Introduction
Cholangiocarcinoma (CCA) is a highly malignant
epithelial neoplasm that arises from the biliary epithelium. It is
a major public health issue in Northeastern Thailand. CCA is highly
lethal due to lack of early detection, resistance to chemotherapy
and high propensity to invade locally and distantly (1). Currently, no effective therapy has
been identified for the disease and alternative therapeutic options
are urgently required.
Increased effort in recent years has been invested
into identifying new cellular targets important for cancer cell
survival and metastasis. Mammalian target of rapamycin (mTOR), a
289-kDa serine/threonine kinase and a downstream effector of the
phosphoinositide 3-kinase (PI3K)/Akt signaling pathway, was
identified as a promising therapeutic target for the treatment of a
number of types of cancer, including CCA (2–4). mTOR
forms the following two distinct multiprotein
complexes:Rapamycin-sensitive mTOR complex 1 (mTORC1); and
rapamycin-insensitive complex 2 (mTORC2). mTORC1, consisting of
raptor and G protein β subunit-like, regulates a number of cellular
processes, including cell proliferation, differentiation and cell
cycle progression by phosphorylating the ribosomal protein S6
kinase (S6K) to stimulate protein translation and ribosome
biogenesis (5). Activation of
mTORC1 also leads to the phosphorylation and inactivation of the
eukaryotic initiation factor 4E binding protein-1 (BP1), promoting
the protein translation (6).
Rapamycin (sirolimus), the first identified mTOR inhibitor, is
extracted from the bacterial strain Streptomyces
hygroscopicus found in soil, and has been used as an antifungal
agent (7). Previous in vitro
and in vivo studies have shown that rapamycin and its
analogs exhibit substantial antitumor activity in a number of types
of cancer (8–10).
Binding of the rapamycin/FK506-binding protein 12
complex to mTOR promotes the dissociation of the scaffold protein,
raptor, from mTORC1, suppressing the mTORC1 function (11). Previous studies have demonstrated
that the treatment of hepatocarcinoma (12) and CCA (13) with rapamycin significantly inhibits
cell growth and induces temporary partial remission or stable
disease. Previously, an orally bioavailable rapamycin derivative,
RAD001 [40-O-(2-hydroxyethyl)-rapamycin or everolimus; Novartis
International AG, Basel, Switzerland], was developed with the aim
of targeting the mTOR pathway. In 2009, RAD001 was approved by the
Food and Drug Administration for the treatment of advanced renal
cell carcinoma (14). However,
whether RAD001 is effective against CCA is unknown.
The present study investigated the effects of RAD001
on the malignant phenotypes of CCA in vitro, using the
RMCCA-1 cell line as a model. It was demonstrated that RAD001
suppresses in vitro invasiveness and alters the actin
cytoskeleton at low, non-toxic concentrations, concomitant with a
significant reduction of p-mTOR and p-extracellular
signal-regulated kinase (ERK)1/2 levels. At high concentrations,
RAD001 exhibited cytotoxic effects, reducing cell proliferation and
inducing apoptosis. Overall, the results of the current study
demonstrated that RAD001 exhibits multiple effects on the CCA
cells, suggesting that it may serve as a potential therapeutic
agent for the treatment of CCA.
Materials and methods
Cell cultures
The RMCCA-1 human intrahepatic CCA cell line
(15) was grown in HAM’s F12 medium
(GIBCO, Grand Island, NY, USA) supplemented with 10% fetal bovine
serum (FBS) at 37°C in a 5% CO2 humidified
atmosphere.
Cell proliferation assay
RMCCA-1 cells were seeded in 96-well culture plates
at a density of 10,000 cells per well overnight prior to the
addition of various concentrations of RAD001 (0–10 μM). After 24
and 48 h, water-soluble tetrazolium salts 1 (WST-1) cell
proliferation assay reagent (Roche Diagnostics, Laval, QC, Canada)
was added to the culture media and incubated for an additional 2 h
before the optical density (OD) at 450 nm was read. The percentage
of proliferation was calculated based on the number of control
[dimethyl sulfoxide (DMSO)-treated] cells. Three independent
experiments were performed, in triplicate.
