Introduction
Pancreatic cancer is one of the most devastating
malignant tumors and the fifth most common cause of cancer-related
mortality in developed countries (1). Due to the difficulties of early
diagnosis and highly aggressive behavior (2), 85% of patients already have local
infiltration or metastasis at the time of diagnosis. Less than 20%
of patients have the option of radical tumor resection following
the initial diagnosis (3). Thus,
the 5-year survival rate of patients with pancreatic cancer is less
than 5% (4,5). Apart from surgery, chemotherapy is an
essential auxiliary treatment for the management of advanced
pancreatic cancer. As the first-line chemotherapy drug for
pancreatic cancer, gemcitabine has been widely used in the clinic
(6). However, due to a high degree
of acquired and inherent resistance to pancreatic cancer
chemotherapy (7), up to 20% of
pancreatic cancer patients show no obvious effect following
treatment with gemcitabine monotherapy (8). Thus, combined therapy with gemcitabine
has gained considerable attention in the attempt to improve the
outcome of pancreatic cancer (9).
Bufalin, an significant active component of the
Chinese medicine chan’su (10), has
widely demonstrated antitumor effects on human leukemia as well as
ovarian, prostate and lung cancer (10–13). A
possible mechanism of the antitumor effect of bufalin may be
through the regulation of the MAPK signaling pathway and activation
of a variety of transcription factors and protein kinases (14–16).
It has been demonstrated that bufalin induces apoptosis in these
cells via the activation of AP-1, the c-Jun N-terminal protein
kinase (JNK), as well as by the induction of bcl-2 and the
inhibition of protein kinase A. However, the effect of bufalin on
pancreatic cancer cells has not yet been thoroughly evaluated.
Apoptosis signal-regulating kinase 1 (ASK1), also
known as mitogen-activated protein kinase kinase kinase 5 (MAP3K5),
a member of the MAPK family, is a serine/threonine protein kinase
which is an upstream activator of JNK and regulates diverse
cellular responses. Previous studies have demonstrated that ASK1
participates in cell differentiation and apoptosis (14). The suppression of ASK1 may provide a
general mechanism for cell survival, and overexpression of ASK1 is
sufficient to cause apoptosis induced by reactive oxygen species
(ROS) in a number of cell lines through many
mitochondrial-dependent apoptotic stimuli including certain
chemotherapeutic agents.
In this study, we investigated whether and how ASK1
becomes activated during cell death induced by bufalin. We found
that bufalin interacts with and positively regulates ASK1 under
various cell death conditions. Additionally, we investigated the
synergistic effect on pancreatic cancer cell apoptosis induced by
gemcitabine combined with bufalin. These findings suggest that ASK1
plays an important role in bufalin-mediated cell death and may
enhance the antitumor effect of gemcitabine.
Materials and methods
Cell culture and reagents
Three human pancreatic cancer cell lines (Bxpc-3,
Mia PaCa-2 and Panc-1) were purchased from American Type Culture
Collection (ATCC, Rockville, MD, USA). The Mia PaCa-2 and Panc-1
cells were cultured in Dulbecco’s modified Eagle’s medium (DMEM;
Gibco, Rockville, MD, USA) supplemented with 10% fetal bovine
serum, 100 U/ml penicillin G and 100 U/ml streptomycin. The Bxpc-3
cells were cultured in RPMI-1640 (Gibco) containing supplements as
above. All three cell lines were incubated at 37°C under 5% CO2 in
air. Bufalin, purchased from Sigma (St. Louis, MO, USA), was
dissolved in dimethyl sulfoxide (DMSO) as a stock solution (10 mM)
and stored at −20°C. The culture media containing different
concentrations of bufalin were all freshly prepared at the time of
each experiment. The final concentration of DMSO was <0.1%.
Gemcitabine was purchased from Ely Lilly (Bad Homburg, Germany) and
dissolved in normal saline to make a 50 mg/ml stock solution.
Cell growth inhibition assay
The MTT [3-(4, 5-dimethylthiazol-2yl)-2,
5-diphenyltetrazolium bromide (Sigma) assay was used to assess the
cellular viability. Briefly, cells were planted on a 96-well plate
at a density of 5x103 cells per well and treated with
drugs at different concentrations for 24, 48 and 72 h. A total of
20 μl MTT solution [5 mg/ml in phosphate-buffered saline
(PBS)] was added to each well, and further incubated for 3–5 h.
Then, the culture medium was removed and the MTT formazan was
dissolved in 150 μl DMSO. The plates were agitated for 10
min, and absorbance was measured using an absorbance reader (BioTek
ELx800, Winooski, VT, USA) at 490 nm.
Flow cytometry and apoptosis
detection
Cells were distributed on a 6-well plate at a
density of 5x105 per well. After treatment with bufalin
and/or gemicitabine for 48 h, cells were harvested and washed with
PBS three times. Then the degree of apoptosis was detected by
Annexin V/FITC binding assay according to the manufacturer’s
instructions (BD Biosciences, Franklin Lakes, NJ, USA). The mixed
solution was gently shaken and stored away from light at room
temperature for 15 min. The stained cells were analyzed directly by
flow cytometry using Cell Quest software (BD Biosciences).
Transfection of siRNA
ASK1 siRNA was designed by GenePharma (Shanghai,
China) with human ASK1 cDNA. The sequences designed against three
separate regions starting from nucleotide 1258, 2025 or 2960 were:
si-ASK1 1258, sense 5′-GGCAGCGAGUAGAUAAUAUTT-3′ and antisense
5′-AUAUUAUCUACUCGCUGCCTT-3′; si-ASK1 2025, sense
5′-GUGGUUAGGUUUCCAGUAUTT-3′ and antisense
5′-AUACUGGAAACCUAACCACTT-3′; si-ASK1 2960, sense
5′-GGGCUGUACAAUCAUUGAATT-3′ and antisense
5′-UUCAAUGAUUGUACAGCCCTT-3′. A non-specific oligonucleotide served
as the negative control. The cells plated in 6-well plates were
transfected with 100 nM siRNA or negative control siRNA using
Lipofectamine™ 2000 (Invitrogen, Carlsbad, CA, USA) following the
manufacturer’s instructions. Briefly, when cells reached 60–70%
confluence, a mixture of Lipofectamine 2000 and OPTI-MEM medium
(Invitrogen) was incubated for 5 min, then incubated with siRNA for
a further 30 min at room temperature to allow the complex
formation. The transfection complex was added to each well ensuring
distribution over the entire plate surface. The OPTI-MEM medium was
replaced with DMEM at 4–6 h after transfection. The cells were
incubated for 48 h prior to being harvested for further
analysis.
