Introduction
Characterized by spontaneous regression, maturation
or aggressive progression, neuroblastoma (NB) is one of the most
common forms of cancer in young children. NB typically presents
with metastatic disease at diagnosis with a poor outcome (1,2). The
most critical step in NB metastasis is that the cells from the
primary tumor, which have enhanced capabilities of migration and
invasion, invade into the surrounding normal tissues (3). To date, a set of genes associated with
tumor progression in NB have been revealed (4). However, the precise mechanisms,
particularly the significant molecular pathways in the transition
from primary to metastatic NB, remain to be completely
understood.
The CDH1 gene encodes the epithelial cell
adhesion molecule, E-cadherin, which forms the core of the adherens
junctions between adjacent epithelial cells (5). The significance of CDH1 inactivation
for metastasis has been demonstrated in a variety of studies
(6–11). However, few studies have analyzed
the potential important roles of CDH1 in metastatic NB tissues.
Previously, a group of non-coding single-stranded
RNAs, ~22 nt in length, which modulate gene expression
post-transcriptionally by interacting with complementary sites
within the 3′ untranslated region (UTR) of target mRNA, have
emerged as important regulators in cancer-related processes,
including metastasis (12–15). In a previous study, Guo et al
observed differential expression of microRNAs (miRNAs) in
metastatic NB compared with primary NB using microarray analysis.
In addition, the potential roles of these miRNAs in the NB
metastatic process were investigated (16). However, the role of miR-23a in
metastatic NB tissues compared with primary NB tissues remains
unclear. Hence, we proposed that the upregulated miR 23a enhances
the metastatic capability of NB cells. Exploring the
miR-23a-mediated NB metastasis molecular mechanism may be useful to
characterize the progression from primary to metastatic NB.
Materials and methods
Tissue samples and cell lines
In total, nine pairs of human primary and metastatic
NB tissues were acquired from the Chengdu Military General Hospital
(Chengdu, China). The study was approved by the ethics committee of
Chengdu Military General Hospital and informed written consent was
obtained from the patients. All the tissues were confirmed by
pathology and immunohistochemistry, frozen in liquid nitrogen and
stored at −80°C. Human NB cell lines, SK-N-SH and GI-LA-N, were
cultured in Eagle’s minimum essential medium (Gibco-BRL, Carlsbad,
CA, USA) supplemented with 10% FBS and 100 U/ml
penicillin/streptomycin. All the cells were placed in a humidified
incubator with 5% CO2 at 37°C.
RNA isolation and qPCR
Total RNA was isolated using TRIzol reagent
(Invitrogen Life Technologies, Carlsbad, CA, USA) according to the
manufacturer’s instructions. The RNA concentration and quality was
measured by a NanoDrop ND-1000 spectrophotometer (Thermo Fisher
Scientific, Waltham, MA, USA). Next, 1 μg RNA was used for reverse
transcription (RT). Specific miR-23a reverse transcription (RT)
primers were used for the cDNA from the miRNA RT reaction, while
the common primer Oligo (dT) was used for the cDNA from total RNA,
for detection of CDH1. qPCR was performed using the SYBR Green PCR
master mix (Applied Biosystems, Inc., Foster City, CA, USA)
according to the following conditions: 95°C for 5 min followed by
40 cycles of amplification at 95°C for 30 sec, 57°C for 30 sec and
72°C for 30 sec. U6 small nuclear B non-coding RNA was used as the
internal control to normalize miR-23a expression and GAPDH was used
as the internal control to normalize CDH1 expression.
Western blot analysis
The NB cells were seeded into six-well plates at a
density of 3×104 cells/well and cells were transfected
once cell density reached ~80% confluence on the second day. At 48
h following transfection, the cells were lysed using RIPA buffer
for 30 min at 4°C. The protein concentration was measured by the
bicinchoninic acid method using a Pierce® BCA Protein
Assay kit (Thermo Fisher Scientific) and then 20 μg protein was
loaded on SDS-PAGE for analysis. The primary antibodies used were
rabbit polyclonal anti-human CDH1 (1:500; Abcam, Cambridge, MA,
USA) and rabbit monoclonal anti-human GAPDH (1:1,000; Abcam). The
secondary antibody used was a goat anti-rabbit IgG conjugated with
horseradish peroxidase (1:1,000; Abcam). The bound antibodies were
detected by ECL Plus Western blotting detection system (GE
Healthcare, Amersham, UK) and the chemiluminescent signals were
detected by high-performance chemiluminescence film (GE
Healthcare).
EGFP reporter assay
The 3′UTR of CDH1 was amplified and cloned
downstream of the pcDNA3/EGFP vector. Next, the mutant 3′UTR of
CDH1 (in which several nucleotides within the binding sites were
deleted) was amplified using pcDNA3/EGFP-CDH1 3′UTR as the template
and cloned downstream of the pcDNA3/EGFP vector. For the EGFP
reporter assay, the cells were co-transfected with miR-23a mimics,
ASO-miR-23a and pcDNA3/EGFP-CDH1 3′UTR or the mutant 3′UTR,
together with the controls. The plasmid expressing RFP was
transfected as the spike-in control. At 48 h following
transfection, the cells were lysed using RIPA buffer, and EGFP and
RFP intensity was measured by an F-4500 fluorescence
spectrophotometer (Hitachi, Ltd., Tokyo, Japan).
