Introduction
Lung cancer is the most common cancer worldwide in
terms of incidence and mortality, and its incidence is rapidly
increasing in developing countries (1–2). The
prognosis of lung cancer remains poor despite recent advances in
chemotherapies and molecular-targeted therapies. The five-year
survival rate of lung cancer is <15%, and ∼90% of mortalities
are caused by metastasis. To improve patient survival, the
elucidation of the regulatory mechanisms that control the tumor
metastatic properties of lung cancer is urgently required.
microRNAs (miRNAs) are small non-coding RNA
molecules that suppress gene expression by interacting with the 3′
untranslated regions (UTRs) of target mRNAs (3). Although miRNAs account for only a
minor fraction of the expressed genome, these RNAs are involved in
modulating a number of cellular pathways, including proliferation
(4), differentiation (5) and apoptosis (6). The deletion or epigenetic silencing of
miRNAs that normally repress the expression of one or more
oncogenes may lead to carcinogenesis (7). Thus, miRNAs have been hypothesized to
function as tumor suppressors or oncogenes, and alterations in
miRNA expression may be critical for tumorigenesis and cancer
progression (8–10). miR-223 is a highly conserved miRNA
and is crucial for triggering the myeloid differentiation of
progenitor cells (11) and for
maintaining granulocyte function (12). Previous studies have reported a
number of significant miR-223 targets associated with malignancy,
including insulin-like growth factor-1 receptor (IGF-1R) (13), myocyte enhancer factor 2C (14), stathmin 1 (15), artemin (16) and FOXO1A (17). miR-223 has been reported to be
markedly downregulated in the lungs of rats exposed to
environmental cigarette smoke for 28 days (18). The reduced serum expression of
miR-223 was found to be associated with cancer-specific mortality
in stage IA/B patients (19). In
our previous study, CXCR4-positive cells from the Lewis lung
carcinoma (LLC) cell line presented with cancer metastatic stem
cell characteristics, and miR-223 expression was reduced compared
with in CXCR4-negative cells (20,21).
However, the mechanism by which miR-223 functions in the
development of NSCLC remains largely unknown and, to date, no
miR-223 targets have been reported in NSCLC. Therefore, in the
present study, the effects of miR-223 transfection in the LLC cell
line were evaluated to determine whether miR-223 functions as a
tumor suppressor of lung cancer.
Materials and methods
Cell line and culture
The LLC cell line was purchased from the American
Type Culture Collection (Manassas, VA, USA). The LLC cells were
cultured in Dulbecco’s modified Eagle’s medium (DMEM) with 10%
fetal calf serum, 100 U/ml streptomycin and 100 U/ml penicillin in
a humidified atmosphere of 95% air and 5% CO2 at
37°C.
Cell transfection
For the functional analysis, miR-223 and
non-targeting miRNA mimics (Thermo Fisher Scientific, Inc.,
Waltham, MA, USA) were used. For the target validation and cell
signaling analysis, the mmu-miRNA-223 expression vector and an
empty pGenesil-1.1 (Wuhan Genesil Biotechnology Co Ltd., Hubei,
China) were used. The mimics were transfected into the appropriate
cells using Lipofectamine 2000 (Invitrogen Life Technologies,
Carlsbad, CA, USA) according to the manufacturer’s
instructions.
Cell survival assays
The effects of miRNA-223 expression on LLC
proliferation were assessed using Cell Counting Kit-8 (CCK-8;
Dojindo Laboratories, Kumamoto, Japan). Briefly, the cells were
plated on 96-well plates. Following transfection, CCK-8 was added
to each well at various times and incubated at 37°C for 1.5 h. The
absorbance at 450 nM was measured using a microplate
spectrophotometer (Tecan Group Ltd, Männedorf, Switzerland).
Anchorage-independent growth ability
assay
In total, 500 cells were trypsinized and suspended
in 2 ml complete medium with 0.3% agar (Sigma-Aldrich, St Louis,
MO, USA). The agar-cell mixture was plated on top of a bottom layer
with 1% complete medium agar mixture. Subsequent to 10 days, the
viable colonies containing >50 cells or those >0.1 mm in size
were counted. The colony size was measured using an ocular
micrometer.
Invasion assay
The LLC measurements were performed in 24-well
matrigel-coated invasion chambers. The lower chambers contained 600
μl DMEM with 10% fetal bovine serum (FBS) as a
chemoattractant. At 24 h post-transfection with miRNA-223 or
non-targeting miRNA mimics, a cell suspension of 5×104
cells in 100 μl DMEM with 0.5% FBS was added to the upper
chamber. Following incubation for 24 h at 37°C in a humidified
incubator with 5% CO2, the invasive cells that had
attached to the lower surface of the membrane insert were fixed by
4% formaldehyde and stained with crystal violet. The number of
cells was then quantified under a microscope.
Wound healing assay
For the wound healing assay, the LLC cells were
grown to confluence on 6-well plates. Next, linear scratch wounds
were created on the confluent monolayer using a pipette tip and the
cells were transfected with 50 nM miR-223 mimics or 50 nM
non-targeting miRNA mimics (control). Floating cells were removed
by gentle washes with culture medium. The healing process was
examined dynamically and was recorded with a digital camera 24 h
after wound generation.