Protein extraction and western blot
analysis
Following treatment as described above, cells were
washed with cold PBS and lysed in pre-chilled lysis buffer [1.0 mM
ethylenediamineteraacetate (EDTA), 50 mM Tris-HCL (pH 7.4), 1%
NP40, 0.1% SDS, 0.5% deoxycholate, 150 mM NaCl and 2% protease
inhibitor cocktail]. Following centrifugation at 13,000 x g for 30
min, the supernatant was collected and quantitated using the BCA
protein assay (Pierce Biotechnology, Inc., Rockford, IL, USA).
Total protein (40 μg) was separated in 8–10%
SDS-polyacrylamide denaturing gels and transferred to
polyvinylidene difluoride membranes. After blocking in TBST (10 mM
Tris-HCL pH 7.4, 150 mM NaCl and 0.1% Tween-20) with 5% non-fat
milk for 1 h, the membranes were incubated with primary antibodies
overnight at 4°C, followed by horseradish peroxidase-conjugated
secondary antibodies. Immunoblotting for bcl-2, cleaved caspase-3,
ASK1 (Cell Signaling Technology, Inc., Danvers, MA, USA), JNK and
p-JNK (Santa Cruz Biotechnology, Inc., Santa Cruz, CA, USA) was
performed. Immunoreactive bands were visualized by enhanced
chemiluminescence (Amersham Biosciences Biotech, Piscataway, NJ,
USA).
Animal experiments
Four-week-old male nu/nu mice were purchased from
the First Affiliated Hospital of Zhejiang University. All animal
experiments were approved by the Laboratory Animal Regulations of
the Ministry of Science and Technology of China. Each mouse was
subcutaneously injected with 6x106 Mia PaCa-2 cells in
the back. Treatment was started when the subcutaneous tumors
reached a minimum size of 100 mm3. The mice were
randomly divided into four treatment groups: a) vehicle alone
(control); b) bufalin (0.1 mg/kg, for 10 days); c) gemcitabine (125
mg/kg, three times/week for 2 weeks); d) bufalin and gemcitabine in
combination. Each group consisted of six animals. The dose of 125
mg/kg twice a week for gemcitabine has been shown to be efficient
in another pancreatic cancer xenograft model (17). To evaluate the tolerable therapeutic
dose of bufalin in this animal model, we performed a preliminary
dose-response experiment. Four times weekly i.p. injections of 1,
0.5, 0.2, 0.1 and 0.05 mg/kg bufalin were performed. Then, we
determined the bufalin dose (0.1 mg/kg) for this study. The tumor
size was measured every four days. The volume was calculated using
the formula: volume = (length x width2) / 2. One month
after the treatment, the xenografts were excised and stocked in 10%
formalin.
Immunohistochemical examination
Tissue sections (4 μm) were prepared using a
microtome and placed on glass slides. For immunohistochemical
examination, endogenous peroxidase was blocked in 3%
H2O2 solution. Sections were incubated at 4°C
with primary antibodies overnight. Following the removal of unbound
antibodies, sections were incubated with biotinylated anti-mouse or
anti-rabbit antibodies for 1 h, and then incubated with horseradish
peroxidase complex for 10 min, followed by counterstaining with
hematoxylin, dehydration and mounting. Sections without primary
antibodies were used as negative controls for immunostaining.
Random images obtained from each of the four groups were captured
and analyzed at x400 magnification. The primary antibodies of
anti-human Ki-67 and ASK1 were purchased from Cell Signaling
Technology.
Statistical analysis
Results are presented as the mean ± standard error
(SE), and all experiments were performed three times independently.
The one-way analysis of variance (ANOVA) and the two-tailed
Student’s t-test for unpaired samples were used to determine the
statistical significance using SPSS 15.0. P<0.05 was considered
to indicate a statistically significant result.
