Introduction
Renal cell carcinoma (RCC) is the most common type
of kidney cancer in adults and the most lethal of all genitourinary
tumors. Nephrectomy remains the main method of curative treatment
for RCC without distant metastasis (1). The other two main types of anticancer
treatment in wide clinical use are radiation and chemotherapy.
However, the majority of RCCs are resistant to radiation therapy
and chemotherapy (2). Current
research is focusing on the development of new methods for RCC
therapy.
ω-3 fatty acids have potential anticancer effects.
The dietary intake of ω-3 fatty acids is associated with a reduced
risk of certain types of cancer in human populations and animal
models (3). In a large
population-based cohort study with data on long-term diet, it was
noted that consumption of ocean fish rich in ω-3 fatty acids
reduced the RCC risk by ∼74% (4,5). In
vitro studies have shown that ω-3 fatty acids inhibit cell
proliferation and induce apoptosis in certain types of cancer cells
(6,7). However, the direct effects of ω-3
fatty acids on RCC in vitro have not been reported.
The anticancer effects of ω-3 fatty acids may be
mediated by multiple pathways, including peroxisome
proliferator-activated receptor-γ (PPAR-γ) activation. PPAR-γ is a
nuclear receptor that regulates lipid homeostasis and is implicated
in the pathology of numerous diseases, including obesity and
diabetes (8,9). Additionally, studies have demonstrated
that PPAR-γ has a significant role in the inhibition of tumor
growth, progression and metastasis (10). PPAR-γ is expressed in a number of
types of cancer including colon, breast, lung and prostate cancer.
PPAR-γ activation inhibits the proliferation of numerous types of
cancer cells in vitro. It has been demonstrated that fatty
acids are natural ligands of PPAR-γ (11,12).
Fatty acids may activate PPAR-γ, then inhibit the growth of cancer
cells. However, the role of PPAR-γ in RCC growth has not been
clarified.
Cyclooxygenase-2 (COX-2) has also been suggested to
be involved in the development of cancers. COX-2 is an inducible
enzyme involved in inflammatory processes. Increasing evidence
indicates that COX-2 inhibition has an important role in the
prevention of cancer and in the delay of progression in established
cancer. For example, ω-3 fatty acids have been shown to
downregulate COX-2 expression and inhibit hepatocellular carcinoma
cell growth (13). It has been
observed that COX-2 is highly expressed in RCC tissues and that it
shows a correlation with pathological features and prognosis in
patients with RCC (14–16). The role of COX-2 in the development
of RCC is not fully understood.
α-linolenic acid (ALA) is an ω-3 fatty acid that
certain diets are rich in and that may be transformed to other ω-3
fatty acids, including eicosapentaenoic acid (EPA) and
docosahexaenoic acid (DHA), in the cells. In the present study, the
effects of ALA on the proliferation of human RCC cell line OS-RC-2
cells were observed in vitro, and the involvement of PPAR-γ
and COX-2 was demonstrated in the effects of ALA.
Materials and methods
Cell culture
The human RCC cell line, OS-RC-2, was obtained from
ATCC (Rockville, MD, USA). The OS-RC-2 cells were cultured in
RPMI-1640 medium (Sigma, St. Louis, MO, USA) supplemented with 10%
heat-inactivated fetal bovine serum (Sigma). The cells were
incubated at 37°C in a 5% CO2 humidified incubator. This
study was approved by the ethics committee of Fourth Military
Medical University, Xi’an, China.
Cell proliferation assay
The cells were seeded into 96-well plates at a
density of 104 cells per well. ALA was added into the
medium the next day after seeding. Following 48 h of treatment,
BrdU was added with ALA into the medium and incubated for one day.
The cells were fixed and denatured by incubation with 100 μl
FixDenat for 30 min at room temperature. Following the aspiration
of FixDenat, the cells were washed with 1 ml blocking buffer [1%
bovine serum albumin (BSA) in phosphate-buffered saline (PBS)],
then incubated for 90 min with 100 μl anti-BrdU
peroxidase-conjugated antibody. The incubation with anti-BrdU
peroxidase-conjugated antibody was terminated by three washes with
blocking buffer, and 100 μl 3,3′,5,5′-tetramethylbenzidine
(TMB) substrates were subsequently added into the wells. Following
5–10 min of incubation with TMB, 25 μl 1 M
H2SO4 was added to stop the reaction. The
absorbance was measured by an enzyme-linked immunosorbent assay
(ELISA) reader at a wavelength of 450 nm (Bio-Rad Laboratories,
Hercules, CA, USA). The BrdU, antibodies, FixDenat and TMB were
provided in the BrdU ELISA kit (Roche, Penzberg, Germany).
PPAR-γ activity assay
The OS-RC-2 cells were seeded into 100-mm dishes and
treated with ALA at various doses for 48 h. Subsequent to the
treatment, the cells were collected, then centrifuged at 300 × g
for 5 min at 4°C. The pellets were washed with ice-cold
PBS/phosphatase inhibitor solution, then treated with hypotonic
buffer for 15 min. NP-40 was added and the samples were centrifuged
for 30 sec at 4°C. The pellets were resuspended in the extraction
buffer for 30 min on ice and centrifuged at 14,000 × g for 10 min
at 4°C. The supernatants containing the nuclear extracts were
collected. The PPAR-γ activity levels of the OS-RC-2 cells were
then measured using a PPAR-γ Transcription Factor kit (Cayman, Ann
Arbor, MI, USA) according to the manufacturer’s instructions.
Briefly, 10 μl nuclear extract from the samples and 90 ml
Complete Transcription Factor buffer were added to the designated
wells. Following an incubation overnight at 4°C, the wells were
washed and the PPAR-γ primary antibodies were added for 1 h at room
temperature, followed by washing and incubation with the secondary
antibody for a further 1 h at room temperature. The developing
solution was added and the reaction was stopped once the blue color
had properly developed. The optical density was read with a
microplate reader (Bio-Rad Laboratories) at 450 nm.
COX-2 assay
The OS-RC-2 cells were seeded into 100-mm dishes and
treated with ALA at various doses for 48 h. Following the
treatment, the cells were harvested and centrifuged at 1,000 × g
for 10 min at 4°C. The cell pellets were homogenized in cold
Tris-HCl buffer and centrifuged at 10,000 × g for 15 min at 4°C.
