Introduction
Neutrophil gelatinase-associated lipocalin (NGAL), a
member of the lipocalin family, was originally identified as a
protein stored in specific granules of the human neutrophil
(1). Besides the neutrophil, NGAL
is expressed in most tissues normally exposed to micro-organisms
and is induced in epithelial cells during inflammation (2). NGAL binds bacterial catecholate-type
ferric siderophores and acts as a potent bacteriostatic agent by
sequestering iron, indicating that NGAL participates in the
iron-depletion strategy of the innate immune system (2). In addition, increased NGAL expression
has been observed in a variety of pathological conditions,
including inflammation, acute ischemic renal injury and various
types of human cancer (3). The
upregulation of NGAL has been found in human tumors of various
organs, including the ovary, colon, pancreas, lung, esophagus and
thyroid (2). Previous studies have
also demonstrated that the association of NGAL with cancer
progression may be due to the ability of NGAL to interact and
protect MMP-9 from degradation, resulting in increased MMP-9
activity (4).
Some findings have been reported concerning the
mechanisms of NGAL transcriptional regulation. The induction
of NGAL expression by the co-stimulation of IL-17 and TNFα
was controlled by IκB-ζ, but not by C/EBPβ or C/EBPδ in lung cancer
A549 cells (5). NGAL was
consistently upregulated in lung cancer A549 cells following IL-1β
stimulation, and in thyroid cancer FRO cells following IκB-ζ
stimulation (6,7). Moreover, the region of the NGAL
promoter from −153 to −90 contained cis-acting elements that
may be significant for the basal expression of NGAL in A549
cells (8). C/EBPε also enhanced the
transcription of NGAL in neutrophilic granulocytes, as
demonstrated by C/EBPε−/− mice experiments (9). We previously studied the
responsiveness of NGAL to TPA stimulus in EC109 cells, an
esophageal squamous cell carcinoma cell line, and found that the
region between −152 and −60 of the NGAL promoter contained
TPA response elements, which were identified to the region required
for basal expression (10). Our
more recent study showed that NGAL was overexpressed in gastric
cancer; the binding of C/EBPβ to the TRE of the NGAL
promoter mediated its TPA-induced overexpression in gastric
carcinoma cells (11). However, to
date the core promoter elements for NGAL basal expression
are uncertain.
To further explore the transcriptional regulation of
NGAL, in the current study, we cloned a 1515-bp fragment (−1431 to
+84) of the NGAL promoter region in lung cancer cells and
generated a series of deletion and mutation constructs. Further
studies using these constructs revealed that the region from −152
to −141 was the core promoter of NGAL and that NGAL
expression was mediated by the binding of C/EBPβ to the −152 and
−141 segments of the NGAL promoter.
Materials and methods
Cell culture
Two cell lines, 95D (a lung carcinoma cell line of
high metastatic propensity) and A549 (type II pneumocyte-derived
cell line), were purchased from the Chinese Academy of Sciences.
They were all epithelial-like lung carcinoma cells and were
cultured in DMEM medium (Invitrogen, Carlsbad, CA, USA) and
maintained in a humidified atmosphere with 5% CO2 at 37°C. For
transfection, the cells were seeded in 96-well plates at
1.5x105 cells/ml.
Immunohistochemical staining
Specimens of human lung carcinomas were obtained
from the Pathology Department of the Medical College of Shantou
University (Shantou, China). Immunohistochemical staining was
performed as previously described (12). The slides were incubated overnight
at 4°C with rat anti-human NGAL monoclonal antibody (R&D
Systems, Minneapolis, MN, USA). Following rinsing with PBS, the
slides were incubated for 20 min at room temperature with
peroxidase-conjugated goat anti-rat antibody (Jackson
Immunoresearch Laboratories, West Grove, PA, USA). Subsequently,
the slides were stained with 0.003% 3, 3′-diaminobenzidine
tetrahydrochloride and 0.005% hydrogen peroxide in 0.05 M Tris HCl
(pH 7.2), counterstained, dehydrated and mounted. Blank controls
were prepared by substituting PBS for the primary antibody.
NGAL-positive samples were defined as those showing brown staining
in the cytoplasm.