Detection of apoptosis by terminal
deoxynucleotidyl transferase-mediated dUTP nick end labeling
(TUNEL) assay
RMCCA-1 cells were grown on sterile coverslips and
allowed to attach for 24 h prior to being treated with 0, 0.5 and 2
μM RAD001 for 24 h. The number of apoptotic cells was determined
using the Apo-BrdU TUNEL assay kit (Invitrogen Life Technologies,
Carlsbad, CA, USA) according to the manufacturer’s instructions.
Briefly, cells were washed with cold phosphate-buffered saline
(PBS; pH 7.4) and fixed with 1% paraformaldehyde and ice-cold 70%
ethanol for 30 min. Fixed cells were then labeled with BrdUTP using
terminal deoxynucleotide transferase in a humidified chamber at
37°C for 1 h, followed by staining with Alexa Fluor 488-conjugated
anti-BrdU antibody for 30 min at room temperature. The number of
apoptotic cells was quantified by counting the number of cells with
green fluorescent dots. Total cell number was determined by
counting the nuclei stained with DAPI in 10 fields under a
fluorescence microscope (magnification, ×20; (Olympus IX71, Olympus
Corporation, Tokyo, Japan). The degree of apoptosis was presented
as the percentage of apoptotic cells compared with the total number
of cells.
In vitro invasion and migration
assay
The in vitro invasion of CCA cells was
assayed using a Transwell chamber with 8-μm pore inserts (24-well
cell culture; Costar, Boston, MA, USA) that were coated with 100 μl
Matrigel (0.3 mg/ml; Becton-Dickinson, Bedford, MA, USA). RMCCA-1
cells were pretreated with 0, 0.1 and 0.5 μM of RAD001 for 6 h in
culture media with 1% FBS at 37°C prior to the harvesting of the
cells. In total, 5×104 cells were seeded into the upper
chamber of the Transwell containing serum-free media, whereas media
with 1% FBS was added into the lower chamber as a chemoattractant.
RAD001 was added to the two chambers. Following incubation for 18
h, the filters were fixed and stained with hematoxylin. The number
of invaded cells per well was counted in 10 fields under the
microscope (magnification, ×10) and presented as a percentage of
control (DMSO-treated) cells. In vitro migration assays were
also performed using the Transwell chambers in a similar manner to
the in vitro invasion assay, with the exception that no
Matrigel coating was applied on the filters.
Detection of actin polymerization
RMCCA-1 cells were grown on sterile coverslips and
allowed to attach overnight prior to being treated with RAD001 or
DMSO for an additional 24 h. The cells were washed twice with PBS,
fixed with 4% paraformaldehyde and permeabilized in 1% Triton X-100
for 15 min. Non-specific binding was blocked with 1% bovine serum
albumin prior to probing the cells with Alexa Fluor 488-conjugated
phalloidin (Molecular Probes, Eugene, OR, Canada) for 30 min.
Following washing with PBS, coverslips were mounted on the slide
glass containing ProLong® Gold Antifade reagent
(Invitrogen Life Technologies) and examined under a confocal
microscope (magnification, ×60; Olympus SV1000, Olympus
Corporation) using Olympus FV10-ASW 1.7 software (Olympus
Corporation).
Gelatin zymography assay
RMCCA-1 cells were starved for 24 h in serum-free
media containing RAD001 or DMSO prior to the collection of the
conditioned medium. The conditioned medium was then mixed with
non-reducing sample buffer [2% SDS, 10% glycerol, 62.5 mM Tris-Cl
(pH 6.8) and 0.01% bromophenol blue] and separated on a 7.5%
polyacrylamide gel containing gelatin (1 mg/ml). Following
electrophoresis, the gel was soaked in two changes of 2.5% Triton
X-100 for 30 min followed by an incubation in 50 mM Tris-Cl (pH
7.5), 10 mM CaCl2, 1 μM ZnCl2 and 1% Triton
X-100 for 18 h at 37°C. The gel was then stained with 0.25% (w/v)
Coomassie blue R250 for 2 h. The gelatinolytic activity was
observed as a clear band on the blue background of the
Coomassie-stained gel and documented using a Bio-Rad GS700 gel
scanner (Bio-Rad, Hercules, CA, USA).