The supernatant was collected for the COX-2 assay. The protein
levels of COX-2 were assayed using a human COX-2 assay kit from IBL
(Gunma, Japan), which is a solid-phase sandwich ELISA. Briefly, the
test samples and diluted standards were added into the appropriate
wells and incubated for 1 h at 37°C. Subsequent to washing, the
labeled antibody solution was added and incubated for 30 min at
37°C. The developing solution was then added and the reaction was
stopped once the blue color had properly developed. The absorbance
of each well was read with the plate reader (Bio-Rad Laboratories)
at 450 nm.
Real-time RT-PCR
The OS-RC-2 cells were seeded into six-well plates.
After reaching 80% confluence, the cells were treated with ALA at
various doses for 24 h. Subsequent to treatment, the total RNA was
extracted from the cells using Trizol reagent according to the
manufacturer’s instructions (Gibco BRL, Grand Island, NY, USA).
Reverse transcription was performed in a reaction system containing
1 μg RNA, reverse transcriptase, DNA polymerase and random
primers. The cDNA obtained was used to run PCR with pairs of
primers. The human primers for the real-time PCR were used as
previously reported (16,17). The sequences were as follows: COX-2
forward, 5′-ATC ATT CAC CAG GCA AAT TGC-3′ and reverse, 5′-GGC TTC
AGC ATA AAG CGT TTG-3′; PPAR-γ forward, 5′-GCC AAG CTG CTC CAG AAA
AT-3′ and reverse, 5′-TGA TCA CCT GCA GTA GCT GCA-3′; and β-actin
forward, 5′-GCT CCT CCT GAG CGC AAG T-3′ and reverse, 5′-TCG TCA
TAC TCC TGC TTG CTG AT-3′. The specificity of the PCR was checked
by analyzing the melting curves. The relative mRNA levels were
determined by comparing the PCR cycle threshold cDNA of PPAR-γ and
COX-2 with that of β-actin.
Statistical analysis
All data are presented as the mean ± SEM, The data
were analyzed by one-way ANOVA. P<0.05 was considered to
indicate a statistically significant difference for all statistical
analyses.
Results
Effects of ALA on human RCC cell line
OS-RC-2 cell proliferation
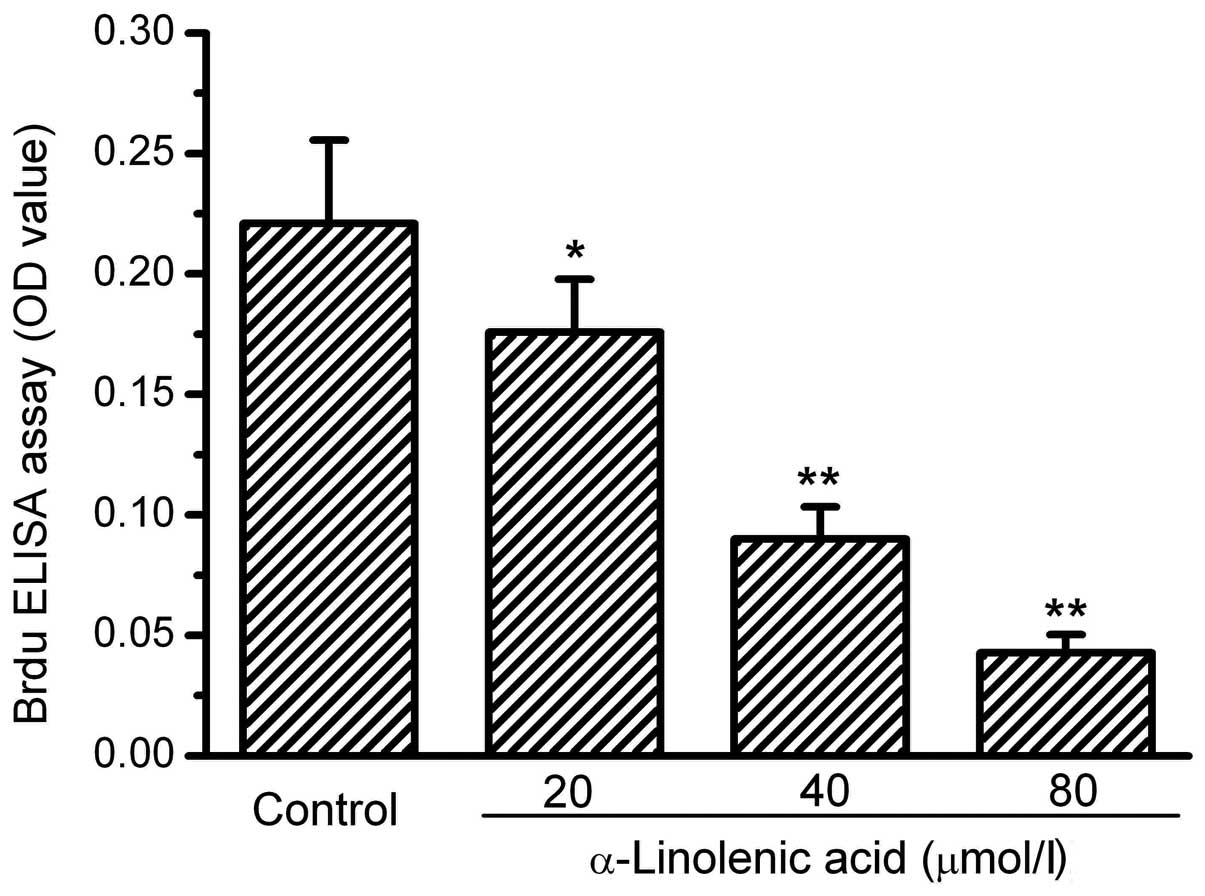
The human RCC OS-RC-2 cells grew well under the
culture conditions. Following 72 h of treatment with ALA at various
doses, the cells exhibited a notable suppression of growth. The
cell numbers in the groups treated with ALA visibly decreased. To
clarify whether the proliferation of the OS-RC-2 cells was affected
by ALA, BrdU incorporation into the cells was measured. The results
of the BrdU incorporation showed that ALA dose-dependently
decreased the incorporation of BrdU into the OS-RC-2 cells,
demonstrating an inhibitory action of ALA on OS-RC-2 cell
proliferation. The assay values of BrdU incorporation in the 20, 40
and 80 mmol/l ALA treatment groups were 81, 40 and 20% of the
control, respectively (Fig. 1).