Transwell migration and invasion
assays
The migration and invasion assays were performed
using the Transwell chamber (Millipore, Billerica, MA, USA). For
the migration assay, following transfection, the cells were seeded
into the upper chamber (2.5×104 cells/well) in 250 μl
serum-free medium, but the bottom chamber was incubated with 750 μl
medium containing 10 or 20% serum. For the invasion assay, the
chamber was coated with Matrigel and the cells were seeded into the
upper chamber (2.5×104 cells/well) in 250 μl serum-free
medium, while the bottom chamber was incubated with 750 μl medium
containing 10 or 20% serum. Following cell migration or invasion
for 20 h, the cells that had not migrated or invaded were scraped
off with a cotton swab. The cells which had migrated or invaded
into the chamber membrane were fixed and then stained with crystal
violet for ~15 min. Finally, images of the cells were captured
under a microscope (ME21 digital microscope, Olympus, Guangzhou,
China) and cells were counted.
Statistics
Data are presented as the mean ± standard deviation
and the difference between groups was determined by a two-tailed
Student’s t-test. P<0.01 or P<0.05 were considered to
indicate a statistically significant difference.
Results
Quantitative analysis of CDH1 expression
in metastatic NB tissues
CDH1 is an important cancer metastasis-related
molecule. To test the expression of CDH1 in metastatic NB tissues
and matched primary NB tissues, qPCR assay was performed in nine
pairs of NB tissues. It was shown that CDH1 mRNA expression levels
were generally and significantly (P<0.01) lower in metastatic NB
tissues than in the matched primary NB tissues (Fig. 1A). In addition, western blot
analysis confirmed that the CDH1 protein expression was lower in
metastatic NB tissues compared with that in primary NB tissues
within the representative patients 3, 6 and 7.
miR-23a is a candidate regulator of CDH1
in NB
miRNAs function as tumor suppressors or oncogenes,
through the direct regulation of associated oncogenes or tumor
suppressor genes (14,17,18).
The oncogene miRNAs (onco-miRs), such as miR-23a, are usually
upregulated in tumors and may cause the loss of expression of tumor
suppressors and contribute to the tumorigenesis of cancer,
including metastasis. The loss of CDH1 expression in metastatic NB
tissues implies that there may be important miRNAs involved in the
regulation of CDH1 in NB metastasis. Greater specificity in miRNA
predictions may be attained by the consensus of multiple
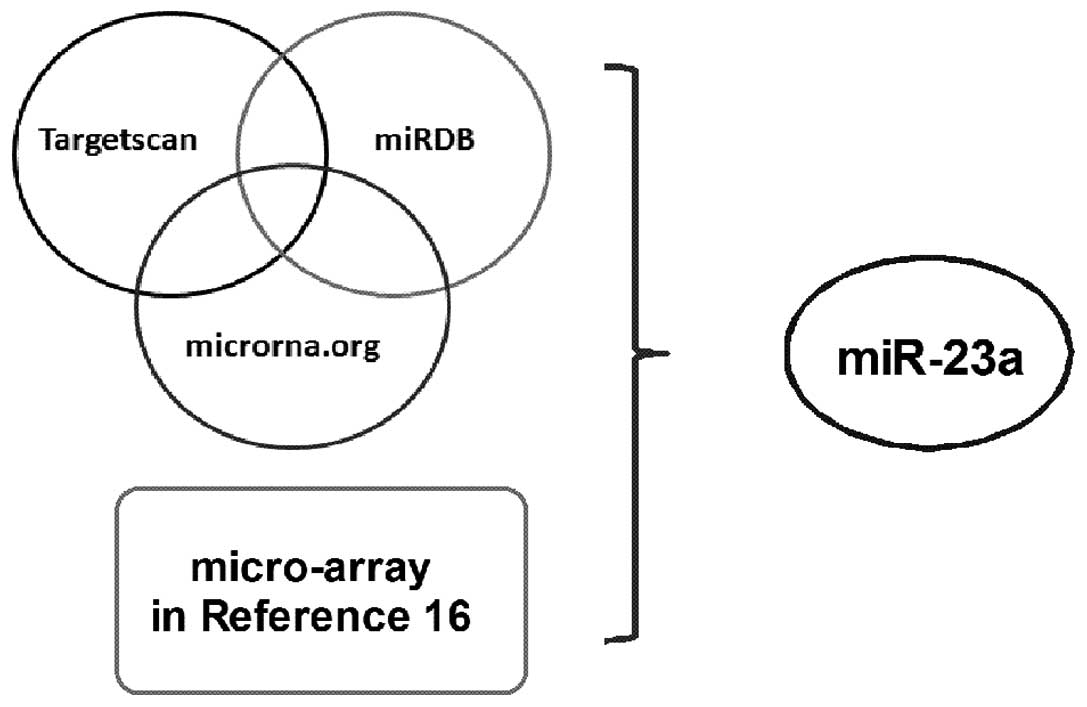
algorithms. Therefore, three programs (TargetScan, microRNA.org and
miRDB) were used for the prediction of which miRNAs target the
3′UTR of CDH1 and regulate its expression. Table I shows the 20 miRNAs that were
identified to target the 3′UTR of CDH1. In addition, Table II shows the 34 downregulated miRNAs
collected from metastatic NB tissues to compare with primary NB
tissues, according to the previous study by Guo et al using
the microarray analysis (16).
