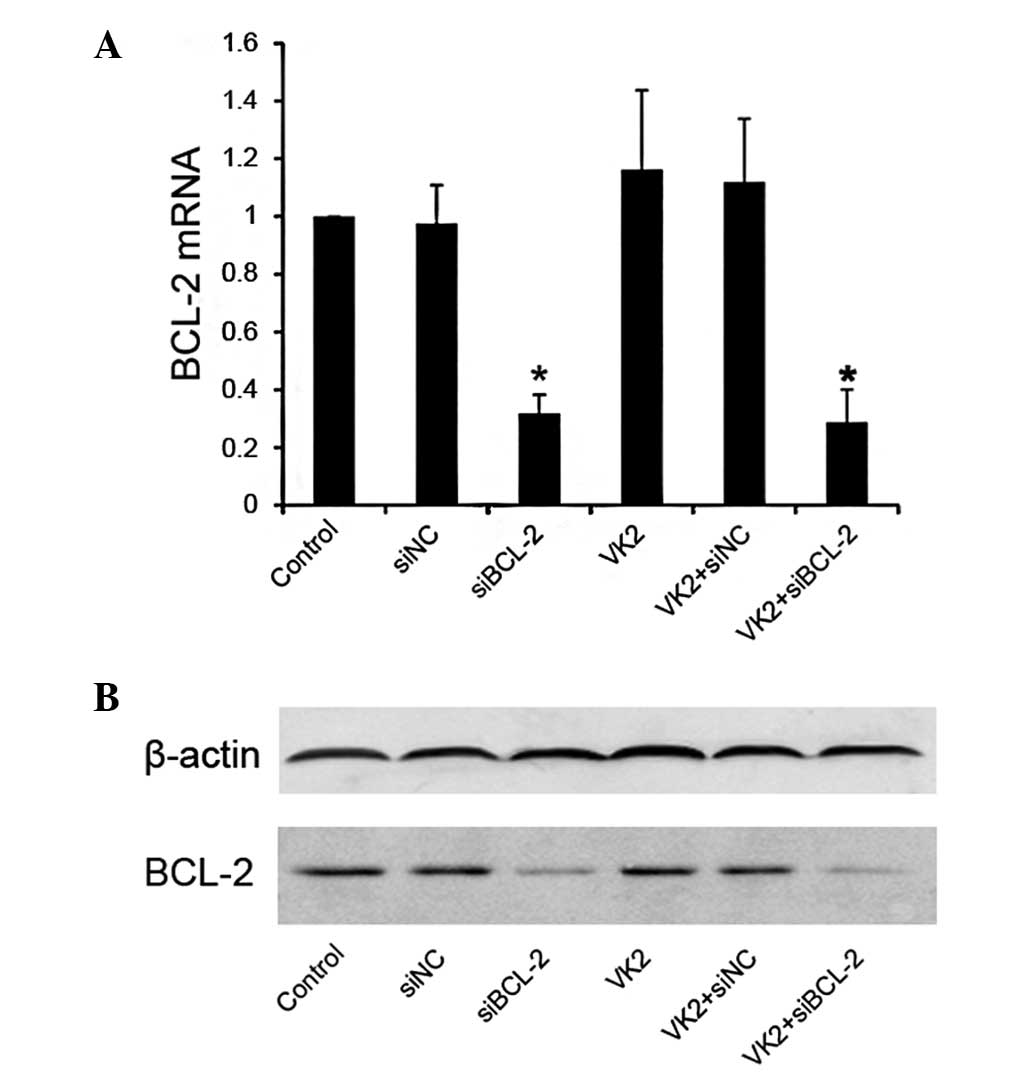
Suppression of BCL-2 by siRNA in
SMMC-7721 cells
The suppression of BCL-2 was examined using its
siRNA in tumor cells. siBCL-2 and control siNC were used to
transfect SMMC-7721 cells, respectively, and the efficiency of
siRNA on SMMC-7721 expression was examined by real-time RT-PCR and
western blot analysis.