Results
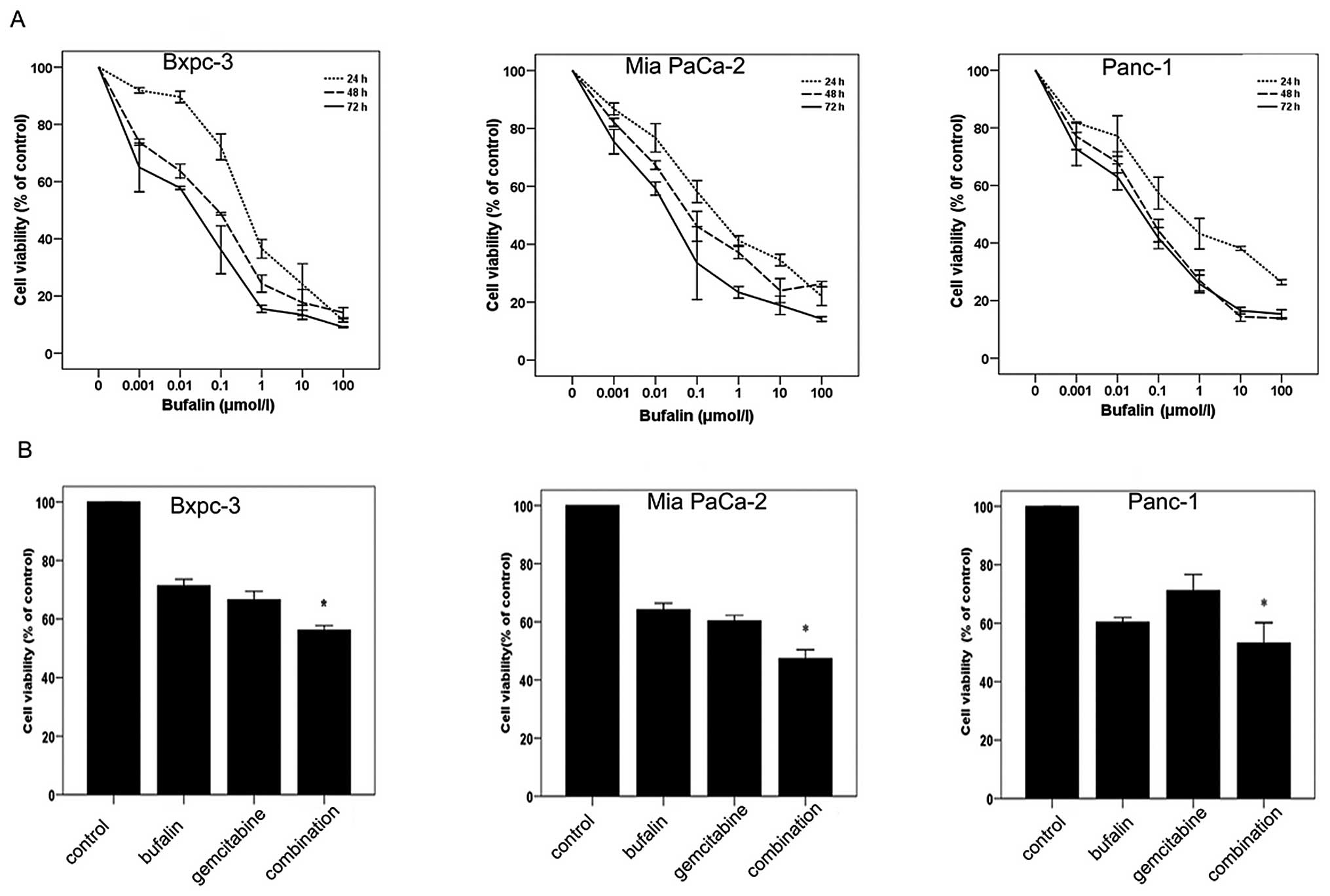
Bufalin potentiates growth inhibition
induced by gemcitabine in pancreatic cancer cell lines
MTT assay was used to examine the cell growth
inhibition efficacy of bufalin on three pancreatic cancer cell
lines (Bxpc-3, Mia PaCa-2 and Panc-1). Cells were treated with
bufalin at different concentrations (0–100 μM) for 24, 48
and 72 h. The results demonstrated that bufalin inhibited the
growth of all three cell lines in a dose- and time-dependent manner
(Fig. 1A). We subsequently
investigated the effect of the combination with bufalin and
gemcitabine on cell viability. Pancreatic cancer cell lines were
treated with bufalin (0.01 μM) and/or gemcitabine (Bxpc-3,
0.5 μg/ml; Mia PaCa-2 and Panc-1, 5 μg/ml) for 48 h
(18). Our results showed that the
combination treatment with bufalin and gemcitabine inhibited the
growth of all three cell lines more than either bufalin or
gemcitabine used alone (Fig.
1B).
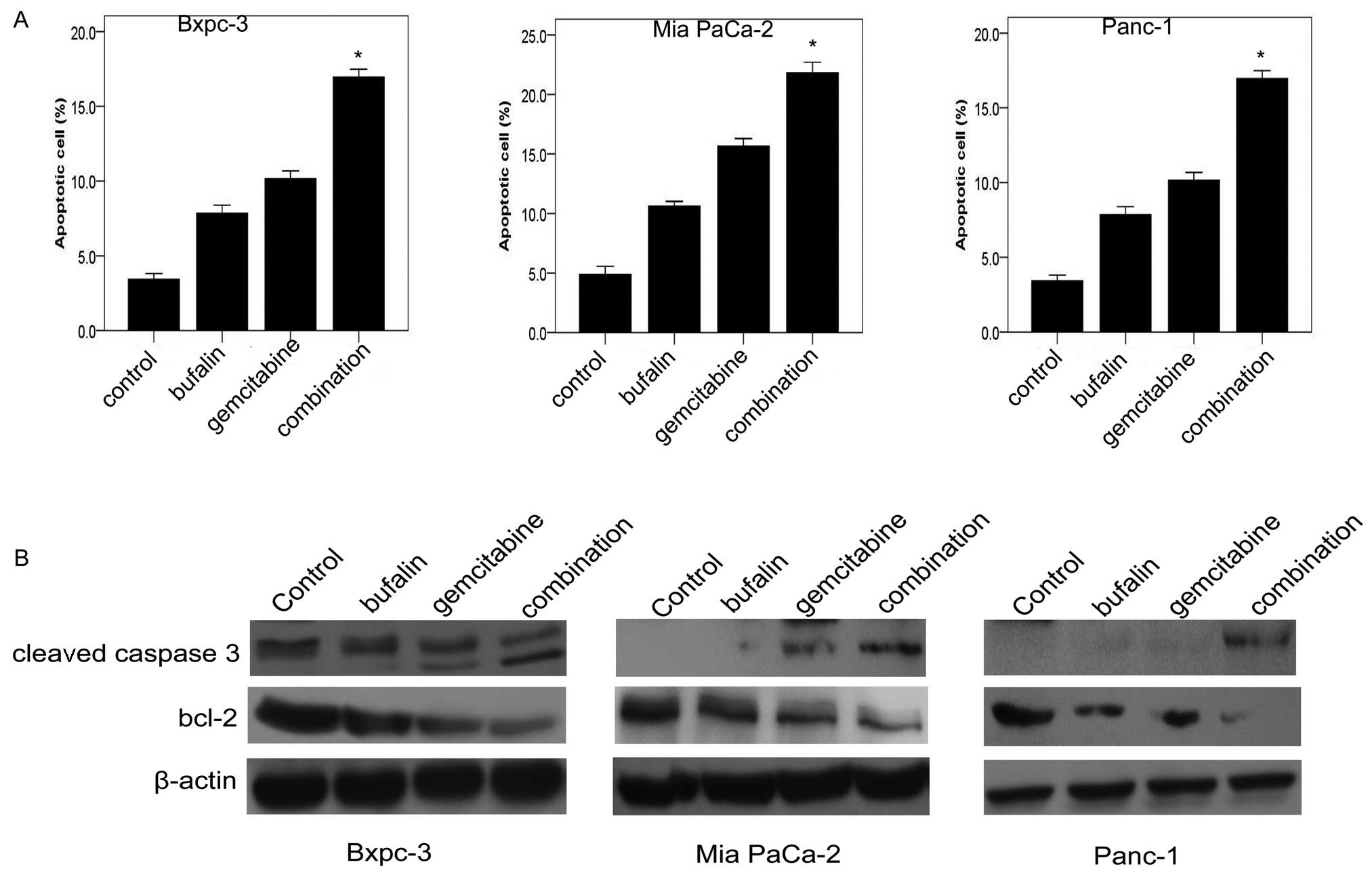
Bufalin enhances the induction of
apoptosis by gemcitabine in pancreatic cancer cells
We investigated whether bufalin was capable of
enhancing gemcitabine-induced apoptosis using flow cytometry
analysis. The three pancreatic cancer cell lines were treated with
different doses of drugs for 48 h in the same way as for the MTT
assay. Exposure to bufalin (0.01 μM) induced apoptosis by up
to 7.8% in Bxpc-3, 11.5% in Mia Paca-21 and 7% in Panc-1. Exposure
to gemcitabine (0.5 or 5 μg/ml) in combination with bufalin