Based on these two tables, miR-23a (Fig. 2) was identified as a candidate for
directly targeting CDH1 whose mRNA 3′UTR contains a putative
binding site. This analysis is consistent with the model in which
tumor onco-miRs were found to promote tumor development by
targeting and negatively regulating tumor suppressors (13).
 | Table IPredicted miRNAs targeting CDH1 by
three algorithms. |
Table I
Predicted miRNAs targeting CDH1 by
three algorithms.
| Gene name | miRNA | microRNA.org | miRDB | TargetScan |
|---|
| CDH1 | hsa-miR-1248 | + | + | + |
| CDH1 | hsa-miR-219-2-3p | + | + | + |
| CDH1 | hsa-miR-504 | + | + | + |
| CDH1 | hsa-miR-340 | + | + | + |
| CDH1 | hsa-miR-580 | + | + | + |
| CDH1 | hsa-miR-296-3p | + | + | + |
| CDH1 | hsa-miR-1299 | + | + | + |
| CDH1 | hsa-miR-1296 | + | + | + |
| CDH1 | hsa-miR-515-5p | + | + | + |
| CDH1 | hsa-miR-1323 | + | + | + |
| CDH1 | hsa-miR-1264 | + | + | + |
| CDH1 | hsa-miR-548o | + | + | + |
| CDH1 | hsa-miR-619 | + | + | + |
| CDH1 | hsa-miR-544 | + | + | + |
| CDH1 | hsa-miR-339-5p | + | + | + |
| CDH1 | hsa-miR-23b | + | + | + |
| CDH1 | hsa-miR-23a | + | + | + |
| CDH1 |
hsa-miR-1207-5p | + | + | + |
| CDH1 | hsa-miR-219-5p | + | + | + |
| CDH1 | hsa-miR-338-3p | + | + | + |
| Table IIDifferential expression of miRNAs in
NB between metastatic and primary tumors (increased expression,
>2-fold). |
Table II
Differential expression of miRNAs in
NB between metastatic and primary tumors (increased expression,
>2-fold).
| miRNA | Fold changes
(metastatic/primary tumors) |
|---|
| hsa-miR-342-3p | 6.2879 |
| hsa-miR-92b | 11.8088 |
| hsa-miR-130b | 2.5771 |
| hsa-miR-129-3p | 3.0205 |
| hsa-miR-483-3p | 9.9583 |
| hsa-miR-345 | 3.1005 |
| hsa-miR-24 | 5.0395 |
| hsa-miR-486-5p | 2.4739 |
| hsa-miR-99b | 3.2086 |
| hsa-miR-145 | 4.9442 |
| hsa-miR-375 | 3.5245 |
| hsa-miR-92a | 5.3560 |
| hsa-miR-361-5p | 4.0653 |
| hsa-miR-484 | 2.9674 |
| hsa-miR-339-5p | 3.8349 |
| hsa-miR-148b | 4.3914 |
| hsa-miR-140-3p | 3.7209 |
| hsa-miR-335 | 2.5920 |
| hsa-miR-483-5p | 3.2567 |
| hsa-miR-23a | 2.5497 |
| hsa-miR-29a | 2.4728 |
| hsa-miR-423-3p | 2.0051 |
| hsa-miR-128a | 2.3610 |
| hsa-miR-130a | 3.1655 |
| hsa-miR-214 | 2.8887 |
| hsa-miR-15a | 2.5875 |
| hsa-miR-191 | 2.3789 |
| hsa-miR-100 | 2.2647 |