As expected, no change occurred in the level of
BCL-2 mRNA in the VK2 group. However, when compared with the siNC
control, the level of BCL-2 mRNA decreased in the siBCL-2 groups,
used alone or in combination with VK2 (Fig. 1A). The same result was found at the
protein level by western blot analysis (Fig. 1B).
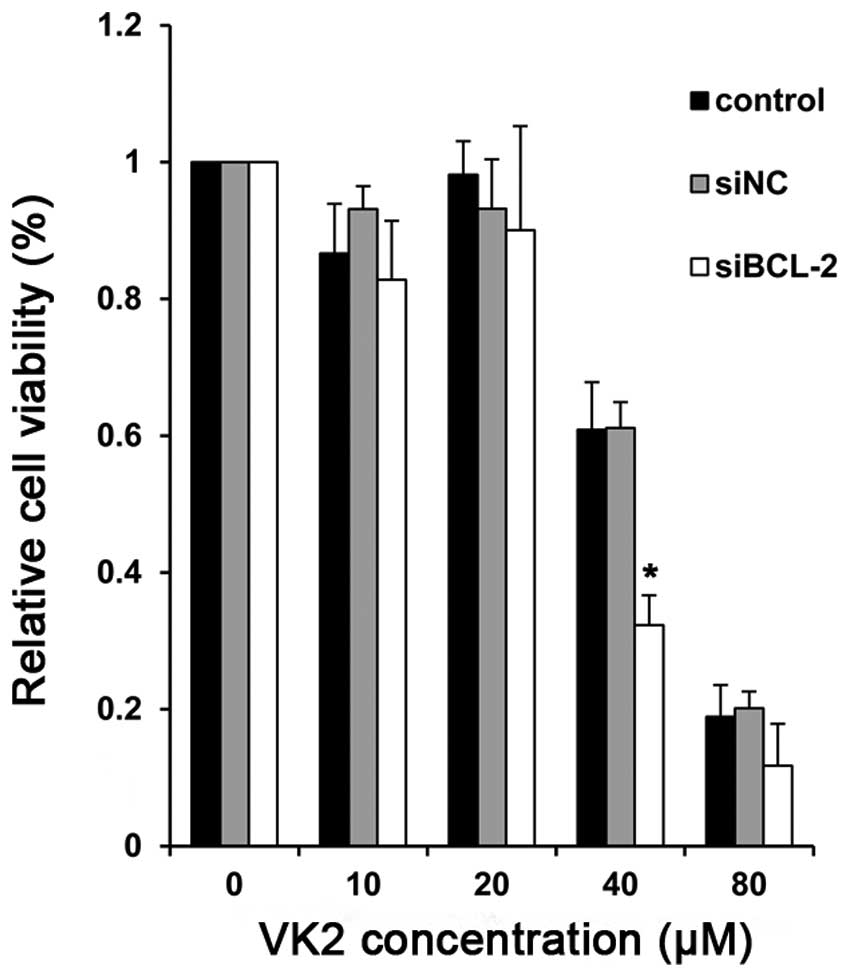
Enhanced cytotoxicity by the combined
treatment with siBCL-2 and VK2
Subsequent to establishing BCL-2-silencing cells, we
determined the proliferation of cells treated with VK2. Compared
with the controls, there was a significant reduction in cell
survival in cells treated with BCL-2 siRNA (siBCL-2) plus VK2. In
particular, at the concentration of 40 μM, the combination group
demonstrated a significant degree of proliferative inhibition after
24 h compared with the negative control and PBS groupσ, indicating
enhanced growth inhibition (Fig.
2).