(0.01 μM) for 48 h induced apoptosis by up to 16.8% in
Bxpc-3, 21.8% in Mia Paca-2 and 17.4% in Panc-1 (Fig. 2A). To further examine the effect of
inducing apoptosis by combination therapy, the expression of
apoptosis-related proteins (bcl-2 and cleaved caspase-3) was
evaluated. As shown in Fig. 2B, the
combined treatment of pancreatic cancer cells with bufalin and
gemcitabine significantly decreased the expression of bcl-2.
Conversely, the expression of cleaved caspase-3 was significantly
upregulated in the combination group compared with the control
group and the bufalin and gemcitabine alone groups (Fig. 2B).
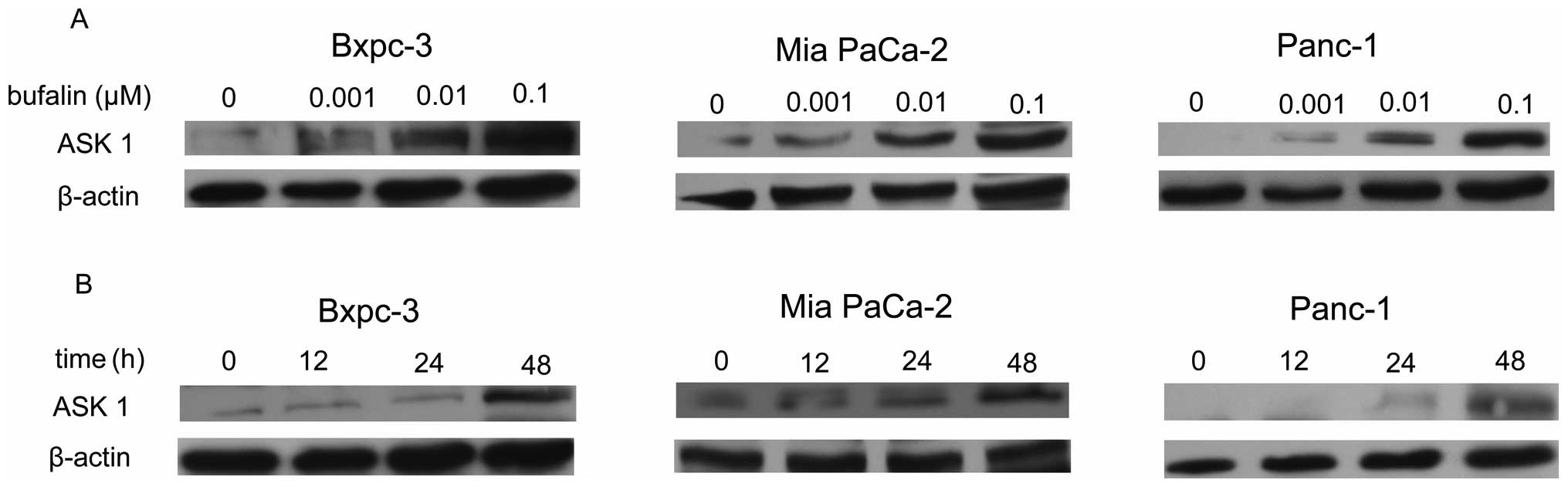
Bufalin upregulates the expression of
ASK1 in three pancreatic cancer cell lines
To further investigate the apoptosis induced by
bufalin, we evaluated the expression of ASK1 in the three
pancreatic cancer cell lines. As shown in Fig. 3A, relative to the control, treatment
with bufalin induced a dose-dependent increasing expression of ASK1
from 0.001 to 0.1 μM in the three pancreatic cancer cell
lines. However, bufalin (0.001 μM) treatment induced a
higher ASK1 level in Bxpc-3 and Mia PaCa-2 cells than in Panc-1
cells (Fig. 3A). When cells were
treated with bufalin at a dose of 0.01 μM, the expression of
ASK1 protein did not differ significantly among the groups. In
addition, ASK1 expression was analyzed at a different treatment
time with 0.01 μM bufalin. A bufalin-induced time-dependent
activation of ASK1 expression was observed in Bxpc-3, Mia PaCa-2
and Panc-1 cells (Fig. 3B).