Matrix metallopeptidase (MMP)9
enzyme-linked immunosorbent assay (ELISA)
Following transfection with miRNA-223 or
non-targeting miRNA mimics, MMP9 protein levels in the culture
supernatants were quantified using an ELISA kit according to the
manufacturer’s instructions (R&D Systems, Minneapolis, MN,
USA). Plates were read at 450 nM using a Synergy HT microplate
reader (BioTek Instruments, Inc., Winooski, VT, USA) and the MMP9
concentration in the samples was calculated using a standard
curve.
Flow cytometric analysis
At 48 h post-transfection with miRNA-223 or
non-targeting miRNA mimics, dissociated LLC cells were stained with
PE-conjugated (PE) rat anti-mouse Sca-1 and the corresponding
isotype controls (1:100; BD Pharmingen, San Diago, CA, USA).
Briefly, the cells were trypsinized; the trypsin was neutralized
with culture medium containing adult bovine serum (ABS) and
centrifuged at 450 × g for 5 min. The cell pellet was re-suspended
in Hank’s buffered salt solution (HBSS) containing 2% ABS
(HBSS+). The cells were incubated with primary
antibodies for 20 min on ice, washed twice with HBSS+
and re-suspended with HBSS+ containing Sytox Blue or
Sytox Red (Invitrogen Life Technologies) to exclude any dead cells.
The cell suspensions were filtered using 40-μm filters and
analyzed on a BD LSRII Fortessa (both BD Biosciences, Franklin
Lakes, NJ, USA). In addition, a cell cycle analysis was performed
as described previously (22).
In vivo tumorigenicity
Female C57BL/6 mice (6–8 weeks old) were obtained
from The Animal Facility of The Third Military Medical University
(Chongqing, China). In vivo experiments were performed in
accordance with institutional guidelines. This study was approved
by the ethics committee of Chongqing Tumor Hospital, Chongqing,
China. Two groups of mice were tested. Group 1 (223-mimic) was
injected with LLC cells transfected with miR-223 mimics and group 2
(mimic-control) was injected with LLC cells transfected with
non-targeting miRNA mimics. The mice were sacrificed at four weeks,
and the tumor volume was calculated using the following formula: [L
× (W)2] / 2, where L is the length and W is the width of
the tumor. For the immunofluorescence labeling, the cells were
blocked with 15% bovine serum albumin (BSA) for 1 h and then
incubated at 37°C for 1 h in PE-conjugated rat anti-mouse Sca-1
(1:100). Following this, the cells were washed with 0.01% PBS and
then stained with 4,6-diamidino-2-phenylindole (Sigma-Aldrich) to
identify the cell nuclei. The cells were then observed under a
fluorescence microscope.
Luciferase reporter plasmid construction
and luciferase assay
The pMIR-REPORT miRNA expression reporter (firefly
luciferase reporter plasmid; Life Technologies, Grand Island, NY,
USA) was used for the plasmid construction. Constructs were
generated using the following primers: IGF-1R 3′-UTR-1,
5′-GGACTAGTAGGGGAGAGCAGGTTG TAACAATCT-3′ and
5′-CGACGCGTGACCTACGGTGTC AGGCAGGTGTAT-3′; IGF-1R 3′-UTR-2,
5′-GGACTAGTC AGTACCTGACAGTAGGCCAATGAT-3′ and 5′-CGACGC
GTAAGATTTGGTCAGTCCTTGTTTAGC-3′; cyclin-dependent kinase (CDK)2
3′-UTR: 5′-GGACTAGTAGCCTTCTGA TGTTTTCTGGCTGTC-3′ and
5′-CGACGCGTGATGAAC AGACCAGAGTGACGTGCA-3′. The 3′-UTR and miR-223
complementary sequence (TGGGGTATTTGACAAACT GACA) were separately
cloned into the pMIR-REPORT plasmid (Life Technologies), according
to the manufacturer’s instructions. Constructs (0.05 μg
each) were cotransfected into 293T with 0.01 μg a Renilla
luciferase control vector using calcium phosphate transfection.
Luciferase activity was measured 36 h after transfection and
normalized against Renilla activity, according to the
manufacturer’s instructions (Dual-Luciferase Reporter Assay System;
Promega Corporation, Madison, WI, USA).
Quantitative (q)PCR
To determine the gene expression levels, qPCR was
performed using the Quantitect SYBR PCR kit (Qiagen, Hilden,
Germany), according to the manufacturer’s instructions. The primers
selected were as follows: IGF-1R forward,
5′-AAGCCGATGTGTGAGAAGACC-3′ and reverse, 5′-ATAGTAGTAGTAGTG
GCGGCAAGC-3′; CDK2 forward, 5′-TTCATGGATGCCTCTGCTCTC-3′ and
reverse, 5′-TCC AAAAGCTCTGGCTAGTCC-3′; MMP9 forward, 5′-GTG
GAGAGTCGAAATCTCTGG-3′ and reverse, 5′-TTTGGA ATCTGCCCAGGTCTG-3′;
GAPDH forward, 5′-TGGTAT CGTGGAAGGACTCATGAC-3′ and reverse,
5′-ATGCCA GTGAGCTTCCCGTTCAGC-3′. ΔΔCT values were
normalized against those obtained from the amplification of GAPDH.
All reactions were performed in triplicate.