Effects of ALA on the activity and
expression levels of PPAR-γ and COX-2 in OS-RC-2 cells
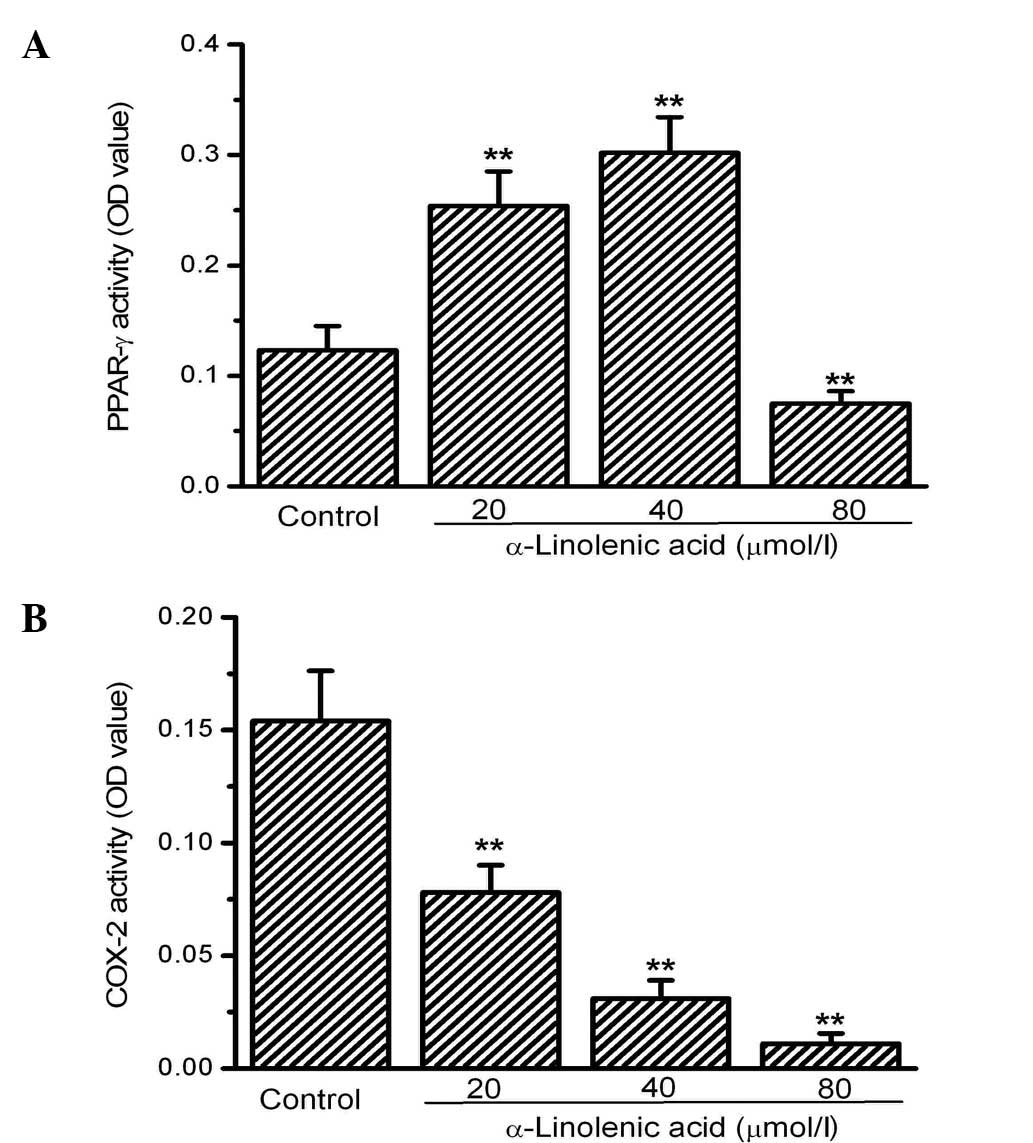
Next, the activity of two factors involved in the
regulation of tumor growth, PPAR-γ and COX-2, was investigated.
PPAR-γ activity was significantly increased by ALA at 20 and 40
mmol/l compared with the control. However, no significant
differences were observed between the 20 and 40 mmol/l ALA groups.
Notably, 80 mM ALA did not induce an increase in PPAR-γ activity
but rather decreased its activity significantly compared with the
control (Fig. 2A). In contrast to
the activity of PPAR-γ, COX-2 activity was significantly inhibited
in the OS-RC-2 cells by ALA. COX-2 activity, measured using an
ELISA kit, dose-dependently decreased in the groups treated with
ALA compared with the control group (Fig. 2B).