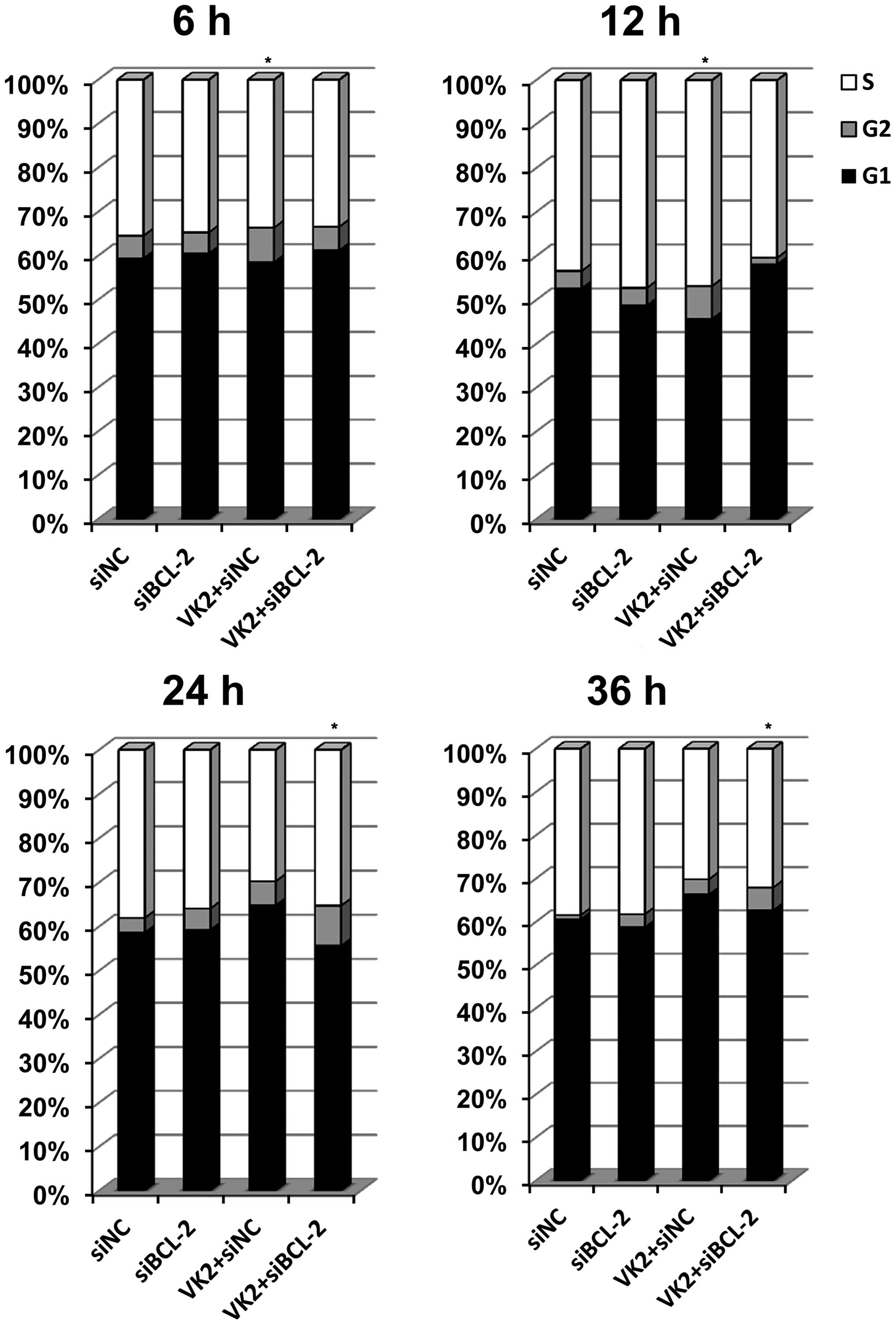
Combined treatment induced changes in the
cell cycle
The cell cycle was evaluated to investigate the
possible mechanism of proliferative inhibition. A significant
arrest was observed in the G2 phase following transfection of the
cells with siBCL-2 for 18 h and treatment with VK2 for 6 h
(Fig. 3). The same result was
observed in cells transfected with siBCL-2 for 24 h and treated
with VK2 for 12 h. However, when the cells were transfected with
siBCL-2 for 36 h and treated with VK2 for 24 h, the combination
treatment group demonstrated a different level of G2 stage
inhibition in comparison to the VK2 treatment, siBCL-2 treatment
and the control groups, and the difference was statistically
significant. In addition, when the cells were transfected with
siBCL-2 for 48 h and treated with VK2 for 36 h, the same result was
observed.