| hsa-miR-10b | 3.1618 |
| hsa-miR-29b | 2.4477 |
| hsa-miR-30c | 2.5702 |
| hsa-miR-324-5p | 2.2561 |
| hsa-miR-602 | 4.4085 |
| hsa-miR-107 | 2.1387 |
miRNA-23a is upregulated in metastatic NB
tissues and regulates NB cell migration and invasion
miR-23a has been previously reported to play a role
as an onco-miR in several types of cancer, and the microarray
analysis identified that miR-23a is upregulated in metastatic NB
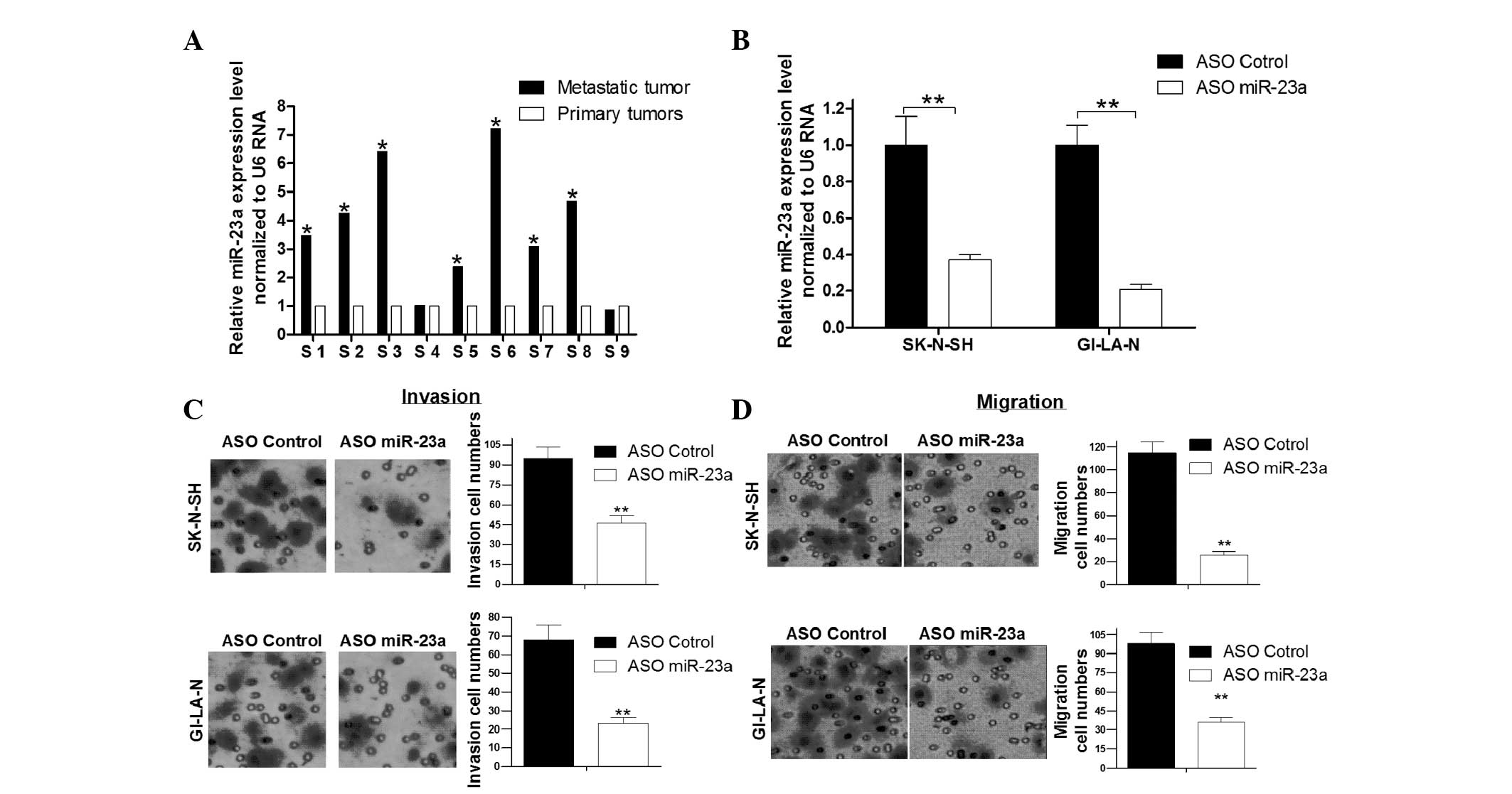
tissues compared with primary NB tissues. Next, qPCR assay was used
to detect the expression of miR-23a in nine pairs of NB samples
(metastatic and matched primary NB tissues). It was shown that
miR-23a expression levels were generally higher in metastatic NB
tissues compared with primary NB tissues (Fig. 3A). Transwell migration (without
Matrigel) and invasion (with Matrigel) assays were used to evaluate
the effect of miR-23a on NB cell migration and invasion. As
Fig. 3C shows, blocking the
expression of miR-23a with miR-23a ASO significantly decreased the
number of invaded SK-N-SH and GI-LA-N cells compared with the ASO
control group. This implies that the upregulated miR-23a promotes
the invasion of NB cells. Consistent with the invasion assay
results, blockage of miR-23a significantly inhibited the migration
of SK-N-SH and GI-LA-N cells compared with the control group
(Fig. 3D). Thus, it was concluded
that miR-23a is overexpressed in metastatic NB tissues and
inhibition of miR-23a suppresses the migration and invasion of NB
cells.
miR-23a directly targets CDH1 expression
in NB cells
To verify that the regulation of CDH1 expression by
miR-23a is direct, an EGFP reporter system was used in SK-N-SH
cells. The alignment of miR-23a with the CDH1 3′UTR insert is
illustrated in Fig. 4A. In
addition, a mutated 3′UTR was constructed by deleting 4 nt in the
seed sequence of miR-23a (asterisk marked in Fig. 4A). The EGFP reporter analysis showed
that with the CDH1-3′UTR insert, the EGFP intensity was
significantly lower than that in the CDH1-3′UTR-mutant group when
treated with control oligo. When miR-23a was blocked by miR-23a
ASO, the EGFP intensity levels were significantly higher than those
in the control group. However, the EGFP intensity with the mutated
3′UTR was not affected by miR-23a (Fig.