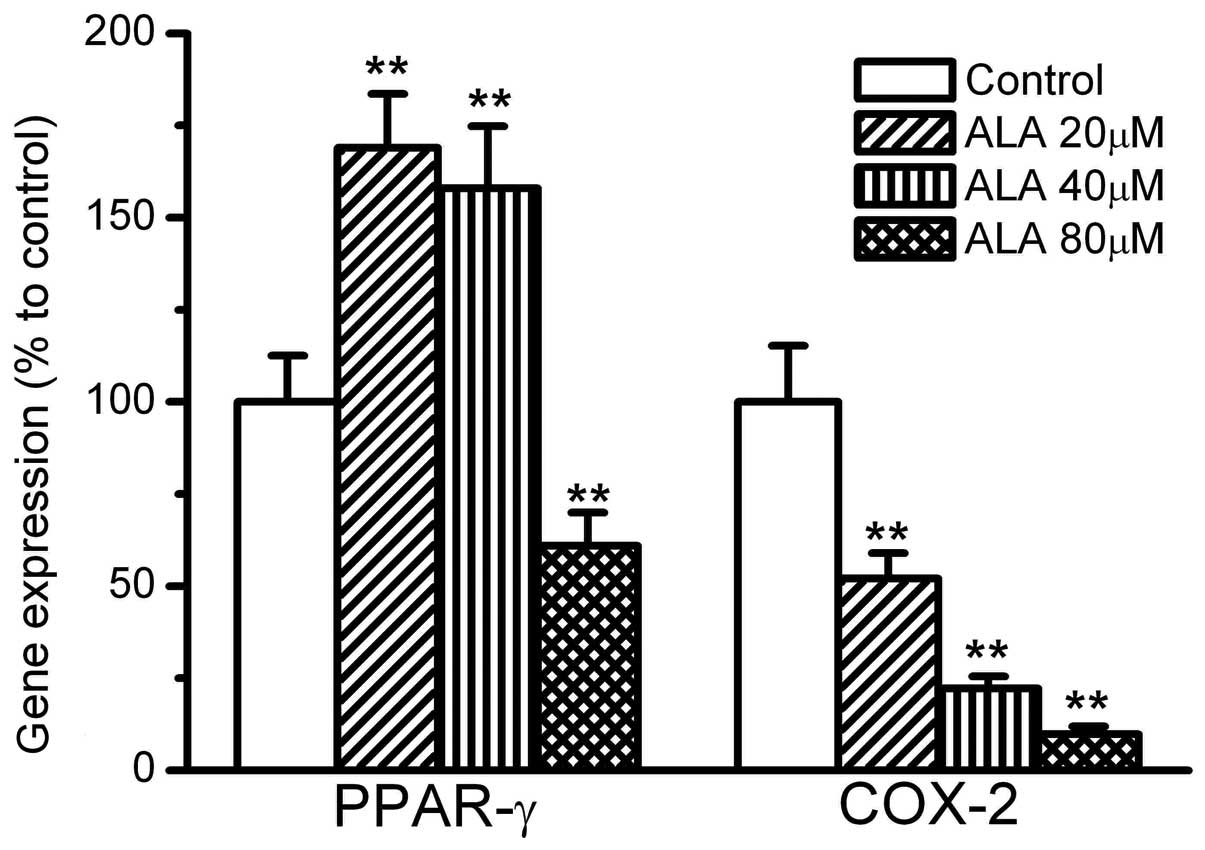
The gene expression levels of PPAR-γ and COX-2 were
also affected by ALA. ALA at 20 and 40 mmol/l induced >60%
greater expression of PPAR-γ compared with the control. However,
ALA at 80 mmol/l did not increase the expression of PPAR-γ, and
instead decreased it to 60% that of the control. By contrast, COX-2
gene expression was significantly suppressed by ALA. ALA at 20, 40
and 80 mmol/l inhibited the expression of COX-2 to 60, 30 and 10%
that of the control, respectively, (Fig. 3).
Role of PPAR-γ and COX-2 in ALA-inhibited
proliferation of OS-RC-2 cells
To clarify the role of PPAR-γ and COX-2 in the
ALA-inhibited proliferation of the OS-RC-2 cells, the effects of a
PPAR-γ agonist on BrdU incorporation into OS-RC-2 cells were
observed. Rosiglitazone (10−5 M), an agonist of PPAR-γ,
significantly inhibited BrdU incorporation, supporting the
hypothesis that PPAR-γ activation is involved in the inhibition of
OS-RC-2 cell proliferation. In combination with PPAR-γ activation
by rosiglitazone, ALA at 40 mmol/l inhibited proliferation,
indicating that PPAR-γ activation is not the only pathway in
ALA-inhibited proliferation. Moreover, at 10−6 mol/l,
N-(3-Pyridyl)indo-methacinamide, a potent and selective COX-2
inhibitor, suppressed the cell proliferation in combination with
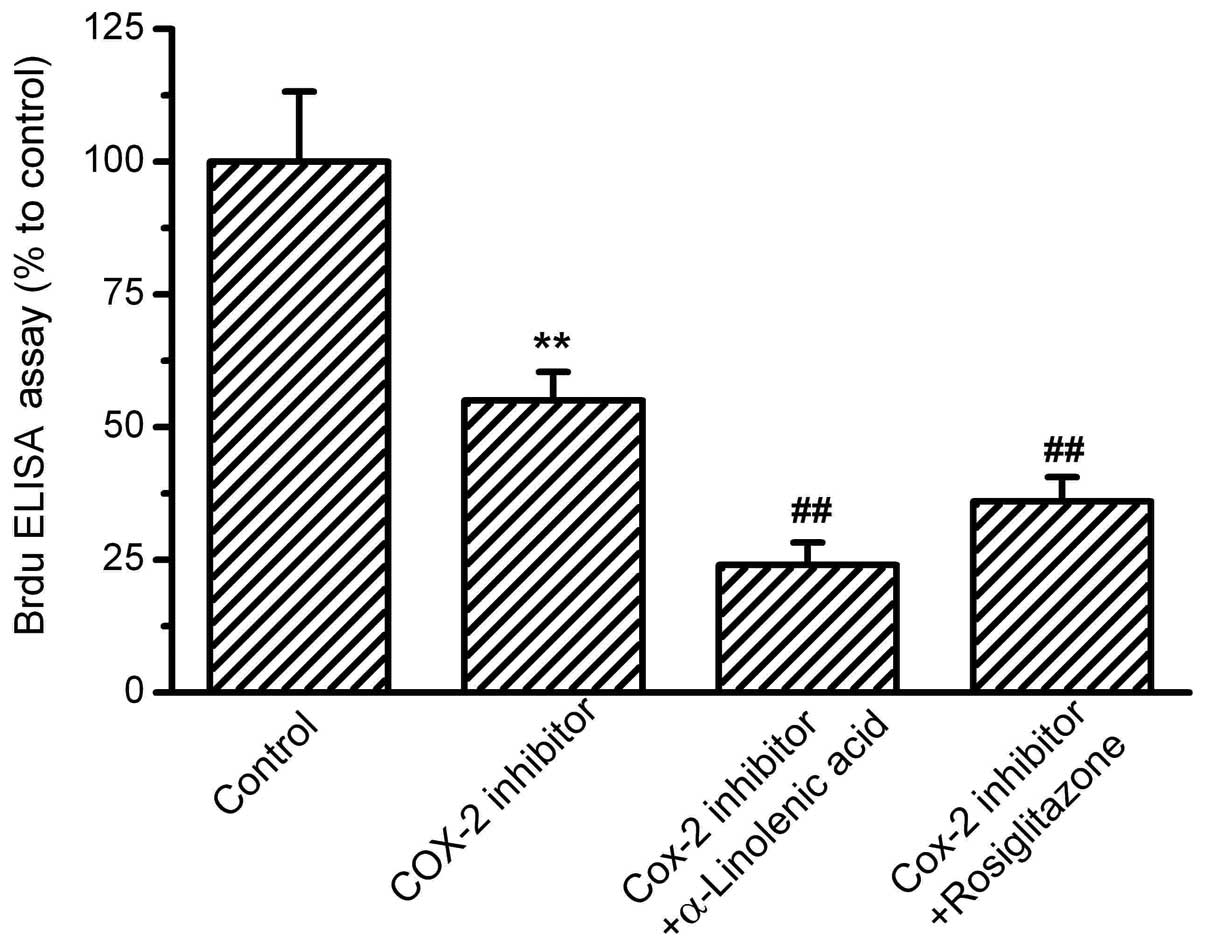
PPAR-γ activation by rosiglitazone (Fig. 4). Next, the effects of COX-2
inhibition on OS-RC-2 cell proliferation were investigated.