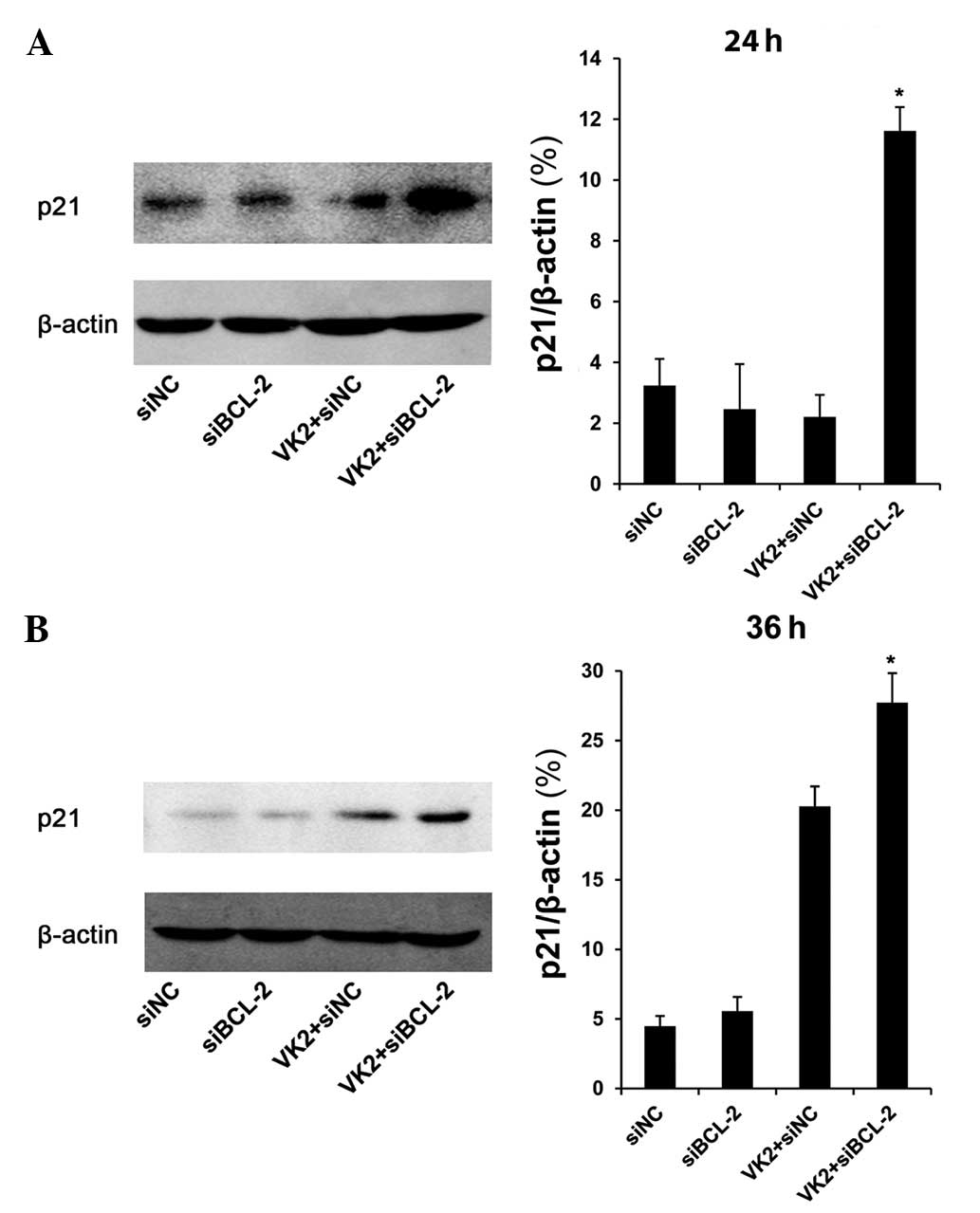
Expression of phosphorylation of p53 and
p21 following combined treatment with siBCL-2 and VK2
In the nucleus, p53 induces cell cycle arrest and
has an ‘extranuclear’ proapoptotic function as it combines with
BCL-2 (13,14). Thus, we determined the protein level
of p53. The protein level of phosphorylation of p53 (Ser 20
phosphorylation) was significantly increased in cells transfected
with siBCL-2 for 36 h and treated with VK2 for 24 h, compared with
other groups (Fig. 4). We then
tested the protein level of p21, a transcriptional target protein
of p53. The data revealed that following transfection with siBCL-2
for 36 and 48 h and treatment with VK2 for 24 and 36 h, there was
an increase in the protein level of p21 in the combination groups
compared with other groups (Fig.