Western blot analysis
Transfected cells in culture were harvested at
various times, washed once with cold PBS and lysed in buffer
containing protease inhibitors. The protein concentrations from
whole cultured cells were measured with the bicinchoninic acid
protein assay kit (Beyotime Institute of Biotechnology, Shanghai,
China), using BSA as the standard. Protein (30 μg) was
separated by SDS-PAGE using a 10% polyacrylamide gel and then
electroblotted onto a nitrocellulose membrane. The membrane was
immunoblotted overnight at 4°C with the primary antibodies. The
following antibodies were used at a 1:1,000 dilution: anti-IGF-IR,
anti-mmp9 (Santa Cruz Technologies, Santa Cruz, CA, USA),
anti-phospho-IGFIR, anti-phospho-Akt, anti-Akt, anti-p44/42 MAPK
(Erk1/2), anti-phospho-Erk1/2 and anti-CDK2 (Cell Signaling
Technology, Beverly, MA, USA). A goat anti-rabbit horseradish
peroxiadse (HRP) secondary antibody (Wuhan Boster Bio-Engineering
Co, Ltd, Wuhan, China) was used at a 1:2,000 dilution. The proteins
were detected using an enhanced chemiluminescence kit (Pierce
Biotechnology, Inc., Rockford, IL, USA), according to the
manufacturer’s instructions. GAPDH was used as an internal
control.
Statistical analysis
Data are presented as the mean ± SD and analyzed
using SPSS 16.0 (SPSS, Inc., Chicago, IL, USA). To assess the
statistical significance of the differences, an unpaired t-test was
performed. P<0.05 was considered to indicate a statistically
significant difference.
Results
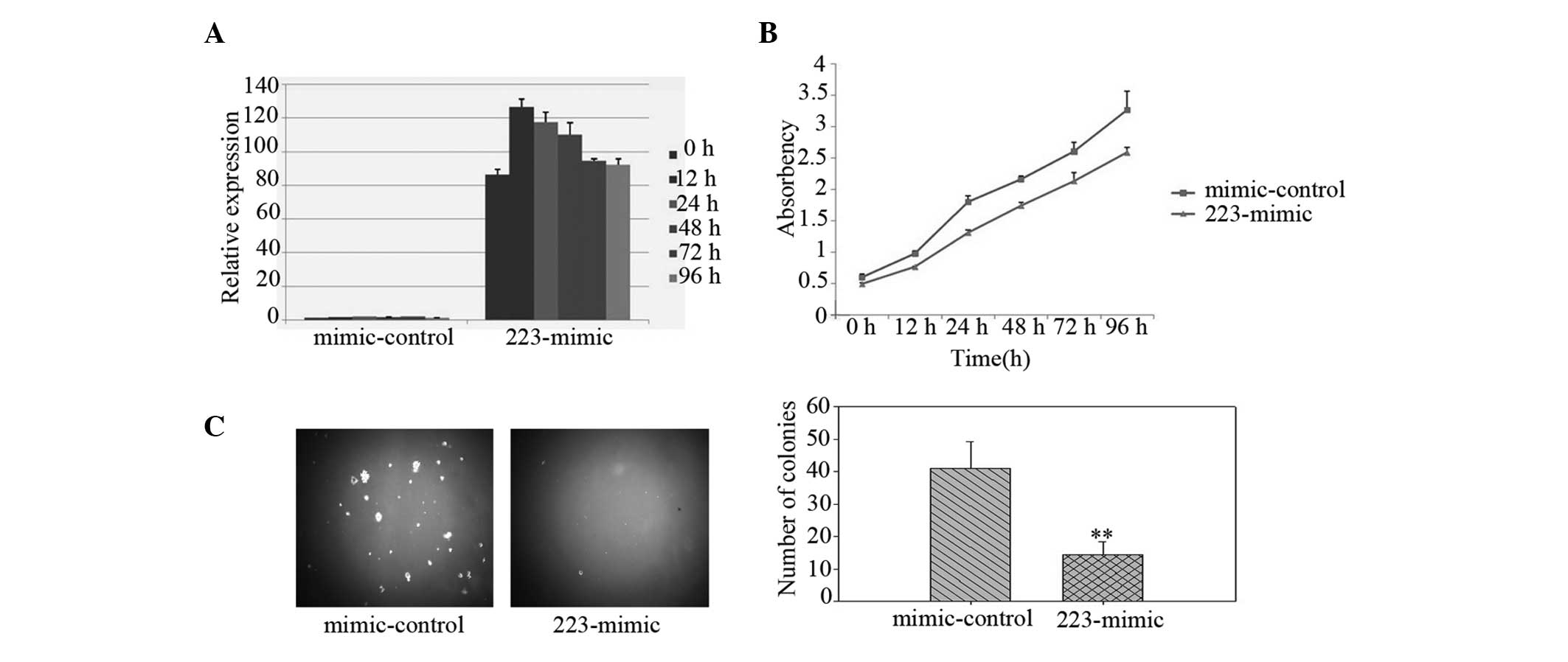
Upregulation of miRNA-223 suppresses
proliferation and tumorigenicity of LLC cells
To investigate the biological role of miR-223
expression in the development and progression of lung cancer, LLC
cells were transfected with miR-223 mimics and the effect on
cellular proliferation was assessed. Following transfection, the
miR-223 levels were increased in the LLC cells, indicating that the
increase was due to miR-223 transfection (Fig. 1A). Using a CCK-8 assay, the
overexpression of miR-223 (223-mimic) was observed to markedly
reduce the growth rate of the LLC cells compared with that of the
non-targeting miRNA mimic-transfected cells (mimic-control;
Fig. 1B). Notably, the LLC cells
ectopically expressing miR-223 were identified to exhibit a
significantly inhibited anchorage-independent growth ability, as
demonstrated by the decrease in colony numbers and sizes (Fig. 1C), indicating that upregulation of
miR-223 reduces the tumorigenicity of lung cancer cells in
vitro.