However, treatment with bufalin (0.01 μM) did not induce
ASK1 expression until after 12 h in Panc-1 cells. The results also
reveal that ASK1 expression was significantly upregulated after 48
h of treatment in the three pancreatic cancer cell lines (Fig. 3B). The Mia PaCa-2 cells treated with
bufalin at 0.01 μM for 48 h were selected for our next
experiment.
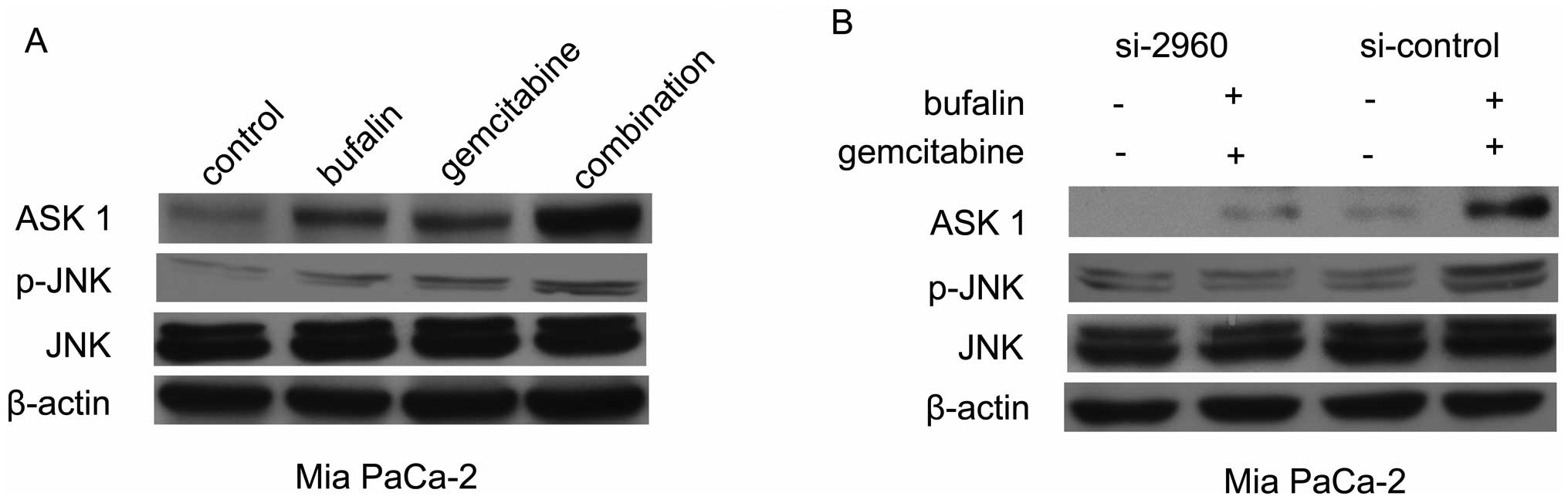
Bufalin increased ASK1 expression induced
by gemcitabine
We analyzed whether gemcitabine could induce ASK1
expression and whether upregulation of ASK1 by bufalin could
eliminate chemoresistance in Mia PaCa-2 cells, resulting in more
marked gemcitabine-induced apoptosis. We found that the level of
ASK1 protein was increased when Mia PaCa-2 cells were treated with
gemcitabine for 48 h (Fig. 4A). We
also tested whether the combined treatment with bufalin for 48 h
could abrogate gemcitabine-induced ASK1 protein expression levels.
Our results revealed that ASK1 expression induced by gemcitabine
increased in the bufalin combination treatment (Fig. 4A).
Bufalin induced apoptosis in pancreatic
cancer cells, possibly via ASK1/JNK pathway
ASK1 activation is a pivotal mechanism in a broad
variety of cell apoptosis. To explore whether the ASK1/JNK pathway
contributes to bufalin-induced apoptosis in pancreatic cancer
cells, the expression of ASK1 and p-JNK proteins were investigated
by western blot analysis. The expression of ASK1 was upregulated
with the combined treatment in Mia PaCa-2 cells. Furthermore, such
effect of p-JNK was also detected in Mia PaCa-2 cell treatment by
bufalin with or without gemcitabine (Fig. 4A). Next, ASK1 siRNA was transfected
into cancer cells. The expression of ASK1 in cells transfected with
si-2096 decreased to 20% of that of the si-control. Immunoblotting
was performed to examine the expression of p-JNK in the treatment
group transfected with si-2096 or with si-control in Mia PaCa-2
cells (Fig. 4B). No difference in
expression was observed with total JNK, demonstrating that bufalin
and gemcitabine have no effect on the total JNK. The expression of
p-JNK was significantly decreased following the combined treatment
in si-2096 Mia PaCa-2 cells, suggesting that the increased level of
p-JNK could be downregulated by si-ASK1 with the combination
treatment in pancreatic cancer cells (Fig. 4B).