5).
Discussion
The anti-proliferative action of VK2 has been
reported in a variety of cancer cells including lung carcinomas
(8), human ovarian cancer cells
(6) and acute myeloid leukemia
cells (4). In leukemia cells, VK2
induces autophagy and apoptosis simultaneously, indicating that the
cell expression levels of BCL-2 appear to determine the phenotype
of cell death (15). In HCC Hep3B
cells, VK2 induced cell cycle arrest at the G1 phase and
involvement of apoptosis was suggested since results of the flow
cytometric analysis revealed the sub-G1 fraction, and nuclear
condensation and fragmentation appeared subsequent to VK2
treatment. VK2 activated extracellular signal-regulated kinase
(ERK)1/2 in a mitogen-activated ERK-regulating kinase
(MEK)-dependent manner in Hep3B (16). In SMMC-7721 HCC cells, VK2 is
capable of inhibiting the growth of SMMC-7721 cells by induction of
apoptosis involving p53 (3). In the
present study, we found that VK2 induced p53 and increased the p21
levels, eventually leading to cell cycle arrest in the G2 phase,
indicating VK2 involvement in tumor suppression.
Malignant tumors are associated with abnormalities
in gene expression derived from genetic and/or epigenetic lesions,
including BCL-2 overexpression (17). As an apoptotic and/or survival
‘switch’, BCL-2 is key in the balance between proapoptotic and
antiapoptotic factors in the intracellular microenvironment.
Overexpression of BCL-2 in tumors critically alters this balance
and results in the permanent survival of tumors. It has also been
reported that overexpression of BCL-2 in a leukemia cell line
resulted in resistance against VK2-induced apoptosis, however,
these cells still underwent differentiation via G1 arrest (18). Thus, downregulation of BCL-2 may
restore this balance and further increase the ability of tumor
cells to respond to the apoptotic signal induced by exogenous
stimuli (19). siBCL-2 treatment
reportedly increased H101 viral replication in both treated cells
and tumor tissues (19), and that
the downregulation of BCL-2 and cyclin D1 may enhance cisplatin
sensitivity in MCF-7 human breast cancer cells (20).
In this study, we investigated the antitumor
efficacy of VK2 in conjunction with siRNA to BCL-2. BCL-2-siRNA
efficiently inhibited the expression of BCL-2 mRNA and protein
(Fig. 1). RNAi activity was not
affected by VK2 treatment. The combined action of BCL-2 knockdown
and treatment with VK2 significantly inhibited tumor growth in
vitro, suggesting an additional effect of the combined tumor
therapy.
The mechanism underlying the additive effect of the
combined therapy remains to be determined. In SMMC-7721 cells
treated with the combination of siBCL-2 and VK2, we found
significant changes in the cell cycle in different groups.
Subsequently, we determined the expression of p53, which is mainly
responsible for arrest of the cell cycle and regulation of
apoptosis. In addition, we determined the level of the protein,
p21, which is a transcriptional target of p53 and plays a crucial
role in mediating growth arrest when cells are exposed to DNA
damaging agents such as doxorubicin and γ-irradiation (21,22).
It has been demonstrated that overexpression of p21 results in G1-,
G2- (23), or S-phase arrest
(24,25). Conversely, p21-deficient cells fail
to undergo cell cycle arrest in response to p53 activation
following DNA damage (26).