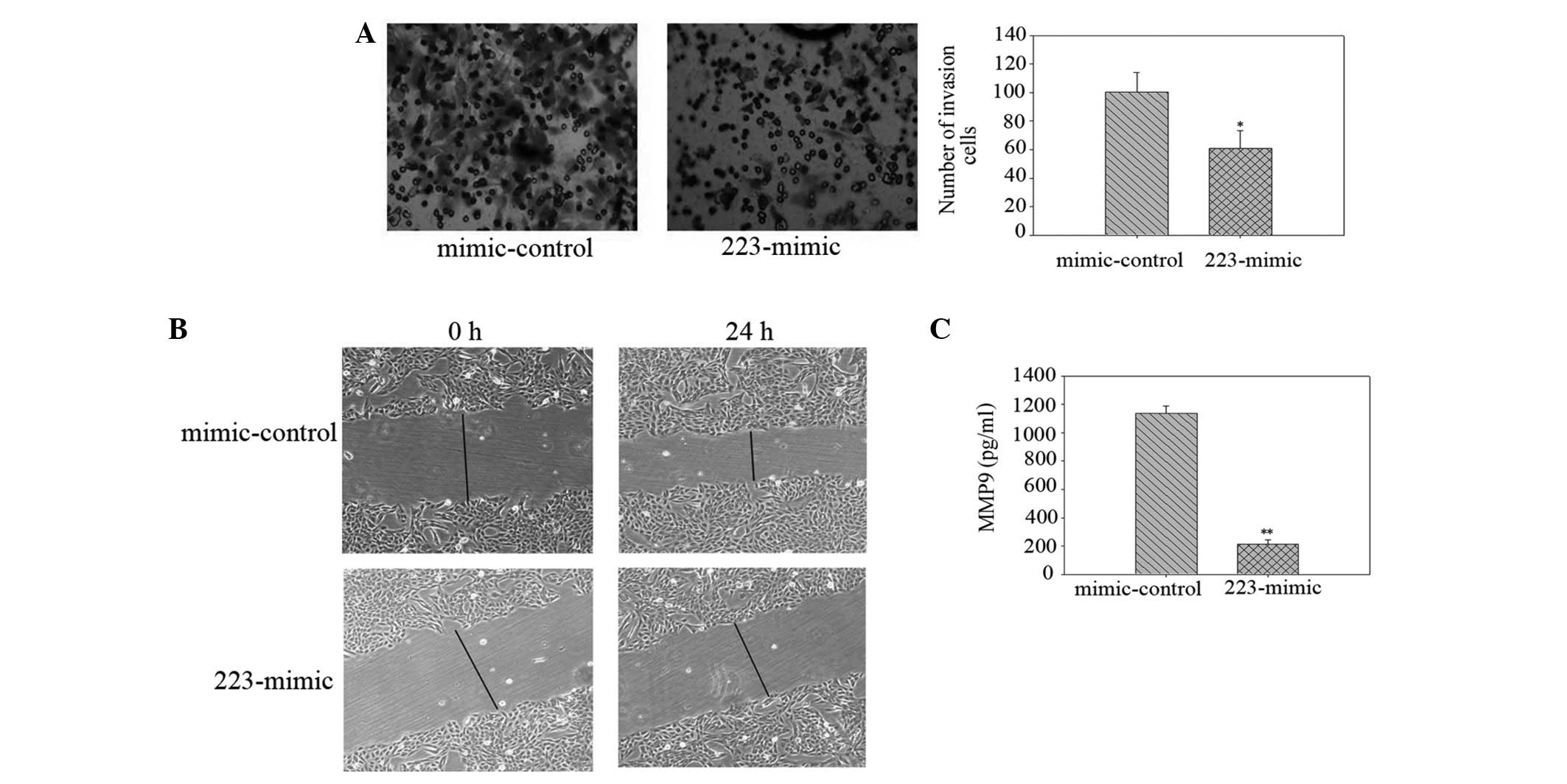
Ectopic expression of miRNA-223 inhibits
LLC invasion
To examine invasion, the LLC cells were transfected
with miR-223 or control mimics and reseeded on top of the insert.
Subsequent to 48 h, the number of transmembrane cells in the
223-mimic group (60.67±12.66) was lower than that of the
mimic-control group (100.33±14.01; P<0.05; Fig. 2A). Next, the LLC cells were
transfected as described, scratch wounds were generated and cell
migration towards the wound was visualised. The wound healing assay
revealed that miR-223 reduced the motility of the LLC cells
(Fig. 2B). To determine whether the
increased invasion observed was correlated with concomitant changes
in MMP levels, the total active MMP9 protein levels were measured
by ELISA in the cultured media. The MMP9 levels in the supernatant
of miR-223-overexpressing cells (214.16±28.18 pg/ml) were reduced
compared with that of the mimic-controls (1,139.14±50.13 pg/ml;
P<0.01; Fig. 2C). These
observations indicated that miR-223 inhibits invasion in LLC
cells.
Overexpression of miR-223 in LLC cells
induces G2/M phase arrest and reduces Sca-1 protein
expression
Propidium iodide staining of miR-223-overexpressing
LLC cells revealed an increase in the G2/M cell
populations (13.3±0.85 vs. .30±0.46%) and a decrease in cells in
the G0/G1 phase populations (45.00±1.11 vs.