4B). Next, it was investigated whether the endogenous CDH1 is
regulated by miR-23a in NB cells. qPCR assay revealed that the CDH1
mRNA levels were inversely correlated with the miR-23a levels
(Fig. 4C). The trend of CDH1
protein levels was similar to that of the mRNA levels, according to
the western blot analysis (Fig.
4D). Therefore, it was concluded that CDH1 is a direct target
of miR-23a in SK-N-SH cells, and that miR-23a regulates the
expression of CDH1 at the mRNA and protein levels.
CDH1 suppresses NB cell migration and
invasion in vitro
Previous studies have shown that the downregulation
of CDH1 is an important feature of a number of transformed cells
(19), and that CDH1 functions as a
tumor suppressor. Accordingly, the present study investigated
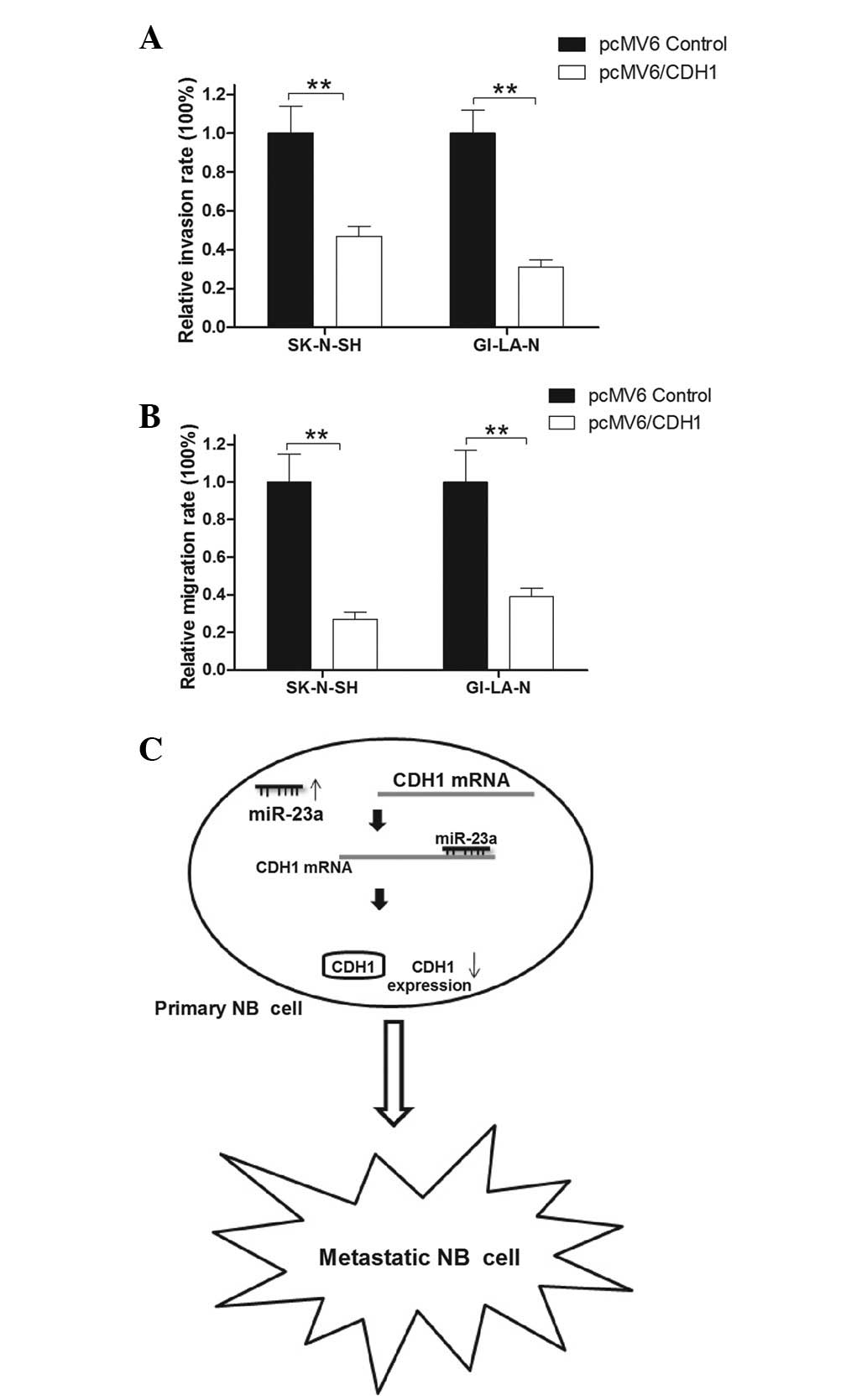
whether CDH1 affects NB cell metastasis. Overexpression plasmids
(pCMV6/CDH1) were constructed that ectopically overexpress the CDH1
protein in NB cells. Subsequently, the effect of CDH1 on cell
metastasis was evaluated by Transwell assay. The results showed
that the invasion rate was reduced by ~50 and ~60% with the
overexpression of CDH1 in SK-N-SH and GI-LA-N cells, respectively,
compared with that of the control group (Fig. 5A). In addition, the migration rate
of the SK-N-SH and GI-LA-N cells transfected with pCMV6/CDH1 was
markedly lower than that of the control group (Fig. 5B). These results provided further
evidence that CDH1 is a tumor suppressor in NB metastasis. The
tumor suppressor role of CDH1 in NB metastasis may explain the
manner in which the upregulation of miR-23a promotes NB cell
migration and invasion and contribute to the transformation of
primary to metastatic NB (Fig.