N-(3-Pyridyl)indomethacinamide at 10−6 mol/l suppressed
the cell proliferation significantly by ∼60% compared with the
control. In combination with the COX-2 inhibitor, ALA and the
PPAR-γ agonist, rosiglitazone, induced further decreases in
proliferation (Fig. 5).
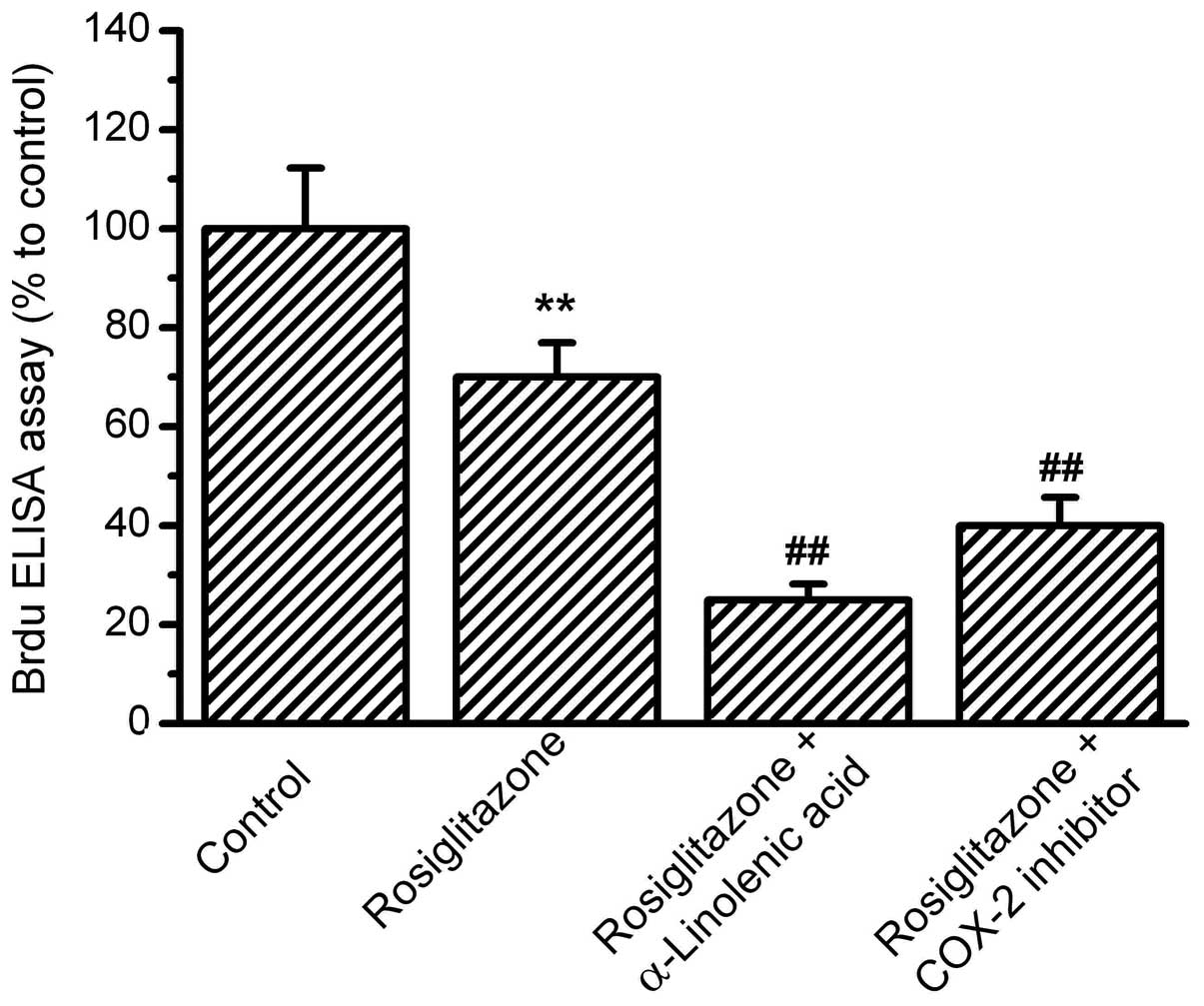 | Figure 4.Effects of PPAR-γ activation on
OS-RC-2 cell proliferation. OS-RC-2 cells were treated with the
agonist of PPAR-γ, rosiglitazone, for 24 h, then treated with
α-linolenic acid (ALA) or the COX-2 inhibitor,
N-(3-Pyridyl)indomethacinamide. BrdU incorporation was measured by
ELISA with absorbance at 450nm. The percentages relative to the
control were analyzed. Rosiglitazone inhibited OS-RC-2
proliferation, and in the presence of rosiglitazone, ALA and COX-2
inhibitors further inhibited OS-RC-2 proliferation.