54.46±0.85%) compared with the mimic-control (P<0.01; Fig. 3A), indicating a block in the
G2/M phase transition of the cell cycle. Notably, the
percentage of Sca-1-positive cells (Sca-1 is a well-known marker in
murine stem cells) was reduced from 39.25±2.36 to 17.47±2.70% in
the miR-223-overexpressing group (P<0.01; Fig. 3B).
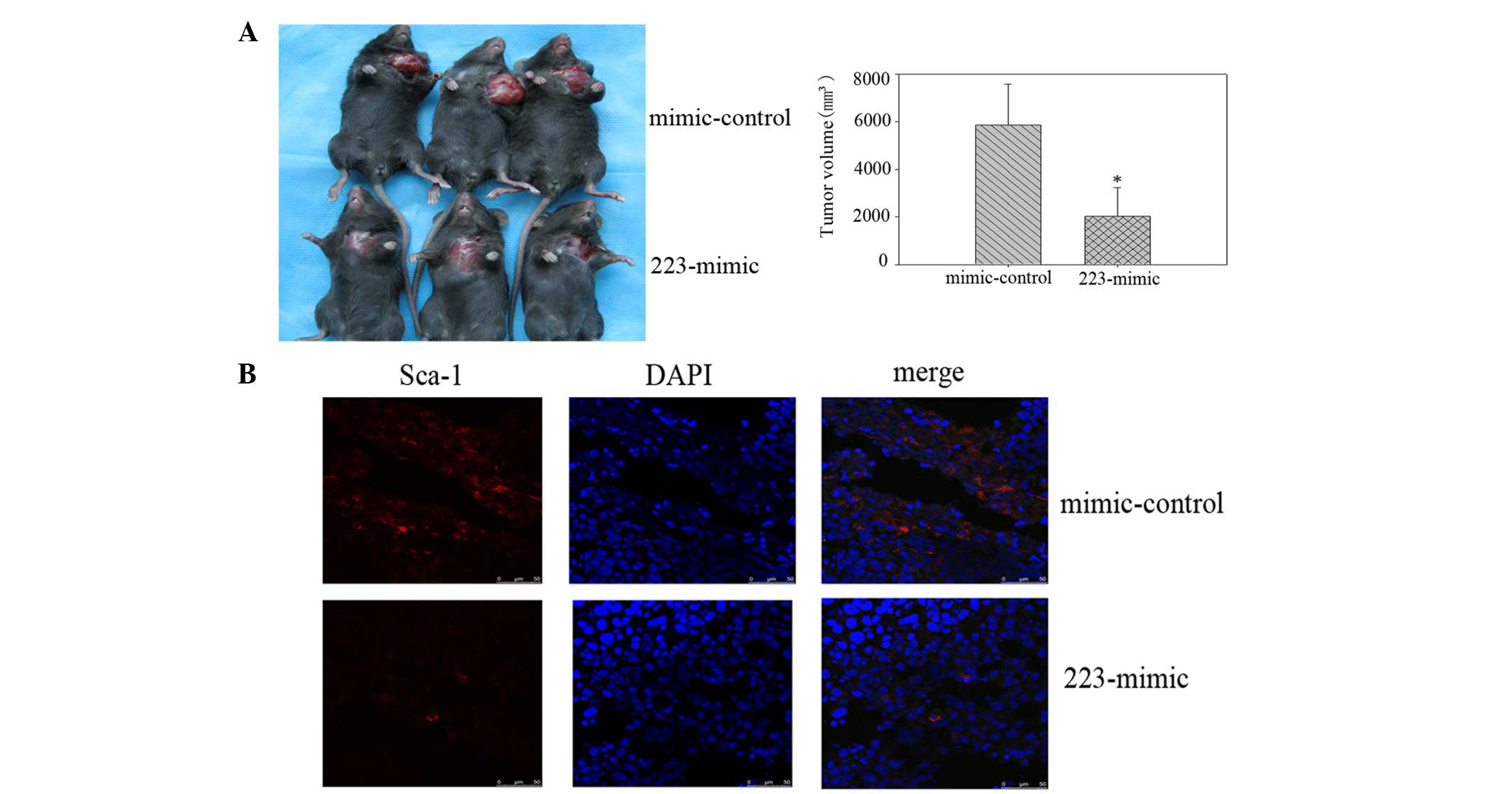
miR-223 expression in LLC inhibits tumor
growth in vivo
At four weeks post-injection, the mice administered
with miR-223 mimics had formed markedly smaller tumors than the
mimic-control group (Fig. 4A). The
tumor volume subsequent to sacrifice in mice injected with miR-223
mimic-transfected cells was 2,034.30±983.99 mm3, whereas
the tumor volume in mice injected with the control
mimic-transfected cells was 5,860.20±692.58 mm3. The
Sca-1 expression in the tumor tissue from the miR-223
mimic-transfected cells was significantly lower than that of the
control mimic-transfected cells, as demonstrated by
immunofluorescence staining (Fig.