RT-PCR
Total RNA was extracted using TRIzol reagent
(Invitrogen) according to the manufacturer’s instructions. Reverse
transcription was performed with 1 μg of total RNA using
Reverse Transcription system (Promega, Madison, WI, USA) according
to the manufacturer’s recommendations. PCR analyses were then
performed and the primer sequences were as follows:
5′-GGATCCGTCAGGACTCCACCTCAGA-3′ (forward primer) and
5′-GGTACCTCAGCCGTCGATACA CTG-3′ (reverse primer) for NGAL
and 5′-GAAGGT GAAGGTCGGAGTC-3′ (forward primer) and 5′-GAAGAT
GGTGATGGGATTTC-3′ (reverse primer) for GAPDH.
Immunofluorescent staining
The cells were seeded on cover-slips and incubated
for 24 h. After washing with PBS, the cells were fixed with 100%
methanol at −20°C for 15 min and then treated with 0.2% Triton
X-100 in PBS for 10 min. The cells were subsequently incubated with
a blocking solution (10% normal goat serum in PBS) for 20 min and
incubated with primary antibody overnight at 4°C. The cells were
washed and incubated with fluorescein-conjugated goat anti-rat IgG
(Zymed, Carlsbad, CA, USA) for 30 min at 37°C. Finally, the cells
were washed and mounted in glycerol and examined using a
fluorescence microscope.
Construction of plasmids
The construction of the original luciferase
expression vector pGLB (Promega), containing the human NGAL
promoter region, has been described previously (10). To further study the putative
cis-acting elements located at −152 to −141, we constructed
mutational plasmids by PCR amplification with mutated primers. The
−152 deletion plasmid was generated by PCR amplification with the
following primers: 5′-TACTCGAGCTGTCTTGCC CAATCCTGAC-3′, containing
a C/EBPs binding site (underlined) and 5′-ATAGATCTGAGACCTAGGGGCATGA
TTT-3′. Then a series of mutants with a −152 deletion mutation were
generated by PCR amplification. The mutations of the C/EBPs binding
site were generated using the following primers: i) 147m,
5′-TACTCGAGCTGTCAACCCGTT
TCCTGAC-3′; ii) 144m, 5′-TACTCGAGCTGTCTTGGA TGGTCCTGAC-3′; iii) 141m,
5′-TACTCGAGCTGTCTT
GCCCGATCCT GAC-3′; iv) CEBP (conc),
5′-TACTCGAGCTGTCTTGCG
CAATCCTGAC-3′. The DNA fragments containing the mutated
region were inserted into the XhoI and BglII sites of
the pGLB vector. Relevant regions of the final constructs were
confirmed by sequencing.
Transient transfection and luciferase
reporter gene assay
The cells in 96-well plates were transfected using
Superfect transfection reagent (Qiagen, Hilden, Germany) with pGLB
promoter constructs. To compensate for differences in transfection
efficiency, the cells were co-transfected with the internal control
plasmid pRL-TK containing Renilla luciferase (Promega). Following
transfection, the cells were cultured at 37°C in a humidified
atmosphere with 5% CO2 for 48 h. Subsequently, the cells
were harvested and the lysates were assayed for luciferase activity
using the Dual Luciferase Reporter Assay system (Promega).
Luciferase activity data are expressed as the mean ± SE of at least
three independent experiments.
Western blot analysis
Nuclear extracts from 95D and A549 cells were
prepared using the method described by Sambrook and Russell
(13). The protein concentration
was estimated using the Bradford method. Equal amounts of nuclear
extracts (100 μg) were separated by SDS-PAGE and transferred
to the PVDF membrane (Millipore, Billerica, MA, USA). The membrane
was blocked in 5% skimmed milk in PBST (phosphate-buffered saline,
containing 0.1% Tween 20) for 1 h at room temperature followed by
the addition of the primary antibody for 2 h at room temperature.
The membrane was subsequently incubated with secondary antibody
(Santa Cruz Biotechnology, Inc., Santa Cruz, CA, USA) for 2 h at
room temperature. The protein-antibody complexes were then
identified by western blot luminol reagent (Santa Cruz
Biotechnology, Inc.).