(**P<0.01 vs. control and ##P<0.01 vs.
rosiglitazone group, n=8). PPAR-γ, peroxisome
proliferator-activated receptor-γ; COX-2, cyclooxygenase-2; ELISA,
enzyme-linked immunosorbent assay. |
Discussion
The present study demonstrated that ALA, an ω-3
fatty acid, inhibited the proliferation of human RCC cells in
vitro, providing direct evidence for the beneficial effect of
ω-3 fatty acids on RCC. A previous study showed that ω-3 fatty
acids reduce the risk of RCC (5),
but no cellular evidence was provided. The present study showed
that ALA treatment significantly inhibited RCC cell proliferation
by the activation of PPAR-γ and the inhibition of COX-2.
The beneficial effects of ω-3 fatty acids in cancer
therapy have been shown in a number of types of cancer. For
example, ω-3 fatty acids have been shown to inhibit the growth of
breast cancer (18,19) and reduce the risk of lung, prostate
and pancreatic cancer (20–23). The present study identified ALA, an
ω-3 fatty acid, as an anticancer agent for RCC.
The present study demonstrated that PPAR-γ
activation is a signaling pathway that is involved in the action of
ALA on OS-RC-2 RCC cells. PPAR-γ is a transcriptional factor that
regulates metabolism and numerous other cell functions (24–26).
Fatty acids are the natural ligands of PPAR-γ, and ALA is able to
activate PPAR-γ (27–29). Therefore, the present study observed
the activity of PPAR-γ in OS-RC-2 cells following ALA treatment. As
expected, PPAR-γ activity and gene expression were markedly
increased by ALA. Furthermore, it was observed that the PPAR-γ
agonist inhibited the proliferation of the OS-RC-2 cells. This is
supported by studies which reported that the PPAR-γ agonist showed
inhibitory effects on cancer growth in certain types of cancers
(10,30–32).
Taken together, it may be suggested that the activation of PPAR-γ
by ALA is one of the signaling pathways for the inhibition of
OS-RC-2 cell proliferation.
In combination with the PPAR-γ agonist, ALA further
inhibited cell proliferation, indicating that other mechanism
besides PPAR-γ were also involved in the action of ALA. The present
study clarified that COX-2 inhibition is another mechanism for the
inhibition of OS-RC-2 cell proliferation by ALA. COX-2 activity and
gene expression were suppressed following ALA treatment. To confirm
the role of COX-2 inhibition in the ALA-inhibited proliferation of
OS-RC-2 cells, the effects of COX-2 inhibitor on the proliferation
of OS-RC-2 cells were investigated. The results showed that the
COX-2 inhibitor significantly inhibited OS-RC-2 cell proliferation.
Therefore, it was demonstrated that COX-2 inhibition led to
decreases in OS-RC-2 cell proliferation. ALA inhibits COX-2
activity, resulting in the inhibition of cell proliferation.
Therefore, COX-2 inhibition is considered to be another signaling
pathway through which ALA inhibits RCC cell proliferation.
The involvement of COX-2 in cancer cell development
has been demonstrated in a number of types of cancer cell (33–35).
For example, it has been reported that COX-2 inhibition led to
decreases in the proliferation of prostate cancer, MDA-MB-435
cancer and PaCa2 pancreatic cancer cells (36–39).
The involvement of COX-2 in RCC has also been suggested. It is
reported that COX-2 is highly activated in RCC tissues (16,40,41).
The present study demonstrated that COX-2 inhibition suppressed the
proliferation of the OS-RC-2 cells, indicating that COX-2
activation is a factor for RCC cell proliferation. Moreover, the
present study showed the inhibition of COX-2 by ALA, an ω-3 fatty
acid. This demonstrated the direct correlation between ω-3 fatty
acid and COX-2.
Therefore, it is suggested that PPAR-γ activation
and COX-2 inhibition serve as two signaling pathways for the
inhibitory effects of ALA in RCC cell proliferation, and that these
two signaling pathways are parallel in the route map. PPAR-γ
activation and COX-2 inhibition have synergistic effects on the
inhibition of OS-RC-2 cell proliferation. In the present study,
following PPAR-γ activation, the COX-2 inhibitor further suppressed
the proliferation of the OS-RC-2 cells in addition to PPAR-γ
activation, suggesting that COX-2 is not a downstream molecule of
PPAR-γ in OS-RC-2 cells. Although it has been suggested that PPAR-γ
regulates COX-2 activity in lung cancer (42,43),
the present results did not support the activation of COX-2 by
PPAR-γ in the OS-RC-2 cells. The discrepancy between the present
and previous studies may be due to tissue-specificity. However, in
combination with the COX-2 inhibitor, PPAR-γ induced further
inhibition of the cell proliferation, indicating that COX-2 is not
the regulator of PPAR-γ action. Therefore, we conclude that PPAR-γ
and COX-2 are two parallel signaling pathways that mediate the
inhibitory effects of ω-3 fatty acid, ALA, on the proliferation of
OS-RC-2 cells.
While ALA increased the activity levels of PPAR-γ at
concentrations of 20 and 40 μM in the present study, high
doses of ALA up to 80 μM inhibited the activity of PPAR-γ.
This inhibition may be due to the inhibition, but not the
activation, of PPAR-γ gene expression by ALA at 80 μM. The
inhibition of gene expression indicates that high doses of ALA have
toxic effects on cell viability. The toxic effects of high doses of
ALA on the OS-RC-2 cells were not clearly observable through its
inhibition of COX-2 expression as the regulated inhibition and
toxic expression overlapped. The inhibition of COX-2 expression by
ALA ≤80 mM observed in the present study may not be purely due to
the regulated inhibitory effects of ALA; the toxic effects of ALA
at high doses may be part of the inhibitory action of ALA. It has
been demonstrated that high concentrations of certain fatty acids
cause cell death via apoptosis or necrosis in numerous types of
cells, such as human melanoma cell lines and pancreatic β-cells
(44–46). The loss of membrane integrity and
the effects on energy metabolism in cells may be two pathways
involved in the toxic action of fatty acids. ALA at high doses may
affect integrity or inhibit the energy metabolism of OS-RC-2 cells.
The exact mechanism of the toxic action of ALA on OS-RC-2 cells
remains to be studied.
In conclusion, the present study demonstrated that
an ω-3 fatty acid, ALA, inhibited the proliferation of OS-RC-2
cells, a type of human RCC cell line. PPAR-γ activation and COX-2
inhibition are two signaling pathways involved in the action of ALA
on OS-RC-2 cells. We suggest that ALA and drugs regulating the
activities of PPAR-γ and COX-2 may be potential targets for RCC
therapy.