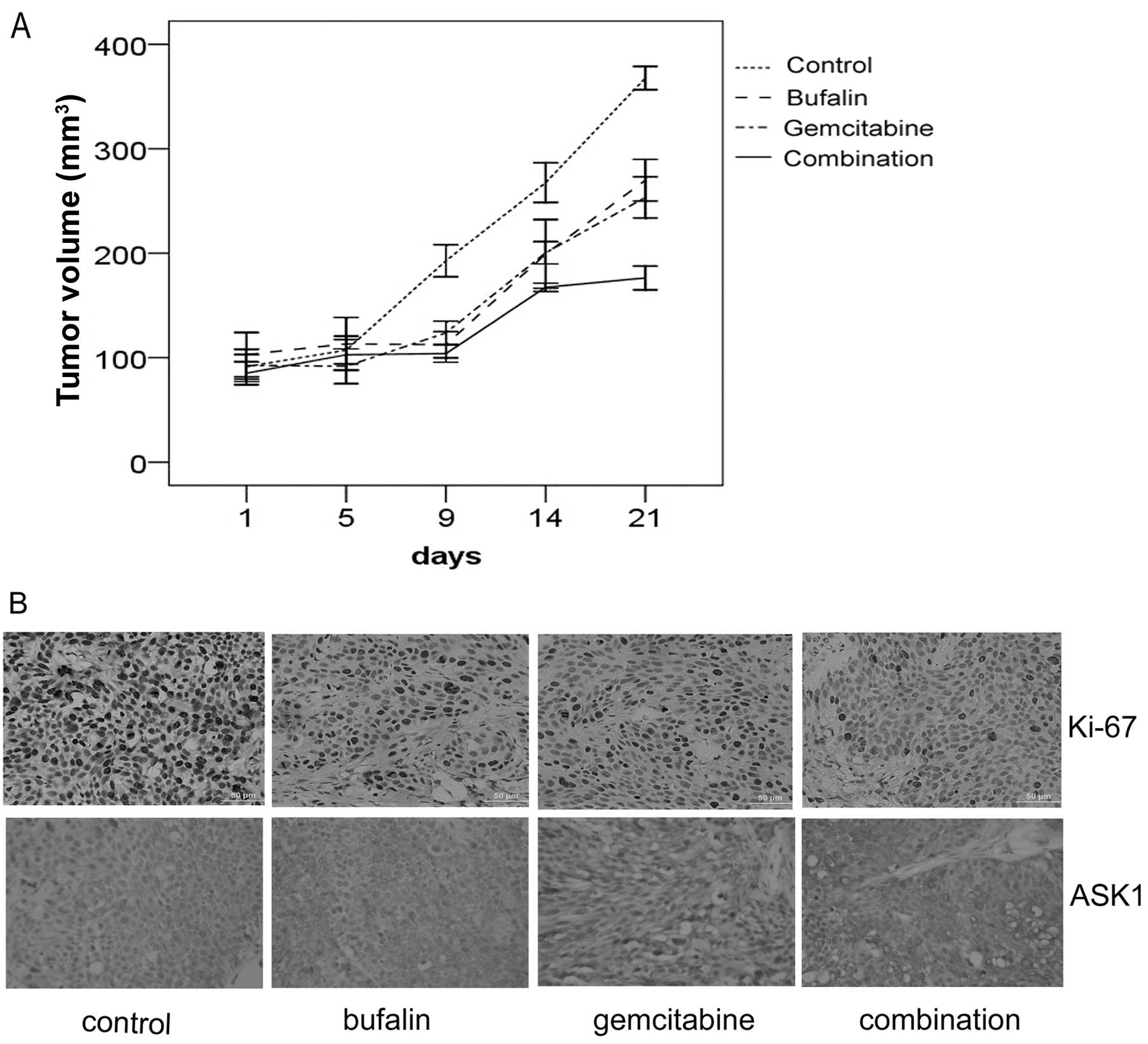
Bufalin potentiates the antitumor effect
of gemcitabine in vivo
We used the Mia PaCa-2 subcutaneous xenograft as an
in vivo model. Following the combination treatment with
gemcitabine and bufalin for two weeks, the tumor volume was
significantly reduced compared with that observed in the groups
treated with control, gemcitabine and bufalin alone (Fig. 5A). Ki-67 nuclear antigen is used to
determine cell proliferation activity, and is a key indicator of
prognosis for certain malignant tumors. It was found to be
significantly decreased in the combination group. In addition, ASK1
was found to be significantly upregulated in the combination group
(Fig. 5B).
Discussion
The survival rate of patients with pancreatic
carcinoma is rather poor, mainly because the disease is frequently
diagnosed at an advanced stage, and is characterized by a
chemoresistant phenotype (19).
Several studies have shown that cytotoxic agents block tumorigenic
cascade activation during cancer initiation and progression
(20). In particular, these
targeted therapies could be used in combination with current
clinical chemotherapeutic drug regimens such as gemcitabine and/or
5-fluorouracil (5-FU) to overcome drug resistance and improve the
efficacy of treatments for patients with locally advanced or
metastatic pancreatic cancer (21).
In the present study, we used bufalin in combination with
gemcitabine to estimate its efficacy against pancreatic cancer
cells.
Bufalin has been reported to play a critical role in
cancer cell apoptosis and differentiation, in ovarian and prostate
cancer (11,12), with little toxic effect on normal
cells at low doses (11). Bufalin
also induces the generation of ROS in lung and colon cancer
(22,23). However, the role of bufalin in
pancreatic cancer cell lines has not been investigated. In our
study, a dose- and time-dependent growth inhibition was observed in
the MTT assay when cells were treated with bufalin. Next, we
examined whether bufalin enhanced the sensitivity of gemcitabine in
pancreatic cancer cell lines. The results revealed that the
combination treatment with gemcitabine and bufalin enhanced tumor
cell growth inhibition compared with either agent alone. By flow
cytometry analysis, potentiation of gemcitabine-induced apoptosis
by bufalin in pancreatic cancer cells was also observed. In
accordance with the results mentioned above, the potentiation of
gemcitabine-induced apoptosis by bufalin in pancreatic cancer cells
was validated by enhancing cleaved caspase-3 activity and
inhibiting bcl-2 protein. These results may be significant in
understanding the role of bufalin in the gemcitabine-induced cell
apoptosis of pancreatic cancer.