4B).
miR-223 regulates IGF-1R and CDK2 levels
by binding to the 3′UTR
Bioinformatic analyses revealed that the IGF-1R
3′UTR contained two putative miR-223 binding sites for miR-223 and
CDK2 contained one putative miR-223 binding site for miR-223
(Fig. 5A). The luciferase
activities of the IGF-1R 3′UTR, CDK2 3′UTR and miR-223
complementary sequence-containing constructs were found to be
further repressed in cells overexpressing miR-223 (Fig. 5B). Next, to examine whether miR-223
affects IGF-1R and CDK2 expression in LLC, the mRNA and protein
expression levels of IGF-1R and CDK2 were analyzed using qPCR and
western blot analysis. Subsequent to 48 h, the expression of IGF-1R
and CDK2 mRNA in the miR-223-expressing group was reduced by ∼17-
and 25-fold of the control vector group, respectively (Fig. 5C). miR-223 also caused a significant
reduction in the IGF-1R and CDK2 protein levels (Fig. 5D). These results indicate that
IGF-1R and CDK-2 are post-transcriptionally regulated by miR-223 in
LLC cells.
Two pathways have been described for IGF-IR, the
phosphatidylinositol-3 kinase-Akt and mitogen-activated protein
kinase pathways (also known as ERKs) (23). To determine the consequences of the
interference of IGF-1R expression by miR-223, the expression of
IGF-1R, Akt, ERK and their active forms (p-IGF-1R, p-Akt and p-ERK)
was measured. The expression of IGF-1R, p-IGF-1R, p-Akt and p-ERK
was reduced, however, the total Akt and ERK levels were unaffected
(Fig. 5D). The downregulation of
MMP9 expression at the mRNA and protein levels was further
supported by qPCR and western blot analysis (Fig. 5C and D). These results indicate that
the IGF-1R-mediated downstream signaling pathway was also affected
by miR-223.
Discussion
The current study is the first study to demonstrate
that miR-223 may be involved in lung cancer stem cell self-renewal
and to show that miR-223 functions as a tumor suppressor in lung
cancer cells at multiple steps of tumorigenesis and progression. In
addition, IGF-1R and CDK2 were demonstrated to represent two
significant targets for miR-223, crucial for mediating the
miR-223-regulated malignant phenotype of LLC cells.
In the present study, IGF-1R expression and its
phosphorylation levels in LLC were investigated and found to be
markedly reduced, while cell proliferation was inhibited, following
miR-223 overexpression. In addition, analyses using a IGF-1R 3′UTR
reporter revealed a significant decrease in luciferase activity. In
conclusion, these observations demonstrate that IGF-1R is the
functional target of miR-223. IGF-1R is a transmembrane receptor
tyrosine kinase encoded by a gene located on chromosome 15q26.3.
IGF-1R is implicated in the promotion of oncogenic transformation,
growth and the survival of cancer cells (24,25).
It has been previously reported that IGF signaling mediates the
transformation of normal lung cells and is involved in tumor
initiation (26,27). In addition, a number of studies in
human lung cancer cell lines have demonstrated that the
downregulation of IGF-IR inhibits lung tumor cell proliferation and
sensitizes lung cancer cells to chemotherapy and radiotherapy
(28,29).
miR-223 has also been found to also target the CDK2
3′UTR. CDK2 is an S-phase cyclin-dependent kinase that is required
for p53-independent G2/M checkpoint control. The
inhibition of cyclin A/cdk2 activation contributes to the
maintenance of G2 phase arrest in response to DNA damage
(30,31). In human leukemia cells, inhibitors
of ERK have been reported to increase the phosphorylation of cdc25c
expression at the G2/M arrest stages and decrease p21
and CDK2 expression at the endoreduplication stages (22). Previous studies have demonstrated
that the cyclolignan, picropodophyllin, downregulates IGF-1R
tyrosine kinase activity and induces a marked accumulation of cells
in the G2/M-phase, as well as increased apoptosis
(32). These observations, together
with the results of the present study, are consistent with a model
in which miR-223 induces G2/M arrest via the
downregulation of IGF-1R and CDK2 by co-targeting their 3′UTR
regions.
The present study primarily focused on whether the
IGF-1R-mediated downstream signaling pathway is affected by
miR-223. The ERK and PI3K/Akt signaling pathways are central to the
regulation of MMP9 expression (33,34).
The present results indicated that miR-223-decreased MMP9 activity
was mediated by the suppression of phospho-ERK1/2 or phospho-Akt.
In addition, the forced expression of miR-223 was found to
downregulate Sca-1 in the LLC cells, as well as a simultaneous
decrease of the cells in the G0/G1 phase and
the induction of the inhibition of anchorage-independent growth.
Sca-1, or Ly6A, is a member of the Ly6 family of glycosyl
phostidylinositol-anchored cell surface proteins that is associated
with murine stem/progenitor cells (35,36).
The MAPK/ERK pathway is essential for Sca-1+ hepatic
progenitor cell proliferation and colony formation (37). In addition, IGF-1 stimulation
directly induces anoikis resistance of a number of varying
epithelial cell types by activating downstream signaling molecules,
including Ras/MAPK and PI3K/Akt (38). In a colon cancer model, the kinase
activities of Akt and ERK1/2 were shown to be significantly
upregulated in CD133+ cells (39). The clonogenic growth of the
CD133+ cells was reduced markedly by inhibiting the
activity of AKT and ERK1/2. In agreement with these results, we
hypothesize that miR-223 induces an aberrant self-renewal capacity
in LLC cells, at least in part, via the inhibition of AKT and ERK
activity. Future studies must be performed to verify this
hypothesis and identify the underlying mechanisms.
In the current study, miR-223 was revealed to
function as a tumor suppressor in lung cancer. The results also
indicate that miR-223 may fine-tune the activity of the IGF-1R
pathway. These observations may provide a basis for novel therapies
targeting IGF-1R in the treatment of NSCLC.