5C).
Discussion
As in the case of various types of cancer
metastasis, the transformation of primary to metastatic NB is a
complex multistep process including cell adhesion, migration,
angiogenesis, immune escape and homing to target organs. The
increased NB cell migration and invasion capabilities are essential
features of the metastatic transformation process. Identifying the
molecules and pathways that control NB cell migration and invasion
is critical to understanding NB metastasis.
Accumulating previous evidence has suggested that
genetic or epigenetic alterations in CDH1 or alterations in their
protein expression, often result in tissue disorder, cellular
dedifferentiation, increased invasiveness of tumor cells and
ultimately metastasis (19–21). It has been previously demonstrated
that the downregulation of CDH1 leads to loss of cell polarity and
derangement of normal tissue architecture (22,23).
In various types of cancer, CDH1-mediated cell-cell adhesion is
lost concomitantly with acquisition of an invasive phenotype, high
tumor grade and low patient survival rates (24–29).
Based on the important role of CDH1 in cancer tumorigenesis, the
investigation of the regulatory mechanisms of CDH1 in the process
of primary to metastatic NB transition may highlight new prognostic
markers and therapy targets for the metastasis of NB in the future.
To date, somatic mutations, chromosomal deletions, proteolytic
cleavage and silencing of the CDH1 promoter have been reported to
cause the loss or reduction of CDH1 expression (30–32).
In a previous study by Hoebeeck et al, the authors tested 42
primary NB tumors and the frequencies of methylation were 8% with
CDH1 (19). This implies that there
may be potential other mechanisms that are involved in the
downregulation of CDH1 in the NB metastatic process. In the present
study, nine pairs of metastatic NB tissues and matched primary NB
tissues were tested, and the mRNA and protein levels of CDH1 were
found to be significantly decreased in the NB metastatic
transformation process (Fig.
1).
Previously, miRNAs have been identified as important
regulators of gene expression. Several miRNAs have been found to
exhibit prometastatic (miR-10b, -21 and -373/520c) or
antimetastatic (miR-34b/c, -126, -148a, -206 and -335) activity
(33–35). In a previous study, miRNA microarray
assay was performed to screen out the miRNAs that were
differentially expressed between metastatic and primary NB tissues
(16). In addition, a previous
study has shown that miR-9, which is directly bound and upregulated
by MYC and MYCN in breast cancer cells, directly targets CDH1,
leading to increased cell motility and invasiveness (26). miR-10b has been found to modulate
breast cancer metastasis by targeting CDH1 and may be a useful
biomarker of advanced progression and metastasis of breast cancer
(36). miR-25 has also been
demonstrated to promote esophageal squamous cell carcinoma cell
migration and invasion by directly targeting CDH1 (37). Recently, Cao et al (38) found that miR-23a regulates
TGF-β-induced epithelial-mesenchymal transition by targeting
E-cadherin (CDH1) in lung cancer cells. However, few studies have
considered the manner in which the miRNAs contribute to the
transformation of primary to metastatic NB through the CDH1-induced
pathway. Therefore, the current study analyzed the microarray
results (Table II) and miRNA
target prediction algorithms (Table
I). miR-23a was identified as a candidate molecule in the
regulation of NB metastasis by the CDH1 pathway (Fig. 2).
miR-23a belongs to the miR-23a/24/27a cluster, which
is located in chromosome 19p13.12 and may be induced by TGF-β. In
several types of human cancer, this gene cluster functions as an
oncogenic miRNA and has been previously reported to be upregulated
in types of human cancer. As one of the most famous members of the
miRNA-cluster, miR-23a has been shown to promote the growth of
gastric adenocarcinoma cells by targeting the interleukin-6
receptor (39,40). miR-23a has also been demonstrated to
promote colon carcinoma cell growth, invasion and metastasis
through inhibition of the MTSS gene (41,42).
The current study revealed that miR-23a is upregulated in
metastatic NB tissues when compared with primary NB tissues. In
addition, blockage of miR-23a may reduce the capability of NB cell
migration and invasion using Transwell assays (Fig. 3). To identify the direct regulation
of CDH1 by miR-23a in NB cells, qPCR, western blot analysis and
EGFP report system were used to demonstrate that CDH1 is a direct
target gene of miR-23a (Fig. 4).
The Transwell assay results by overexpression of CDH1 in SK-N-SH
and GI-LA-N cells showed that the CDH1 may inhibit the capability
of NB cell migration and invasion (Fig.
5). This was found to correlate with the blocking of the
expression of miR-23a. These results suggest that there exists a
miR-23a/CDH1 pathway, which is important in the regulation of NB,
particularly in the process of primary to metastatic NB transition
(Fig. 5C).