It is well-known that the bcl-2 protein is an
anti-apoptotic factor, which confers resistance to gemcitabine in
pancreatic cancer cells. SiRNA-mediated silencing of bcl-2 enhances
gemcitabine sensitivity in human pancreatic cancer cells (24). A previous study has reported that
decreasing bcl-2 levels were associated with bufalin-induced
apoptosis (10). In our study, the
level of bcl-2 was downregulated in the combination group compared
with bufalin or gemcitabine used alone, suggesting that bufalin may
enhance the effect of gemcitabine by down-regulating the levels of
bcl-2 in pancreatic cancer cells.
ASK1 is a ROS-sensitive protein, which is involved
in the activation of AP-1, Rac1, cdc2 kinase and JNK, as well as
the inhibition of protein kinases A and C (25–27).
It constitutes a pivotal signaling pathway in cytokine- and
stress-induced apoptosis (28,29).
Activation of the JNK family is involved in various physiological
and pathological processes, including cell apoptosis, inflammatory
response and cytokine production (30–32).
JNKs are activated following dual phosphorylation of threonine and
tyrosine specifically by MKK4 and MKK7 (33). Overexpression of ASK1 may induce
cytochrome c release from the mitochondria and activate caspase-9
and caspase-3 (28). Furthermore,
Yu et al reported that the ASK1/JNK signaling cascade
contributed to denbinobin-induced apoptosis in A549 cells (34). Yamamoto et al demonstrated
that ASK1-mediated JNK activation phosphorylated bcl-2, leading to
a reduction in its anti-apoptotic activity (35). However, whether the ASK1/JNK
signaling pathway participates in bufalin-induced apoptosis in
pancreatic cancer has not previously been demonstrated. In this
study, we found that treatment of pancreatic cancer cells with
bufalin caused the activation of ASK1 and p-JNK. Furthermore,
treatment with the combination therapy significantly increased the
expression of ASK1/JNK in Mia PaCa-2 cells. When ASK1 was knocked
down, the level of p-JNK was decreased in cells with combined
treatment of bufalin and gemcitabine. These results suggest that
bufalin may, at least partially, enhance the antitumor effect of
gemcitabine in pancreatic cancer by activating ASK1 to induce JNK
activation, which ultimately leads to bcl-2 expression in
pancreatic cancer cells. In the tumor-bearing animal model, the
results were replicated in vitro. The final tumor volumes in
the combination group were significantly reduced compared with the
control group and the gemcitabine alone group. The expression of
Ki-67 was notably reduced in tumor tissue treated with the
combination therapy. More importantly, the expression of ASK1
increased in tumor tissues with the combined treatment.
In conclusion, the results from the present study
demonstrated for the first time that it was possible to enhance the
chemosensitivity of pancreatic cancer cells through treatment with
bufalin. Apoptosis may be mediated by the upregulation of the
ASK1/JNK pathway, which eventually induces bcl-2 expression in
human pancreatic cancer. Our results provide a mechanism linking
bufalin and apoptosis kinase ASK1 and provide support for the
development of therapeutic strategies to overcome the resistance to
gemcitabine in pancreatic cancer chemotherapy.
Acknowledgements
This study was supported by grants
from the National Natural Science Foundation of China (nos.
30872531 and 81001094), and the Ministry of Science and Technology
of the People’s Republic of China (no. 2007AA02Z476).
References
|
1.
|
A JemalR SiegelE WardCancer statistics,
2006CA Cancer J Clin56106130200610.3322/canjclin.56.2.106
|
|
2.
|
AN ShahJM SummyJ ZhangSI ParkNU ParikhGE
GallickDevelopment and characterization of gemcitabine-resistant
pancreatic tumor cellsAnn Surg
Oncol1436293637200710.1245/s10434-007-9583-517909916
|
|
3.
|
A JemalT MurrayA SamuelsA GhafoorE WardMJ
ThunCancer statistics, 2003CA Cancer J
Clin53526200310.3322/canjclin.53.1.5
|
|
4.
|
TS RiallWH NealonJS GoodwinPancreatic
cancer in the general population: Improvements in survival over the
last decadeJ Gastrointest Surg1012121223discussion 1223–1214,
2006.
|
|
5.
|
HL PearceM Alice MillerThe evolution of
cancer research and drug discovery at Lilly Research
LaboratoriesAdv Enzyme
Regul45229255200510.1016/j.advenzreg.2005.02.01716143373
|
|
6.
|
Y ShenM CaiW XiaFTY720, a synthetic
compound from Isaria sinclairii, inhibits proliferation and induces
apoptosis in pancreatic cancer cellsCancer
Lett254288297200710.1016/j.canlet.2007.03.01317462818
|
|
7.
|
SH LeeJK RyuKY LeeEnhanced anti-tumor
effect of combination therapy with gemcitabine and apigenin in
pancreatic cancerCancer
Lett2593949200810.1016/j.canlet.2007.09.01517967505
|
|
8.
|
HA Burris IIIMJ MooreJ
AndersenImprovements in survival and clinical benefit with
gemcitabine as first-line therapy for patients with advanced
pancreas cancer: a randomized trialJ Clin
Oncol152403241319979196156
|
|
9.
|
K KimuraT SawadaM KomatsuAntitumor effect
of trastuzumab for pancreatic cancer with high HER-2 expression and
enhancement of effect by combined therapy with gemcitabineClin
Cancer Res1249254932200610.1158/1078-0432.CCR-06-054416914581
|
|
10.
|
M WatabeN KawazoeY MasudaS NakajoK
NakayaBcl-2 protein inhibits bufalin-induced apoptosis through
inhibition of mitogen-activated protein kinase activation in human
leukemia U937 cellsCancer Res57309731001997
|
|
11.
|
N TakaiT UedaM NishidaK NasuH
NaraharaBufalin induces growth inhibition, cell cycle arrest and
apoptosis in human endometrial and ovarian cancer cellsInt J Mol
Med21637643200818425357
|
|
12.
|
CH YuSF KanHF PuE Jea ChienPS
WangApoptotic signaling in bufalin- and cinobufagin-treated
androgen-dependent and -independent human prostate cancer
cellsCancer
Sci9924672476200810.1111/j.1349-7006.2008.00966.x19037992
|
|
13.