Electrophoretic mobility shift assay
(EMSA) and supershift analysis
The sequences of the oligonucleotides used in the
EMSA are listed in Table I and the
mutated bases are presented in italics. Complementary
oligonucleotides were annealed and labeled with DIG-ddUTP by
terminal transferase using DIG Gel Shift kit (Roche Diagnostics,
Mannheim, Germany). Nuclear extracts were incubated with labeled
probes for 30 min at room temperature in a 20 μl reaction
mixture containing 20 mM Hepes (pH 7.9), 1 mM EDTA (pH 8.0), 1 mM
dithiotreitol, 10 mM (NH4)2SO4,
0.2% Tween-20 (w/v), 30 mM KCl, 1 μg poly-[d(I-C)] and 0.1
μg poly L-lysine. Then, complexes were resolved on 6%
nondenatured polyacrylamide gel (acrylamide/bis-acrylamide ratio of
29:1) in 0.5X TBE at 80 V for 100 min at 4°C. The gels were
transferred to a positively-charged nylon membrane. Alkaline
phosphatase-conjugated anti-digoxigenin antibody and the
chemiluminescent substrate CSPD were used to detect digoxigenin
according to the manufacturer’s instructions (Dig Gel Shift kit,
Roche Diagnostics) and immunoreactive bands were photographed and
analyzed by FluorChemTMIS-8900 (Alpha Innotech, Santa Clara, CA,
USA). For supershift analysis, 2 μl anti-C/EBPβ antibodies
(Santa Cruz Biotechnology, Inc.) were incubated with nuclear
extracts for 20 min prior to the addition of labeled probes. In the
competition experiments, different folds molar excess of unlabeled
oligonucleotide was added to the binding reactions.
 | Table IOligonucleotides used in EMSA
analysis. |
Table I
Oligonucleotides used in EMSA
analysis.
| Name | Sequence
(5′-3′) |
|---|
| −152/−114 |
CTGTCTTGCCCAATCCTGACCAGGTGCAGAAATCTTGCCGGCAAGATTTCTGCACCTGGTCAGGATTGGGCAAGACAG |
| −147/−140m |
CTGTCGACTAGTCTCCTGACCAGGTGCAGAAATCTTGCCGGCAAGATTTCTGCACCTGGTCAGGAGACTAGTCGACAG |
| −145/−143m |
CTGTCTTTAACAATCCTGACCAGGTGCAGAAATCTTGCCGGCAAGATTTCTGCACCTGGTCAGGATTGTTAAAGACAG |
Results
Expression of NGAL in human lung
carcinoma tissues
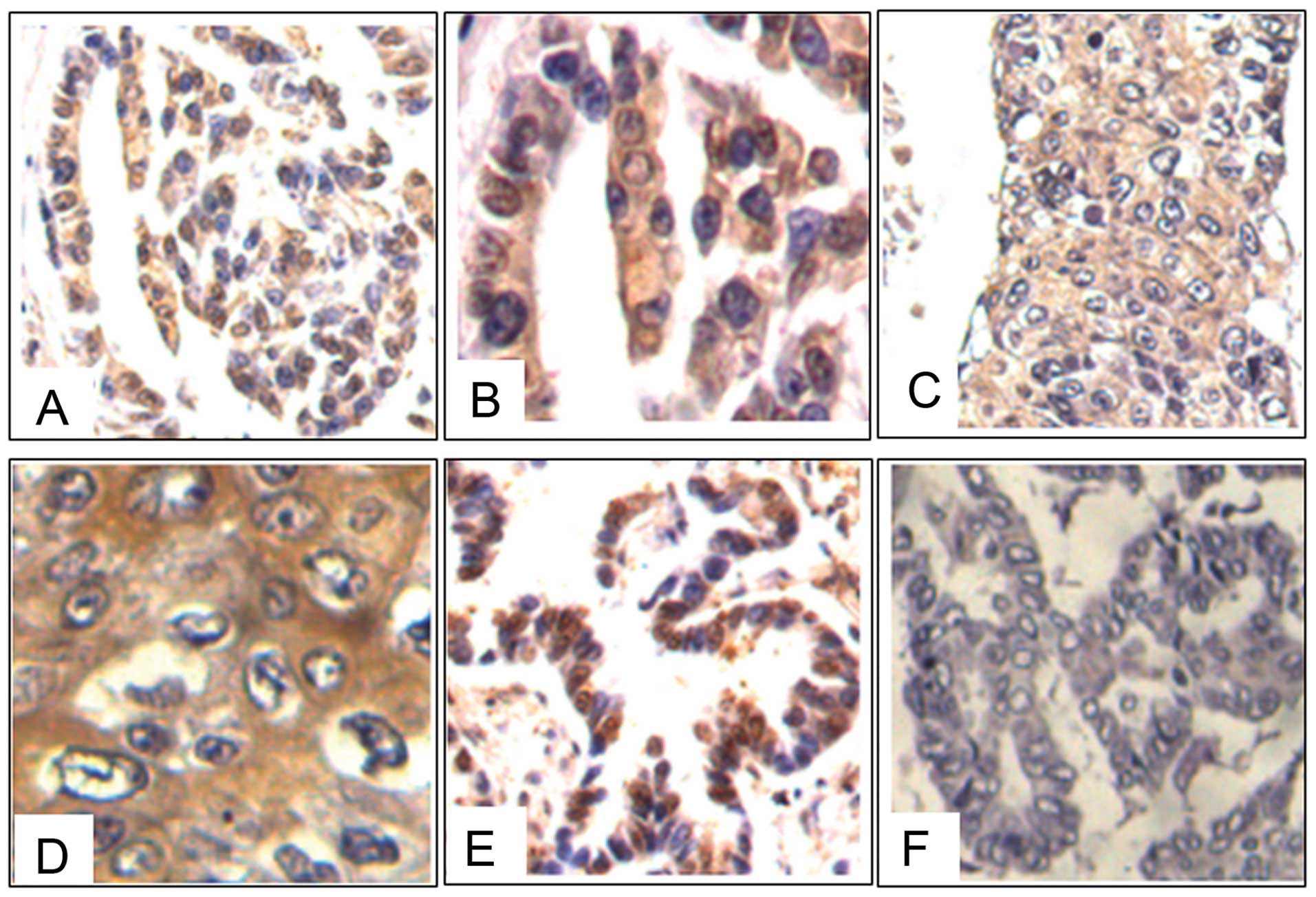
Previous studies have reported that in lung cancer
tissues, NGAL showed moderate to strong positive expression in
adenocarcinomas; whilst staining for the protein was negative or
weakly positive in squamous cell or large cell carcinomas (14). To further evaluate the expression of