In conclusion, the results of the present study may
be summarized by the following six major observations: i) CDH1 was
inactive in the metastatic NB tissues compared with the primary NB
tissues at the mRNA and protein levels; ii) bioinformatic software,
including TargetScan, microRNA.org and miRDB algorithms, were used
and miR-23a was identified as a candidate regulator of CDH1 in the
transformation process of primary to metastatic NB; iii) miR-23a
was found to be upregulated in metastatic NB tissues compared with
primary NB tissues; iv) CDH1 is negatively regulated by miR-23a at
the mRNA and protein levels and is a direct target in NB; v)
inhibition of miR-23a suppresses the migration and invasiveness of
SK-N-SH and GI-LA-N cells; and vi) there exists a miR-23a/CDH1
pathway in the transition of primary to metastatic NB. However,
whether miR-23a may become a new prognostic marker and therapy
target for the metastasis of NB must be explored in future
studies.
Acknowledgements
The current study was supported by the National
Natural Science Foundation of China (grant nos. 81071037 and
81271395).
References
|
1
|
Maris JM, Hogarty MD, Bagatell R and Cohn
SL: Neuroblastoma. Lancet. 369:2106–2120. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Maris JM: Recent advances in
neuroblastoma. N Engl J Med. 362:2202–2211. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Steeg PS: Tumor metastasis: mechanistic
insights and clinical challenges. Nat Med. 12:895–904. 2006.
View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Nevo I, Oberthuer A, Botzer E, et al:
Gene-expression-based analysis of local and metastatic
neuroblastoma variants reveals a set of genes associated with tumor
progression in neuroblastoma patients. Int J Cancer. 126:1570–1581.
2010.
|
|
5
|
Gumbiner BM: Regulation of
cadherin-mediated adhesion in morphogenesis. Nat Rev Mol Cell Biol.
6:622–634. 2005. View
Article : Google Scholar : PubMed/NCBI
|
|
6
|
Ceteci F, Ceteci S, Karreman C, et al:
Disruption of tumor cell adhesion promotes angiogenic switch and
progression to micrometastasis in RAF-driven murine lung cancer.
Cancer Cell. 12:145–159. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Derksen PW, Liu X, Saridin F, et al:
Somatic inactivation of E-cadherin and p53 in mice leads to
metastatic lobular mammary carcinoma through induction of anoikis
resistance and angiogenesis. Cancer Cell. 10:437–449. 2006.
View Article : Google Scholar
|
|
8
|
Onder TT, Gupta PB, Mani SA, Yang J,
Lander ES and Weinberg RA: Loss of E-cadherin promotes metastasis
via multiple downstream transcriptional pathways. Cancer Res.
68:3645–3654. 2008. View Article : Google Scholar
|
|
9
|
Vleminckx K, Vakaet L Jr, Mareel M, Fiers
W and van Roy F: Genetic manipulation of E-cadherin expression by
epithelial tumor cells reveals an invasion suppressor role. Cell.
66:107–119. 1991. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Frixen UH, Behrens J, Sachs M, et al:
E-cadherin-mediated cell-cell adhesion prevents invasiveness of
human carcinoma cells. J Cell Biol. 113:173–185. 1991. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Perl AK, Wilgenbus P, Dahl U, Semb H and
Christofori G: A causal role for E-cadherin in the transition from
adenoma to carcinoma. Nature. 392:190–193. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Bartel DP: MicroRNAs: genomics,
biogenesis, mechanism, and function. Cell. 116:281–297. 2004.
View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Gabriely G, Wurdinger T, Kesari S, et al:
MicroRNA 21 promotes glioma invasion by targeting matrix
metalloproteinase regulators. Mol Cell Biol. 28:5369–5380. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
14
|
O’Donnell KA, Wentzel EA, Zeller KI, Dang
CV and Mendell JT: c-Myc-regulated microRNAs modulate E2F1
expression. Nature. 435:839–843. 2005.PubMed/NCBI
|
|
15
|
Pillai RS, Bhattacharyya SN and Filipowicz
W: Repression of protein synthesis by miRNAs: how many mechanisms?
Trends Cell Biol. 17:118–126. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Guo J, Dong Q, Fang Z, et al:
Identification of miRNAs that are associated with tumor metastasis
in neuroblastoma. Cancer Biol Ther. 9:446–452. 2010. View Article : Google Scholar
|
|
17
|
Johnson SM, Grosshans H, Shingara J, et
al: RAS is regulated by the let-7 microRNA family. Cell.
120:635–647. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Cimmino A, Calin GA, Fabbri M, et al:
miR-15 and miR-16 induce apoptosis by targeting BCL2. Proc Natl
Acad Sci USA. 102:13944–13949. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Hoebeeck J, Michels E, Pattyn F, et al:
Aberrant methylation of candidate tumor suppressor genes in
neuroblastoma. Cancer Lett. 273:336–346. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Paredes J, Figueiredo J, Albergaria A, et
al: Epithelial E- and P-cadherins: role and clinical significance
in cancer. Biochim Biophys Acta. 1826:297–311. 2012.PubMed/NCBI
|
|
21
|
Jiang T, Jiang H, Su XM, et al:
Expressions of E-cadherin and alpha-catenin in benign, malignant
and metastatic prostate tumors. Zhonghua Nan Ke Xue. 18:499–503.