Acknowledgements
The present study was supported by
grants from the National Basic Research Program of China
(2009CB521705) and the Natural Scientific Foundation of China
(81172420 and 81172146).
References
|
1.
|
Motzer RJ, Bander NH and Nanus DM:
Renal-cell carcinoma. N Engl J Med. 335:865–875. 1996. View Article : Google Scholar
|
|
2.
|
Rini BI, Rathmell WK and Godley P: Renal
cell carcinoma. Curr Opin Oncol. 20:300–306. 2008. View Article : Google Scholar
|
|
3.
|
Rose DP and Connolly JM: Omega-3 fatty
acids as cancer chemopreventive agents. Pharmacol Ther. 83:217–244.
1999. View Article : Google Scholar : PubMed/NCBI
|
|
4.
|
Rashidkhani B, Akesson A, Lindblad P and
Wolk A: Major dietary patterns and risk of renal cell carcinoma in
a prospective cohort of Swedish women. J Nutr. 135:1757–1762.
2005.PubMed/NCBI
|
|
5.
|
Wolk A, Larsson SC, Johansson JE and Ekman
P: Long-term fatty fish consumption and renal cell carcinoma
incidence in women. JAMA. 296:1371–1376. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
6.
|
Spencer L, Mann C, Metcalfe M, Webb M,
Pollard C, Spencer D, Berry D, Steward W and Dennison A: The effect
of omega-3 FAs on tumour angiogenesis and their therapeutic
potential. Eur J Cancer. 45:2077–2086. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
7.
|
Wendel M and Heller AR: Anticancer actions
of omega-3 fatty acids - current state and future perspectives.
Anticancer Agents Med Chem. 9:457–470. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
8.
|
Desvergne B and Wahli W: Peroxisome
proliferator-activated receptors: Nuclear control of metabolism.
Endocr Rev. 20:649–688. 1999.PubMed/NCBI
|
|
9.
|
Feige JN, Gelman L, Michalik L, Desvergne
B and Wahli W: From molecular action to physiological outputs:
peroxisome proliferator-activated receptors are nuclear receptors
at the crossroads of key cellular functions. Prog Lipid Res.
45:120–159. 2006. View Article : Google Scholar
|
|
10.
|
Krishnan A, Nair SA and Pillai MR: Biology
of PPAR gamma in cancer: a critical review on existing lacunae.
Curr Mol Med. 7:532–540. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
11.
|
Kliewer SA, Sundseth SS, Jones SA, Brown
PJ, Wisely GB, Koble CS, Devchand P, Wahli W, Willson TM, Lenhard
JM and Lehmann JM: Fatty acids and eicosanoids regulate gene
expression through direct interactions with peroxisome
proliferator-activated receptors alpha and gamma. Proc Natl Acad
Sci USA. 94:4318–4323. 1997. View Article : Google Scholar
|
|
12.
|
Xu HE, Lambert MH, Montana VG, Parks DJ,
Blanchard SG, Brown PJ, Sternbach DD, Lehmann JM, Wisely GB,
Willson TM, Kliewer SA and Milburn MV: Molecular recognition of
fatty acids by peroxisome proliferator-activated receptors. Mol
Cell. 3:397–403. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
13.
|
Lim K, Han C, Dai Y, Shen M and Wu T:
Omega-3 polyunsaturated fatty acids inhibit hepatocellular
carcinoma cell growth through blocking beta-catenin and
cyclooxygenase-2. Mol Cancer Ther. 8:3046–3055. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
14.
|
Hashimoto Y, Kondo Y, Kimura G, Matsuzawa
I, Sato S, Ishizaki M, Imura N, Akimoto M and Hara S:
Cyclooxygenase-2 expression and relationship to tumour progression
in human renal cell carcinoma. Histopathology. 44:353–359. 2004.
View Article : Google Scholar : PubMed/NCBI
|
|
15.
|
Khan KN, Stanfield KM, Trajkovic D and
Knapp DW: Expression of cyclooxygenase-2 in canine renal cell
carcinoma. Vet Pathol. 38:116–119. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
16.
|
Chen Q, Shinohara N, Abe T, Watanabe T,
Nonomura K and Koyanagi T: Significance of COX-2 expression in
human renal cell carcinoma cell lines. Int J Cancer. 108:825–832.
2004. View Article : Google Scholar : PubMed/NCBI
|
|
17.
|
Xin X, Yang S, Kowalski J and Gerritsen
ME: Peroxisome proliferator-activated receptor gamma ligands are
potent inhibitors of angiogenesis in vitro and in vivo. J Biol
Chem. 274:9116–9121. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
18.
|
Rose DP and Connolly JM: Effects of fatty
acids and inhibitors of eicosanoid synthesis on the growth of a
human breast cancer cell line in culture. Cancer Res. 50:7139–7144.
1990.PubMed/NCBI
|
|
19.
|
Rose DP, Connolly JM, Rayburn J and
Coleman M: Influence of diets containing eicosapentaenoic or
docosahexaenoic acid on growth and metastasis of breast cancer
cells in nude mice. J Natl Cancer Inst. 87:587–592. 1995.
View Article : Google Scholar : PubMed/NCBI
|
|
20.
|
Karmali RA, Reichel P, Cohen LA, Terano T,
Hirai A, Tamura Y and Yoshida S: The effects of dietary omega-3
fatty acids on the du-145 transplantable human prostatic tumor.
Anticancer Res. 7:1173–1179. 1987.PubMed/NCBI
|
|
21.
|
Prentice RL and Sheppard L: Dietary fat
and cancer: consistency of the epidemiologic data, and disease
prevention that may follow from a practical reduction in fat
consumption. Cancer Causes Control. 1:81–109. 1990. View Article : Google Scholar : PubMed/NCBI
|
|
22.
|
Stolzenberg-Solomon RZ, Pietinen P, Taylor
PR, Virtamo J and Albanes D: Prospective study of diet and
pancreatic cancer in male smokers. Am J Epidemiol. 155:783–792.
2002. View Article : Google Scholar : PubMed/NCBI
|
|
23.
|
Takezaki T, Inoue M, Kataoka H, Ikeda S,
Yoshida M, Ohashi Y, Tajima K and Tominaga S: Diet and lung cancer
risk from a 14-year population-based prospective study in Japan:
with special reference to fish consumption. Nutr Cancer.