NGAL in lung carcinomas, 23 tissue samples were examined by
immunohistochemical staining. The samples consisted of a spectrum
of tissues ranging from low- to high-grade human lung squamous
carcinomas, adenocarcinomas, adenosquamous carcinomas and bronchial
alveolar cell carcinomas. The results showed that 82.61% (19/23) of
the cases were positive for NGAL staining. All the positive cases
revealed a diffuse cytoplasmic distribution of NGAL in the cancer
cells (Fig. 1).
Expression of NGAL in 95D and A549
cells
The mRNA level of NGAL was analyzed by RT-PCR. A
band of ∼537 bp was detected in 95D and A549 cells, suggesting that
NGAL was expressed in these cells (Fig.
2A). The distribution of NGAL in lung carcinoma cells was
determined by means of immunofluorescent staining. NGAL was
localized to the cytoplasm of these cells, showing a diffuse to
granular pattern (Fig. 2B).
Region −152 to −141 was the core promoter
of NGAL
To determine the core promoter of human NGAL
and to identify regulatory elements within the promoter, a 1515-bp
fragment (−1431 to +84) of the NGAL promoter region was
cloned and a series of deletion and mutation constructs were
obtained. These constructs were then co-transfected with the pRL-TK
plasmid into 95D and A549 cells and subjected to the luciferase
reporter assay. It was shown that, when truncated from −152 to
−140, the promoter activity of NGAL was decreased by ∼70%
(Fig. 3A), indicating that the −152
to −141 region was essential for the basal activity of the
NGAL promoter and this region was identified as the core
promoter of NGAL. We analyzed the sequence using TESS
software (http://www.cbil.upenn.edu/cgi-bin/tess) and found
several putative binding sites of transcription factors, including
C/EBPs, in this region. We then generated a series of mutants with
a −152 deletion plasmid by PCR amplification with the mutated
primers (see Materials and methods). The relative activity of the
−152 mutations, including 147m, 144m and 141m, were reduced by ∼50%
compared with the −152 deletion plasmid, and the activity of the
CEBP(conc) was slightly increased, suggesting that C/EBPs binding
sites were responsible for promoter activity (Fig. 3B).