2012.(In Chinese).
|
|
22
|
Larue L, Ohsugi M, Hirchenhain J and
Kemler R: E-cadherin null mutant embryos fail to form a
trophectoderm epithelium. Proc Natl Acad Sci USA. 91:8263–8267.
1994. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Karayiannakis AJ, Syrigos KN, Savva A,
Polychronidis A, Karatzas G and Simopoulos C: Serum E-cadherin
concentrations and their response during laparoscopic and open
cholecystectomy. Surg Endosc. 16:1551–1554. 2002. View Article : Google Scholar
|
|
24
|
Alexiou D, Karayiannakis AJ, Syrigos KN,
et al: Serum levels of E-selectin, ICAM-1 and VCAM-1 in colorectal
cancer patients: correlations with clinicopathological features,
patient survival and tumour surgery. Eur J Cancer. 37:2392–2397.
2001. View Article : Google Scholar
|
|
25
|
Cavallaro U and Christofori G: Cell
adhesion and signalling by cadherins and Ig-CAMs in cancer. Nat Rev
Cancer. 4:118–132. 2004. View
Article : Google Scholar : PubMed/NCBI
|
|
26
|
Karatzas G, Karayiannakis AJ, Syrigos KN,
et al: E-cadherin expression correlates with tumor differentiation
in colorectal cancer. Hepatogastroenterology. 46:232–235.
1999.PubMed/NCBI
|
|
27
|
Karayiannakis AJ, Syrigos KN, Chatzigianni
E, et al: Aberrant E-cadherin expression associated with loss of
differentiation and advanced stage in human pancreatic cancer.
Anticancer Res. 18:4177–4180. 1998.PubMed/NCBI
|
|
28
|
Karayiannakis AJ, Syrigos KN, Chatzigianni
E, Papanikolaou S and Karatzas G: E-cadherin expression as a
differentiation marker in gastric cancer. Hepatogastroenterology.
45:2437–2442. 1998.PubMed/NCBI
|
|
29
|
Syrigos KN, Krausz T, Waxman J, et al:
E-cadherin expression in bladder cancer using formalin-fixed,
paraffin-embedded tissues: correlation with histopathological
grade, tumour stage and survival. Int J Cancer. 64:367–370. 1995.
View Article : Google Scholar
|
|
30
|
Berx G, Becker KF, Höfler H and van Roy F:
Mutations of the human E-cadherin (CDH1) gene. Hum Mutat.
12:226–237. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Maretzky T, Reiss K, Ludwig A, et al:
ADAM10 mediates E-cadherin shedding and regulates epithelial
cell-cell adhesion, migration, and beta-catenin translocation. Proc
Natl Acad Sci USA. 102:9182–9187. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Strathdee G: Epigenetic versus genetic
alterations in the inactivation of E-cadherin. Semin Cancer Biol.
12:373–379. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Voorhoeve PM, le Sage C, Schrier M, et al:
A genetic screen implicates miRNA-372 and miRNA-373 as oncogenes in
testicular germ cell tumors. Cell. 124:1169–1181. 2006. View Article : Google Scholar
|
|
34
|
Zhu S, Si ML, Wu H and Mo YY: MicroRNA-21
targets the tumor suppressor gene tropomyosin 1 (TPM1). J Biol
Chem. 282:14328–14336. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Ma L, Teruya-Feldstein J and Weinberg RA:
Tumour invasion and metastasis initiated by microRNA-10b in breast
cancer. Nature. 449:682–688. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Liu Y, Zhao J, Zhang PY, et al:
MicroRNA-10b targets E-cadherin and modulates breast cancer
metastasis. Med Sci Monit. 18:BR299–308. 2012.PubMed/NCBI
|
|
37
|
Xu X, Chen Z, Zhao X, et al: MicroRNA-25
promotes cell migration and invasion in esophageal squamous cell
carcinoma. Biochem Biophys Res Commun. 421:640–645. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Cao M, Seike M, Soeno C, et al: MiR-23a
regulates TGF-β-induced epithelial-mesenchymal transition by
targeting E-cadherin in lung cancer cells. Int J Oncol. 41:869–875.
2012.
|
|
39
|
Zhu LH, Liu T, Tang H, et al: MicroRNA-23a
promotes the growth of gastric adenocarcinoma cell line MGC803 and
downregulates interleukin-6 receptor. FEBS J. 277:3726–3734. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Huang S, He X, Ding J, et al: Upregulation
of miR-23a approximately 27a approximately 24 decreases
transforming growth factor-beta-induced tumor-suppressive
activities in human hepatocellular carcinoma cells. Int J Cancer.
123:972–978. 2008. View Article : Google Scholar
|
|
41
|
Jahid S, Sun J, Edwards RA, et al: miR-23a
promotes the transition from indolent to invasive colorectal
cancer. Cancer Discov. 2:540–553. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Wang Z, Wei W and Sarkar FH: miR-23a, a
critical regulator of ‘migR’ation and metastasis in colorectal
cancer. Cancer Discov. 2:489–491. 2012.
|