Western blot analysis
RMCCA-1 cells (~5×105) were seeded in a
100-mm culture plate overnight prior to the addition of various
concentrations of RAD001. DMSO-treated cells were used as a
control. Following incubation, cells were collected, washed with
PBS and lysed in lysis buffer. Next, protein samples were mixed
with SDS sample buffer and β-mercaptoethanol, boiled and separated
using 7.5% SDS-PAGE. The gel was run for 1.5 h at 180 V prior to
transferring the proteins onto a nitrocellulose membrane (Bio-Rad,
Hercules, CA, USA) by electroblotting for 2.5 h at 120 V and 4°C.
Non-specific binding was blocked using 5% skimmed milk for 1 h at
room temperature. The blot was probed with rabbit anti-phospho-mTOR
(Ser 2448) antibody and rabbit anti-cleaved caspase 7 antibody,
prior to being stripped and reprobed with rabbit anti-mTOR and
rabbit anti-caspase 7 antibody, respectively. The antibodies were
purchased from Cell Signaling Technology (Beverly, MA, USA).
Quantification of matrix metalloproteinase (MMP)-14 was determined
by using specific antibody against MMP-14 (Abcam, Cambridge, MA,
USA), whereas, β-actin (Cell Signaling Technology) was used as a
loading control. Anti-rabbit IgG secondary antibody conjugated with
horseradish peroxidase (HRP; Cell Signaling Technology) was used as
a secondary antibody. The immunoreactive bands were detected using
enhanced chemiluminescence (Amersham Pharmacia Biotech, Amersham,
UK) and visualized by exposure to X-ray film.
Detection of intracellular signaling
The PathScan® intracellular signaling
array kit (Cell Signaling Technology) was used, according to the
manufacturer’s instructions, to simultaneously detect 18
significant and well-characterized signaling molecules, when
phosphorylated or cleaved. Briefly, cells were washed with ice-cold
PBS and lysed in cell lysis buffer. The array blocking buffer was
added to each well and incubated for 15 min at room temperature.
Then, the lysate was added to each well and incubated for 2 h at
room temperature. Following washing, the detection antibody
cocktail was added to each well and incubated for 1 h at room
temperature. The HRP-linked streptavidin was added to each well and
incubated for 30 min at room temperature. The slide was then
covered with LumiGLO/Peroxide reagent (Cell Signaling Technology)
and exposed to film for 2–30 sec.
Statistical analysis
All experiments were performed in triplicate and
data are presented as the mean ± standard error of the mean.
Statistical comparisons were performed using Student’s t-test.
P<0.05 was considered to indicate a statistically significant
difference.
Results
Effect of RAD001 on mTOR
phosphorylation
To determine whether RAD001 affects the
phosphorylation of mTOR, an antibody specific for phosphorylated
Ser 2448, a Rheb-catalyzed phosphorylation site that has been shown
to associate predominantly, but not exclusively, with mTORC1, was
used (16). As shown in Fig. 1A, treatment with 0.1 μM RAD001 for 1
h significantly reduced p-mTOR, whereas treatment for 24 h
suppressed the phosphorylation of mTOR almost completely.
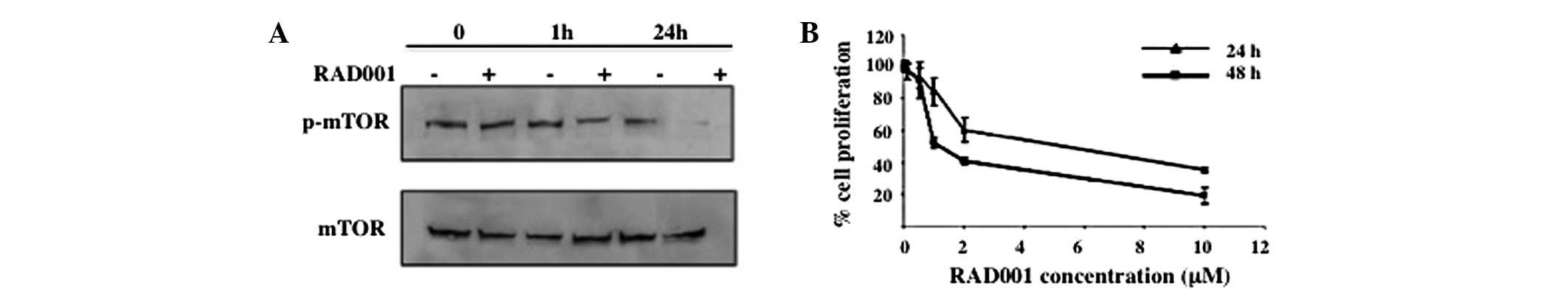 | Figure 1Effect of RAD001 on mTOR
phosphorylation and cell proliferation. (A) Suppression of mTOR
phosphorylation by RAD001 was determined by western blot analysis.