Acknowledgements
The present study was supported, in
part, by grants from the National Natural Science Foundation of
China (no. 30901790) and the Chongqing Natural Science Foundation
(no. 2008BB5117).
References
|
1.
|
Jemal A, Center MM, DeSantis C and Ward
EM: Global patterns of cancer incidence and mortality rates and
trends. Cancer Epidemiol Biomarkers Prev. 19:1893–1907. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
2.
|
Jemal A, Bray F, Center MM, Ferlay J, Ward
E and Forman D: Global cancer statistics. CA Cancer J Clin.
61:69–90. 2011. View Article : Google Scholar
|
|
3.
|
Bartel DP: MicroRNAs: genomics,
biogenesis, mechanism and function. Cell. 116:281–297. 2004.
View Article : Google Scholar : PubMed/NCBI
|
|
4.
|
Bueno MJ, Pérez de Castro I and Malumbres
M: Control of cell proliferation pathways by microRNAs. Cell Cycle.
7:3143–3148. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
5.
|
Lee CT, Risom T and Strauss WM: MicroRNAs
in mammalian development. Birth Defects Res C Embryo Today.
78:129–139. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
6.
|
Jovanovic M and Hengartner MO: miRNAs and
apoptosis: RNAs to die for. Oncogene. 25:6176–6187. 2006.
View Article : Google Scholar : PubMed/NCBI
|
|
7.
|
Mendell JT: miRiad roles for the miR-17-92
cluster in development and disease. Cell. 133:217–222. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
8.
|
Esquela-Kerscher A and Slack FJ: Oncomirs
- microRNAs with a role in cancer. Nat Rev Cancer. 6:259–269. 2006.
View Article : Google Scholar
|
|
9.
|
Kent OA and Mendell JT: A small piece in
the cancer puzzle: microRNAs as tumor suppressors and oncogenes.
Oncogene. 25:6188–6196. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
10.
|
Kumar MS, Erkeland SJ and Pester RE:
Suppression of non-small cell lung tumor development by the let-7
microRNA family. Proc Natl Acad Sci USA. 105:3903–3908. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
11.
|
Fazi F, Rosa A, Fatica A, Gelmetti V, De
Marchis ML, Nervi C and Bozzoni I: Aminicircuitry comprised of
microRNA-223 and transcription factors NFI-A and C/EBPalpha
regulates human granulopoiesis. Cell. 123:819–831. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
12.
|
Johnnidis JB, Harris MH, Wheeler RT,
Stehling-Sun S, Lam MH and Kirak O: Regulation of progenitor cell
proliferation and granulocyte function by microRNA-223. Nature.
451:1125–1129. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
13.
|
Jia CY, Li HH, Zhu XC, Dong YW, Fu D, Zhao
QL, Wu W and Wu XZ: MiR-223 suppresses cell proliferation by
targeting IGF-1R. Plos One. 6:e270082011. View Article : Google Scholar : PubMed/NCBI
|
|
14.
|
Liu Q, Zhang M, Jiang X, Zhang Z, Dai L,
Min S, Wu X, He Q, Liu J, Zhang Y, Zhang Z and Yang R: miR-223
suppresses differentiation of tumor-induced CD11b+ Gr1+
myeloid-derived suppressor cells from bone marrow cells. Int J
Cancer. 129:2662–2673. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
15.
|
Kang W, Tong JH, Chan AW, Lung RW, Chau
SL, Wong QW, Wong N, Yu J, Cheng AS and To KF: Stathmin1 plays
oncogenic role and is a target of microRNA-223 in gastric cancer.
PLoS One. 7:e339192012. View Article : Google Scholar : PubMed/NCBI
|
|
16.
|
Li S, Li Z, Guo F, Qin X, Liu B, Lei Z,
Song Z, Sun L, Zhang HT, You J and Zhou Q: miR-223 regulates
migration and invasion by targeting Artemin in human esophageal
carcinoma. J Biomed Sci. 18:242011. View Article : Google Scholar : PubMed/NCBI
|
|
17.
|
Wu L, Li H, Jia CY, Cheng W, Yu M, Peng M,
Zhu Y, Zhao Q, Dong YW, Shao K, Wu A and Wu XZ: MicroRNA-223
regulates FOXO1 expression and cell proliferation. FEBS Lett.
586:1038–1043. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
18.
|
Izzotti A, Calin GA, Arrigo P, Steele VE,
Croce CM and De Flora S: Downregulation of microRNA expression in
the lungs of rats exposed to cigarette smoke. FASEB J. 23:806–812.
2009. View Article : Google Scholar : PubMed/NCBI
|
|
19.
|
Heegaard NH, Schetter AJ, Welsh JA, Yoneda
M, Bowman ED and Harris CC: Circulating micro-RNA expression
profiles in early stage nonsmall cell lung cancer. Int J Cancer.
130:1378–1386. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
20.
|
Nian WQ, Chen FL, Ao XJ and Chen ZT: CXCR4
positive cells from Lewis lung carcinoma cell line have cancer
metastatic stem cell characteristics. Mol Cell Biochem.