Furthermore, p21 and p53 are essential in sustaining the G2
checkpoint following DNA damage in human cells (27). p53-mediated signaling plays an
integral role in the maintenance of the G2 checkpoint delay
following activation of the checkpoint. p53 is believed to exert G2
checkpoint responses through the transcriptional upregulation of
the downstream target genes p21, 14-3-3 and GADD45. p21 is capable
of binding to and inhibiting the cyclin B1/cdc2 complex and
inhibiting cyclin-activated kinase-mediated cdc2 activation
(28).
The combined treatment with siBCL-2 and VK2
down-regulated the expression of the antiapoptotic protein BCL-2,
whereas the protein, p53, was overexpressed 24 h following
treatment. This transcription factor plays a significant role in
the regulation of the cell cycle to prevent mutations and cancer
(29), as well as to control the
functions of p21. It regulates the progression of the cell cycle,
which was clearly increased at 24 and 36 h as a consequence of the
increased p53 activity.
In conclusion, our study proved that the antitumor
effect of VK2 may be improved by silencing BCL-2 expression in HCC
SMMC-7721. This finding provides support for the combined use of
VK2 and siBCL-2 as a promising approach in cancer gene therapy.
Acknowledgements
We thank Lu Li from the Department of Biochemistry
and Molecular Biology, Anhui University of Traditional Chinese
Medicine, for experimental guidance and assistance. This study was
supported by the National Key Program for Basic Research of China
(2010CB529902), the National Natural Science Foundation of China
(10979034 and 81001008); the Science and Technology Commission of
Shanghai (10JC1409100), the Shanghai Leading Academic Discipline
Project (S30205); and the Shanghai Rising-Star Program
(11QA1404000).
References
|
1
|
Conly JM and Stein K: The production of
menaquinones (vitamin K2) by intestinal bacteria and their role in
maintaining coagulation homeostasis. Prog Food Nutr Sci.
16:307–343. 1992.PubMed/NCBI
|
|
2
|
Shiraki M, Shiraki Y, Aoki C and Miura M:
Vitamin K2 (menatetrenone) effectively prevents fractures and
sustains lumbar bone mineral density in osteoporosis. J Bone Miner
Res. 15:515–521. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Li L, Qi Z, Qian J, et al: Induction of
apoptosis in hepatocellular carcinoma Smmc-7721 cells by vitamin
K(2) is associated with p53 and independent of the intrinsic
apoptotic pathway. Mol Cell Biochem. 342:125–131. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Meek DW: Tumour suppression by p53: a role
for the DNA damage response? Nat Rev Cancer. 9:714–723.
2009.PubMed/NCBI
|
|
5
|
Smeenk L and Lohrum M: Behind the scenes:
Unravelling the molecular mechanisms of p53 target gene selectivity
(Review). Int J Oncol. 37:1061–1070. 2010.PubMed/NCBI
|
|
6
|
Wu JN, Huang J, Yang J, Tashiro S, Onodera
S and Ikejima T: Caspase inhibition augmented oridonin-induced cell
death in murine fibrosarcoma l929 by enhancing reactive oxygen
species generation. J Pharmacol Sci. 108:32–39. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Kim R, Tanabe K, Emi M, Uchida Y and Toge
T: Potential roles of antisense therapy in the molecular targeting
of genes involved in cancer (Review). Int J Oncol. 24:5–17.
2004.PubMed/NCBI
|
|
8
|
Zusman I, Gurevich P, Gurevich E and
Ben-Hur H: The immune system, apoptosis and apoptosis-related
proteins in human ovarian tumors (A review). Int J Oncol.
18:965–972. 2001.PubMed/NCBI
|
|
9
|
Reed JC: Dysregulation of apoptosis in
cancer. J Clin Oncol. 17:2941–2953. 1999.PubMed/NCBI
|
|
10
|
Futami T, Miyagishi M, Seki M and Taira K:
Induction of apoptosis in HeLa cells with siRNA expression vector
targeted against bcl-2. Nucleic Acids Res. (Suppl): 251–252. 2002.