45:160–167. 2003.PubMed/NCBI
|
|
24.
|
Savage DB: PPAR gamma as a metabolic
regulator: insights from genomics and pharmacology. Expert Rev Mol
Med. 7:1–16. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
25.
|
Knouff C and Auwerx J: Peroxisome
proliferator-activated receptor-gamma calls for activation in
moderation: lessons from genetics and pharmacology. Endocr Rev.
25:899–918. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
26.
|
Picard F and Auwerx J: PPAR(gamma) and
glucose homeostasis. Annu Rev Nutr. 22:167–197. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
27.
|
Itoh T, Fairall L, Amin K, Inaba Y, Szanto
A, Balint BL, Nagy L, Yamamoto K and Schwabe JW: Structural basis
for the activation of PPARgamma by oxidized fatty acids. Nat Struct
Mol Biol. 15:924–931. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
28.
|
Edwards IJ and O’Flaherty JT: Omega-3
fatty acids and PPARgamma in cancer. PPAR Res. 2008:3580522008.
View Article : Google Scholar : PubMed/NCBI
|
|
29.
|
Gani OA: Are fish oil omega-3 long-chain
fatty acids and their derivatives peroxisome proliferator-activated
receptor agonists? Cardiovasc Diabetol. 7:62008. View Article : Google Scholar : PubMed/NCBI
|
|
30.
|
Mansure JJ, Nassim R and Kassouf W:
Peroxisome proliferator-activated receptor gamma in bladder cancer:
a promising therapeutic target. Cancer Biol Ther. 8:6–15. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
31.
|
Roman J: Peroxisome proliferator-activated
receptor gamma and lung cancer biology: implications for therapy. J
Investig Med. 56:528–533. 2008.PubMed/NCBI
|
|
32.
|
Voutsadakis IA: Peroxisome
proliferator-activated receptor gamma (PPARgamma) and colorectal
carcinogenesis. J Cancer Res Clin Oncol. 133:917–928. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
33.
|
de Souza Pereira R: Selective
cyclooxygenase-2 (COX-2) inhibitors used for preventing or
regressing cancer. Recent Pat Anticancer Drug Discov. 4:157–163.
2009.
|
|
34.
|
Harris RE: Cyclooxygenase-2 (cox-2)
blockade in the chemo-prevention of cancers of the colon, breast,
prostate, and lung. Inflammopharmacology. 17:55–67. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
35.
|
Fujimura T, Ohta T, Oyama K, Miyashita T
and Miwa K: Cyclooxygenase-2 (COX-2) in carcinogenesis and
selective COX-2 inhibitors for chemoprevention in gastrointestinal
cancers. J Gastrointest Cancer. 38:78–82. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
36.
|
Gilhooly EM and Rose DP: The association
between a mutated ras gene and cyclooxygenase-2 expression in human
breast cancer cell lines. Int J Oncol. 15:267–270. 1999.PubMed/NCBI
|
|
37.
|
Hussain T, Gupta S and Mukhtar H:
Cyclooxygenase-2 and prostate carcinogenesis. Cancer Lett.
191:125–135. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
38.
|
Lee KS, Lee HJ, Ahn KS and Kim SH, Nam D,
Kim DK, Choi DY, Ahn KS, Lu J and Kim SH:
Cyclooxygenase-2/prostaglandin e2 pathway mediates icariside II
induced apoptosis in human PC-3 prostate cancer cells. Cancer Lett.
280:93–100. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
39.
|
Sun WH, Chen GS, Ou XL, Yang Y, Luo C,
Zhang Y, Shao Y, Xu HC, Xiao B, Xue YP, Zhou SM, Zhao QS and Ding
GX: Inhibition of COX-2 and activation of peroxisome
proliferator-activated receptor gamma synergistically inhibits
proliferation and induces apoptosis of human pancreatic carcinoma
cells. Cancer Lett. 275:247–255. 2009. View Article : Google Scholar
|
|
40.
|
Chen Q, Shinohara N, Abe T, Harabayashi T
and Nonomura K: Impact of cyclooxygenase-2 gene expression on tumor
invasiveness in a human renal cell carcinoma cell line. J Urol.
172:2153–2157. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
41.
|
Hemmerlein B, Galuschka L, Putzer N,
Zischkau S and Heuser M: Comparative analysis of COX-2, vascular
endothelial growth factor and microvessel density in human renal
cell carcinomas. Histopathology. 45:603–611. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
42.
|
Bren-Mattison Y, Meyer AM, Van Putten V,
Li H, Kuhn K, Stearman R, Weiser-Evans M, Winn RA, Heasley LE and
Nemenoff RA: Antitumorigenic effects of peroxisome
proliferator-activated receptor-gamma in non-small-cell lung cancer
cells are mediated by suppression of cyclooxygenase-2 via
inhibition of nuclear factor-kappaB. Mol Pharmacol. 73:709–717.
2008. View Article : Google Scholar
|
|
43.
|
Chêne G, Dubourdeau M, Balard P,
Escoubet-Lozach L, Orfila C, Berry A, Bernad J, Aries MF, Charveron
M and Pipy B: n-3 and n-6 polyunsaturated fatty acids induce the
expression of COX-2 via PPARgamma activation in human keratinocyte
HaCaT cells. Biochim Biophys Acta. 1771:576–589. 2007.PubMed/NCBI
|
|
44.
|
Andrade LN, de Lima TM, Curi R and
Castrucci AM: Toxicity of fatty acids on murine and human melanoma
cell lines. Toxicol In Vitro. 19:553–560. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
45.
|
Azevedo-Martins AK, Monteiro AP, Lima CL,
Lenzen S and Curi R: Fatty acid-induced toxicity and neutral lipid
accumulation in insulin-producing RINm5F cells. Toxicol In Vitro.
20:1106–1113. 2006. View Article : Google Scholar
|
|
46.
|
Otton R and Curi R: Toxicity of a mixture
of fatty acids on human blood lymphocytes and leukaemia cell lines.
Toxicol In Vitro. 19:749–755. 2005. View Article : Google Scholar : PubMed/NCBI
|