|
K NasuM NishidaT UedaBufalin induces
apoptosis and the G0/G1 cell cycle arrest of endometriotic stromal
cells: a promising agent for the treatment of endometriosisMol Hum
Reprod11817823200510.1093/molehr/gah24916390854
|
|
14.
|
CT KuoBC ChenCC YuApoptosis
signal-regulating kinase 1 mediates denbinobin-induced apoptosis in
human lung adenocarcinoma cellsJ Biomed
Sci1643200910.1186/1423-0127-16-4319405983
|
|
15.
|
W SchonerG Scheiner-BobisEndogenous and
exogenous cardiac glycosides: their roles in hypertension, salt
metabolism, and cell growthAm J Physiol Cell
Physiol293C509536200710.1152/ajpcell.00098.200717494630
|
|
16.
|
Y AmanoY ChoM MatsunawaK KomiyamaM
MakishimaIncreased nuclear expression and transactivation of
vitamin D receptor by the cardiotonic steroid bufalin in human
myeloid leukemia cellsJ Steroid Biochem Mol
Biol114144151200910.1016/j.jsbmb.2009.01.022
|
|
17.
|
ZY TangYL WuSL GaoHW ShenEffects of the
proteasome inhibitor bortezomib on gene expression profiles of
pancreatic cancer cellsJ Surg
Res145111123200810.1016/j.jss.2007.03.06117714734
|
|
18.
|
Q GuoY ChenB ZhangM KangQ XieY
WuPotentiation of the effect of gemcitabine by emodin in pancreatic
cancer is associated with survivin inhibitionBiochem
Pharmacol7716741683200910.1016/j.bcp.2009.02.02119428321
|
|
19.
|
M KornmannHG BegerKH LinkChemosensitivity
testing and test-directed chemotherapy in human pancreatic
cancerRecent Results Cancer
Res161180195200310.1007/978-3-642-19022-3_1512528808
|
|
20.
|
A ZalatnaiJ MolnarReview. Molecular
background of chemoresistance in pancreatic cancerIn
Vivo21339347200717436586
|
|
21.
|
M MimeaultR HaukeSK BatraRecent advances
on the molecular mechanisms involved in the drug resistance of
cancer cells and novel targeting therapiesClin Pharmacol
Ther83673691200810.1038/sj.clpt.610029617786164
|
|
22.
|
L SunT ChenX WangY ChenX WeiBufalin
induces reactive oxygen species dependent bax translocation and
apoptosis in ASTC-a-1 cellsEvid Based Complement Alternat
Med200919592481
|
|
23.
|
CM XieWY ChanS YuJ ZhaoCH ChengBufalin
induces autophagy-mediated cell death in human colon cancer cells
through reactive oxygen species generation and JNK activationFree
Radic Biol Med5113651375201110.1016/j.freeradbiomed.2011.06.016
|
|
24.
|
K OkamotoM OckerD Neureiterbcl-2-specific
siRNAs restore gemcitabine sensitivity in human pancreatic cancer
cellsJ Cell Mol
Med11349361200710.1111/j.1582-4934.2007.00013.x17378914
|
|
25.
|
M KurosawaS NumazawaY TaniT YoshidaERK
signaling mediates the induction of inflammatory cytokines by
bufalin in human monocytic cellsAm J Physiol Cell
Physiol278C500508200010712238
|
|
26.
|
N KawazoeM WatabeY MasudaS NakajoK
NakayaTiam1 is involved in the regulation of bufalin-induced
apoptosis in human leukemia
cellsOncogene1824132421199910.1038/sj.onc.120255510229192
|
|
27.
|
M KurosawaY TaniS NishimuraS NumazawaT
YoshidaDistinct PKC isozymes regulate bufalin-induced
differentiation and apoptosis in human monocytic cellsAm J Physiol
Cell Physiol280C459C464200111171564
|
|
28.
|
T HataiA MatsuzawaS InoshitaExecution of
apoptosis signal-regulating kinase 1 (ASK1)-induced apoptosis by
the mitochondria-dependent caspase activationJ Biol
Chem2752657626581200010.1074/jbc.M00341220010849426
|
|
29.
|
H IchijoE NishidaK IrieInduction of
apoptosis by ASK1, a mammalian MAPKKK that activates SAPK/JNK and
p38 signaling
pathwaysScience2759094199710.1126/science.275.5296.908974401
|
|
30.
|
S NumazawaMA ShinokiH ItoT YoshidaY
KuroiwaInvolvement of Na+,K(+)-ATPase inhibition in K562 cell
differentiation induced by bufalinJ Cell Physiol1601131201994
|
|
31.
|
Y JingM WatabeS HashimotoS NakajoK
NakayaCell cycle arrest and protein kinase modulating effect of
bufalin on human leukemia ML1 cellsAnticancer
Res141193119819948074471
|
|
32.
|
Y MasudaN KawazoeS NakajoT YoshidaY
KuroiwaK NakayaBufalin induces apoptosis and influences the
expression of apoptosis-related genes in human leukemia cellsLeuk
Res19549556199510.1016/0145-2126(95)00031-I7658701
|
|
33.
|
F SuH LiC YanB JiaY ZhangX ChenDepleting
MEKK1 expression inhibits the ability of invasion and migration of
human pancreatic cancer cellsJ Cancer Res Clin
Oncol13516551663200910.1007/s00432-009-0612-619513748
|
|
34.
|
CC YuMJ HsuML KuoThrombin-induced
connective tissue growth factor expression in human lung
fibroblasts requires the ASK1/JNK/AP-1 pathwayJ
Immunol18279167927200910.4049/jimmunol.080158219494316
|
|
35.
|
K YamamotoH IchijoSJ KorsmeyerBCL-2 is
phosphorylated and inactivated by an ASK1/Jun N-terminal protein
kinase pathway normally activated at G(2)/MMol Cell
Biol1984698478199910567572
|