C/EBPβ was probably the transcription
factor regulating NGAL basal expression
According to the results of the luciferase reporter
assay, we presumed that C/EBPs regulated the basal expression of
NGAL. Nuclear extracts from 95D and A549 cells were
collected and the levels of expression of C/EBPs were analyzed
using western blotting. The results showed that C/EBPα and γ were
undetected (data not shown), but C/EBPβ could be detected in
different isoforms (42, 35 and 20 kDa; Fig. 4A). In order to further confirm that
C/EBPβ played a critical role in regulating the activity of the
NGAL promoter, we synthesized oligonucleotide probes
corresponding to C/EBPs binding sites, and simultaneously,
oligonucleotide probes with a mutated C/EBPs binding site served as
non-specific competitors (Table I).
We then examined nuclear extracts from 95D and A549 cells by EMSA,
using the C/EBPs site of the NGAL promoter (−152/−114) as a
probe. Similar DNA-protein complexes were found in the two types of
cells (Fig. 4B and C, lane 1). To
determine the specificity of these binding complexes, we added 125-
to 725-fold molar excess of unlabeled specific competitor to the
binding reaction. With the increasing molarity of unlabeled
oligonucleotide, the complex disappeared gradually (Fig. 4B and C, lanes 5–7). However, when
the unlabeled mutated oligonucleotide was added to the reaction,
the complex remained prominent [Fig. 4B
and C, lane 3 (−147/−140m): TTGCCCAA→GACTAGTC; lane 4
(−145/−143m): GCC→TAA]. In supershift analysis, anti-C/EBPβ
antidody led to the disappearance of the DNA-protein complex and
later a migrating band appeared (Fig.
4B and C, lane 2). These results suggest that C/EBPβ binds to
the core promoter of NGAL in lung carcinoma cells.
Discussion
NGAL has been identified in a variety of normal and
pathological human tissues (14).
The expression of NGAL has been demonstrated in several types of
cancer, including carcinoma of the colon, lung, pancreas, breast
and esophagus (2). NGAL also
promotes breast tumor growth by enhancing MMP-9 activity and
facilitating tumor progression. The detection of urinary MMP-9 and
NGAL complexes in breast cancer patients may serve as new
independent predictors of disease status (4). Also, our previous studies have
indicated that NGAL was overexpressed in esophageal squamous cell
carcinoma and gastric carcinoma, and that altered NGAL expression
may play a significant role in the transformation and progression
of esophageal squamous cell carcinoma (10,11,15).
In the current study, we further showed that NGAL was expressed in
lung squamous carcinoma and adenocarcinoma tissues.
The significant correlation between NGAL and tumor
progression led to studies concerning the expression regulation
mechanisms of NGAL. Increasing evidence suggests that
certain transcription factors, including IκB-ζ, NF-κB, C/EBPα,
C/EBPβ and C/EBPε, play dominant roles in the induction of
NGAL expression within tumor cells (5). Our previous study in esophageal
squamous cell carcinoma cells indicated that there was a TPA
response element located at the −152 and −60 regions of the
NGAL promoter which mediated TPA stimulation (10). More recently, in gastric cancer, we
found that the binding of C/EBPβ to the TRE of the NGAL
promoter mediates its TPA-induced overexpression (11). In lung cancer, it has been reported
that NGAL was induced by IL-1β through the NF-κB and IκB-ζ
pathway in A549 cells (6,8). However, the mechanisms for NGAL
basal expression have not been thoroughly investigated. In the
current study, we aimed to find the cis-acting elements
which regulate NGAL basal expression. We identified the core
promoter region of NGAL, which contained C/EBPs binding
sites located at −152 to −141 and showed that the C/EBPs binding
sites were crucial for the basal expression of NGAL.