View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Zhang J, Huang S, Zhang H, Wang H, Guo H,
Qian G, Fan X, Lu J, Hoffman AR, Hu JF and Ge S: Targeted knockdown
of Bcl2 in tumor cells using a synthetic TRAIL 3′-UTR microRNA. Int
J Cancer. 126:2229–2239. 2010.PubMed/NCBI
|
|
12
|
Huang SL, Wu Y, Yu H, et al: Inhibition of
Bcl-2 expression by a novel tumor-specific RNA interference system
increases chemosensitivity to 5-fluorouracil in Hela cells. Acta
Pharmacol Sin. 27:242–248. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Mihara M, Erster S, Zaika A, et al: p53
has a direct apoptogenic role at the mitochondria. Mol Cell.
11:577–590. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Marchenko ND, Zaika A and Moll UM: Death
signal-induced localization of p53 protein to mitochondria. A
potential role in apoptotic signaling. J Biol Chem.
275:16202–16212. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Yokoyama T, Miyazawa K, Naito M, et al:
Vitamin K2 induces autophagy and apoptosis simultaneously in
leukemia cells. Autophagy. 4:629–640. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Matsumoto K, Okano J, Nagahara T and
Murawaki Y: Apoptosis of liver cancer cells by vitamin K2 and
enhancement by MEK inhibition. Int J Oncol. 29:1501–1508.
2006.PubMed/NCBI
|
|
17
|
Reed JC: Apoptosis-based therapies. Nat
Rev Drug Discov. 1:111–121. 2002. View
Article : Google Scholar
|
|
18
|
Miyazawa K, Yaguchi M, Funato K, et al:
Apoptosis/differentiation-inducing effects of vitamin K2 on HL-60
cells: dichotomous nature of vitamin K2 in leukemia cells.
Leukemia. 15:1111–1117. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Zhang H, Wang H, Zhang J, et al: Enhanced
therapeutic efficacy by simultaneously targeting two genetic
defects in tumors. Mol Ther. 17:57–64. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Yde CW and Issinger OG: Enhancing
cisplatin sensitivity in MCF-7 human breast cancer cells by
down-regulation of Bcl-2 and cyclin D1. Int J Oncol. 29:1397–1404.
2006.PubMed/NCBI
|
|
21
|
El-Deiry WS, Tokino T, Velculescu VE, et
al: WAF1, a potential mediator of p53 tumor suppression. Cell.
75:817–825. 1993. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
El-Deiry WS, Harper JW, O’Connor PM, et
al: WAF1/CIP1 is induced in p53-mediated G1 arrest and apoptosis.
Cancer Res. 54:1169–1174. 1994.PubMed/NCBI
|
|
23
|
Niculescu AB III, Chen X, Smeets M, Hengst
L, Prives C and Reed SI: Effects of p21(Cip1/Waf1) at both the G1/S
and the G2/M cell cycle transitions: pRb is a critical determinant
in blocking DNA replication and in preventing endoreduplication.
Mol Cell Biol. 18:629–643. 1998.PubMed/NCBI
|
|
24
|
Ogryzko VV, Wong P and Howard BH: WAF1
retards S-phase progression primarily by inhibition of
cyclin-dependent kinases. Mol Cell Biol. 17:4877–4882.
1997.PubMed/NCBI
|
|
25
|
Radhakrishnan SK, Feliciano CS, Najmabadi
F, et al: Constitutive expression of E2F-1 leads to p21-dependent
cell cycle arrest in S phase of the cell cycle. Oncogene.
23:4173–4176. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Waldman T, Kinzler KW and Vogelstein B:
p21 is necessary for the p53-mediated G1 arrest in human cancer
cells. Cancer Res. 55:5187–5190. 1995.PubMed/NCBI
|
|
27
|
Bunz F, Dutriaux A, Lengauer C, et al:
Requirement for p53 and p21 to sustain G2 arrest after DNA damage.
Science. 282:1497–1501. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Smits VA, Klompmaker R, Arnaud L, Rijksen
G, Nigg EA and Medema RH: Polo-like kinase-1 is a target of the DNA
damage checkpoint. Nat Cell Biol. 2:672–676. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Harms KL and Chen X: The C terminus of p53
family proteins is a cell fate determinant. Mol Cell Biol.
25:2014–2030. 2005. View Article : Google Scholar : PubMed/NCBI
|