355:241–248. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
21.
|
Nian WQ, Chen FL, Ao XJ and Chen ZT: Lowly
expression of miR-223 in CXCR4 positive cells from Lewis lung
carcinoma cell line and its target gene prediction. Di San Jun Yi
Da Xue Xue Bao. 31:2202–2205. 2009.(In Chinese).
|
|
22.
|
Moon DO, Kim MO, Kang SH, Lee KJ, Heo MS,
Choi KS, Choi YH and Kim GY: Induction of G2/M arrest,
endoreduplication and apoptosis by actin depolymerization agent
pextenotoxin-2 in human leukemia cells, involving activation of ERK
and JNK. Biochem Pharmacol. 76:312–321. 2008.
|
|
23.
|
Dufourny B, Alblas J and van Teeffelen HA:
Mitogenic signaling of insulin-like growth factor I in MCF-7 human
breast cancer cells requires phosphatidylinositol 3-kinase and is
independent of mitogen-activated protein kinase. J Biol Chem.
272:31163–31171. 1997. View Article : Google Scholar
|
|
24.
|
Khandwala HM, McCutcheon IE and Flyvbjerg
A: The effects of insulin-like growth factors on tumorigenesis and
neoplastic growth. Endocr Rev. 21:215–244. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
25.
|
Blakesley VA, Stannard BS and Kalebic T:
Role of the IGF-I receptor in mutagenesis and tumor promotion. J
Endocrinol. 152:339–344. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
26.
|
Moats-Staats BM, Price WA, Xu L, Jarvis HW
and Stiles AD: Regulation of the insulin-like growth factor system
during normal rat lung development. Am J Respir Cell Mol Biol.
12:56–64. 1995. View Article : Google Scholar : PubMed/NCBI
|
|
27.
|
Linnerth NM, Siwicky MD, Campbell CI,
Watson KL, Petrik JJ, Whitsett JA and Moorehead RA: Type I
insulin-like growth factor receptor induces pulmonary
tumorigenesis. Neoplasia. 11:672–682. 2009.PubMed/NCBI
|
|
28.
|
Goetsch L, Gonzalez A, Leger O, Beck A,
Pauwels PJ, Haeuw JF and Corvaia N: A recombinant humanized
anti-insulin-like growth factor receptor type I antibody (h7C10)
enhances the antitumor activity of vinorelbine and anti-epidermal
growth factor receptor therapy against human cancer xenografts. Int
J Cancer. 113:316–328. 2005. View Article : Google Scholar
|
|
29.
|
Cosaceanu D, Carapancea M, Castro J,
Ekedahl J, Kanter L, Lewensohn R and Dricu A: Modulation of
response to radiation of human lung cancer cells following
insulin-like growth factor 1 receptor inactivation. Cancer Lett.
222:173–181. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
30.
|
Chung JH and Bunz F: Cdk2 Is required for
p53-independent G2/M checkpoint control. PLoS Genet.
6:e10008632010. View Article : Google Scholar : PubMed/NCBI
|
|
31.
|
Goldstone S, Pavey S, Forrest A, Sinnamon
J and Gabrielli B: Cdc25-dependent activation of cyclin A/cdk2 is
blocked in G2 phase arrested cells independently of
ATM/ATR. Oncogene. 209:921–932. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
32.
|
Strömberg T, Ekman S, Girnita L, Dimberg
LY, Larsson O, Axelson M, Lennartsson J, Hellman U, Carlson K,
Osterborg A, Vanderkerken K, Nilsson K and Jernberg-Wiklund H:
IGF-1 receptor tyrosine kinase inhibition by the cyclolignan PPP
induces G2/M-phase accumulation and apoptosis in
multiple myeloma cells. Blood. 107:669–678. 2006.PubMed/NCBI
|
|
33.
|
Lin CC, Kuo CT, Cheng CY, Wu CY, Lee CW,
Hsieh HL, Lee IT and Yang CM: IL-1β promotes A549 cell migration
via MAPKs/AP-1- and NF-κB-dependent matrix metalloproteinase-9
expression. Cell Signal. 21:1652–1662. 2009.
|
|
34.
|
Ellerbroek SM, Halbleib JM, Benavidez M,
Warmka JK, Wattenberg EV, Stack MS and Hudson LG:
Phosphatidylinositol 3-kinase activity in epidermal growth
factor-stimulated matrix metalloproteinase-9 production and cell
surface association. Cancer Res. 61:1855–1861. 2001.
|
|
35.
|
Wu X, Pang L, Lei W, Lu W, Li J, Li Z,
Frassica FJ, Chen X, Wan M and Cao X: Inhibition of Sca-1-positive
skeletal stem cell recruitment by alendronate blunts the anabolic
effects of parathyroid hormone on bone remodeling. Cell Stem Cell.
7:571–580. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
36.
|
Lu G, Haider HK, Jiang S and Ashraf M:
Sca-1+ stem cell survival and engraftment in the
infarcted heart: dual role for preconditioning induced connexin-43.
Circulation. 119:2587–2596. 2009.
|
|
37.
|
Jin C, Samuelson L, Cui CB, Sun Y and
Gerber DA: MAPK/ERK and Wnt/β-catenin pathways are synergistically
involved in proliferation of Sca-1 positive hepatic progenitor
cells. Biochem Biophys Res Commun. 409:803–807. 2011.
|
|
38.
|
Valentinis B, Morrione A, Peruzzi F,
Prisco M and Reiss K: Antiapoptotic signaling of the IGF-I receptor
in fibroblasts following loss of matrix adhesion. Oncogene.
18:1827–1836. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
39.
|
Wang YK, Zhu YL, Qiu FM, Zhang T, Chen ZG,
Zheng S and Huang J: Activation of Akt and MAPK pathways enhances
the tumorigenicity of CD133+ primary colon cancer cells.
Carcinogenesis. 31:1376–1380. 2010. View Article : Google Scholar : PubMed/NCBI
|