C/EBPβ belongs to the C/EBP family of basic region
leucine zipper (bZIP) transcription factors that consists of six
members: C/EBPα, C/EBPβ, C/EBPγ, C/EBPδ, C/EBPε and C/EBPζ. With
the exception of C/EBPζ, which lacks a canonical basic region, each
protein contains a similar basic region and leucine zipper
sequences at its C-terminus, which mediate DNA binding and
dimerization, respectively (16).
C/EBPs form both homo- and heterodimers and may interact with other
non-bZIP transcription factors (17). C/EBPs play significant roles in a
number of cellular processes, including metabolism and inflammatory
response, and are specifically involved in the differentiation of
adipocytes, myeloid cells, hepatocytes, mammary epithelial cells,
intestinal epithelial cells, keratinocytes and ovarian luteal cells
(18). Therefore, C/EBPs lead to
not only granulocytic differentiation, but also the expression of
granulocyte-specific genes, including the expression of
NGAL, which is stored in specific granules of the human
neutrophil (1,19). In the present study, we demonstrated
that C/EBPβ exists as different isoforms in 95D and A549 cells.
C/EBPβ binds to the C/EBPs binding site of the NGAL promoter
and regulates the basal expression of NGAL. C/EBPβ has been
shown to function as a pro-oncogenic transcription factor that
promotes the proliferation and/or survival of certain tumor cells,
and its levels were increased in a number of tumors, including lung
cancer (20). This may be the
reason for NGAL overexpression in these tumors. Moreover, NGAL has
been implicated in epithelial cell differentiation (21). We presume that C/EBPβ promotes NGAL
expression in the cell differentiation process.
In summary, we identified the core promoter of
NGAL in lung carcinoma cells and revealed the regulation
mechanism of NGAL basal expression by the binding of
transcriptional factor C/EBPβ to this core promoter.
Acknowledgements
This study was supported by grants
from the National High Technology Research and Development Program
of China (No. 2006AA02A403), the Natural Science Foundation of
China-Guangdong Joint Fund (No. U0932001) and the National Natural
Science Foundation of China (No. 30772485; No. 31000347).
References
|
1.
|
L KjeldsenAH JohnsenH SengeløvN
BorregaardIsolation and primary structure of NGAL, a novel protein
associated with human neutrophil gelatinaseJ Biol
Chem268104251043219937683678
|
|
2.
|
D BolignanoV DonatoA LacquanitiMR FazioC
BonoG CoppolinoM BuemiNeutrophil gelatinase-associated lipocalin
(NGAL) in human neoplasias: a new protein enters the sceneCancer
Lett2881016201010.1016/j.canlet.2009.05.02719540040
|
|
3.
|
K MoriHT LeeD RapoportIR DrexlerK FosterJ
YangKM Schmidt-OttX ChenJY LiS WeissEndocytic delivery of
lipocalin-siderophore-iron complex rescues the kidney from
ischemia-reperfusion injuryJ Clin
Invest115610621200510.1172/JCI2305615711640
|
|
4.
|
CA FernándezL YanG LouisJ YangJL KutokMA
MosesThe matrix metalloproteinase-9/neutrophil
gelatinase-associated lipocalin complex plays a role in breast
tumor growth and is present in the urine of breast cancer
patientsClin Cancer Res11539053952005
|
|
5.
|
JR KarlsenN BorregaardJB CowlandInduction
of neutrophil gelatinase-associated lipocalin expression by
co-stimulation with interleukin-17 and tumor necrosis factor-alpha
is controlled by IkappaB-zeta but neither by C/EBP-beta nor
C/EBP-deltaJ Biol Chem2851408814100201010.1074/jbc.M109.017129
|
|
6.
|
JB CowlandT MutaN
BorregaardIL-1beta-specific up-regulation of neutrophil
gelatinase-associated lipocalin is controlled by IkappaB-zetaJ
Immunol17655595566200610.4049/jimmunol.176.9.555916622025
|
|
7.
|
A IannettiF PacificoR AcquavivaA LavorgnaE
CrescenziC VascottoG TellAM SalzanoA ScaloniE VuttarielloThe
neutrophil gelatinase-associated lipocalin (NGAL), a
NF-kappaB-regulated gene, is a survival factor for thyroid
neoplastic cellsProc Natl Acad Sci
USA1051405814063200810.1073/pnas.071084610518768801
|
|
8.
|
JB CowlandOE SørensenM SehestedN
BorregaardNeutrophil gelatinase-associated lipocalin is
up-regulated in human epithelial cells by IL-1 beta, but not by
TNF-alphaJ
Immunol17166306639200310.4049/jimmunol.171.12.663014662866
|
|
9.
|
AF GombartSH KwokKL AndersonY YamaguchiBE
TorbettHP KoefflerRegulation of neutrophil and eosinophil secondary
granule gene expression by transcription factors C/EBP epsilon and
PU.1Blood10132653273200310.1182/blood-2002-04-103912515729
|
|
10.
|
LY XuEM LiYD NiuWJ CaiHM YuanJX ChangZY
ShenY ZengThere are TPA response elements in −152∼-60 position of
NGAL gene 5′ flanking region in the esophageal cancer cells
EC109Progress in Biochem and Biophys331401482006(In Chinese)
|
|
11.
|
ZP DuHM YuanBL WuJX ChangZ LvJ ShenJY WuHB
ChenEM LiLY XuNeutrophil gelatinase-associated lipocalin in gastric
carcinoma cells and its induction by TPA are controlled by
C/EBPβBiochem Cell Biol89314324201121612443
|
|
12.
|
ZP DuZ LvBL WuZY WuJH ShenJY WuXE XuQ
HuangJ ShenHB ChenNeutrophil gelatinase-associated lipocalin and
its receptor: independent prognostic factors of esophageal squamous
cell carcinomaJ Clin
Pathol646974201110.1136/jcp.2010.08390721109701
|
|
13.
|
J SambrookDW RussellMolecular Cloning: A
Laboratory Manual3rd editionCold Spring Harbor Laboratory PressNew
York2001
|
|
14.
|
A FriedlSP StoeszP BuckleyMN
GouldNeutrophil gelatinase-associated lipocalin in normal and
neoplastic human tissues. Cell type-specific pattern of
expressionHistochem
J31433441199910.1023/A:100370880893410475571
|
|
15.
|
H ZhangL XuD XiaoJ XieH ZengZ WangX ZhangY
NiuZ ShenJ ShenUpregulation of neutrophil gelatinase-associated
lipocalin in esophageal squamous cell carcinoma: significant
correlation with cell differentiation and tumour invasionJ Clin
Pathol60555561200710.1136/jcp.2006.039297
|
|
16.
|
MP HollandSP BlissKA BerghornMS RobersonA
role for CCAAT/enhancer-binding protein beta in the basal
regulation of the distal-less 3 gene promoter in placental
cellsEndocrinology14510961105200410.1210/en.2003-077714670999
|
|
17.
|
YH LeeSC WilliamsM BaerE SterneckFJ
GonzalezPF JohnsonThe ability of C/EBP beta but not C/EBP alpha to
synergize with an Sp1 protein is specified by the leucine zipper
and activation domainMol Cell Biol172038204719979121452
|
|
18.
|
J Lekstrom-HimesKG XanthopoulosBiological
role of the CCAAT/enhancer-binding protein family of transcription
factorsJ Biol
Chem2732854528548199910.1074/jbc.273.44.285459786841
|
|
19.
|
A NumataK ShimodaK KamezakiT HaroH
KakumitsuK ShideK KatoT MiyamotoY YamashitaY OshimaSignal
transducers and activators of transcription 3 augments the
transcriptional activity of CCAAT/enhancer-binding protein alpha in
granulocyte colony-stimulating factor signaling pathwayJ Biol
Chem2801262112629200510.1074/jbc.M408442200
|
|
20.
|
PF JohnsonMolecular stop signs: regulation
of cell-cycle arrest by C/EBP transcription factorsJ Cell
Sci11825452555200510.1242/jcs.0245915944395
|
|
21.
|
L MallbrisKP O’BrienA HulthénB SandstedtJB
CowlandN BorregaardM Ståhle-BäckdahlNeutrophil
gelatinase-associated lipocalin is a marker for dysregulated
keratinocyte differentiation in human skinExp
Dermatol11584591200210.1034/j.1600-0625.2002.110611.x12473066
|