RMCCA-1 cells were treated with 0.1 μM RAD001 for 0, 1 and 24 h,
and cells treated with DMSO were used as negative control.
Phosphorylation of mTOR was determined using an antibody specific
for phosphorylated mTOR at Ser 2448, the Rheb phosphorylation site.
Total mTOR was used as a loading control. A representative of three
independent experiments is shown. (B) Effect of RAD001 on cell
proliferation. RMCCA-1 was treated with 0, 0.1, 0.5, 1, 2 and 10 μM
RAD001 for 24 and 48 h, prior to the assessment of cell
proliferation by water-soluble tetrazolium salts 1 assay. The
absorbance at 450 nm was measured. Data are presented as the mean ±
SEM of the results presented as a percentage of the control
(DMSO-treated) cells from three independent experiments, each
performed in triplicate. mTOR, mammalian target of rapamycin; DMSO,
dimethyl sulfoxide. |
Effect of RAD001 on cell
proliferation
To identify whether RAD001 has an effect on the cell
proliferation of RMCCA-1 cells, a WST-1 assay was performed.
RMCCA-1 cells were treated with 0–10 μM RAD001 or DMSO for 24 and
48 h, respectively, prior to being subjected to the WST-1 assay.
The OD at 450 nm was measured and was presented as a percentage of
the control (DMSO-treated) cells. Treatment with RAD001 resulted in
a dose-dependent inhibition of cell proliferation compared with the
control at the two time points. A 50% reduction of cell
proliferation was achieved at ~5.2 and ~1.3 μM RAD001 at 24 and 48
h, respectively (Fig. 1B).
Effect of RAD001 on apoptosis
Apoptosis, a programed cell death, has been shown to
be important in the maintenance of the cell population. Impairment
of apoptosis checkpoints has been shown to correlate with cancer
progression (17). Since the
results of the current study demonstrated that treatment with
RAD001 results in a reduction of RMCCA-1 cell proliferation, it was
further determined if this reduction is associated with the
induction of apoptosis using TUNEL assay. RMCCA-1 cells were
cultured on coverslips in the presence of various concentrations of
RAD001 for 24 h prior to subjecting the cells to TUNEL assay. Cells
treated with DMSO (the diluent of RAD001) were used as negative
control. Treatment of the RMCCA-1 cells with RAD001 resulted in an
induction of apoptosis as shown by the bright green fluorescent
dots (Fig. 2A). The proportion of
apoptotic cells increased from 4.2±0.9 to 27.5±7.4% with the
increasing concentrations of RAD001 from 0.5 to 2 μM (Fig. 2B), respectively.
Apoptosis may be triggered by various extracellular
or intracellular stimuli, inducing the extrinsic and intrinsic
death signaling pathways, respectively. These diverse death
signals, however, eventually activate a common set of executioner
caspases, including caspase 3, 6 and 7, leading to apoptotic cell
death (18). Caspase 7 activities
were assessed by western blot analysis to identify whether they are
affected by RAD001 using an antibody against the cleaved
(activated) form of caspase 7 (20 kDa). The amount of caspase 7 in
each sample was normalized using an antibody against the full
length (inactive) caspase 7 (35 kDa). The results showed that a
faint band of 20 kDa, representing the cleaved, active form of
caspase 7, was present in the DMSO-treated cells. However, the
intensity of this band was markedly increased compared with the
35-kDa band identified in cells treated with 0.5 and 2 M RAD001
(Fig. 2C).
Effect of RAD001 on invasion and
migration
Metastasis is a process by which cancer cells spread
from the primary tumor to distant locations. This process depends
on the ability of the cancer cells to migrate and invade the
surrounding tissues (19). Although
a number of previous studies have demonstrated the suppressive
effect of RAD001 on cancer growth in vitro, few studies have
analyzed the effect of RAD001 on its ability to induce the invasion
and migration of cancer cells. The effect of RAD001 on in
vitro invasion and migration was determined by exposing RMCCA-1
cells to 0.1 and 0.5 μM RAD001 for a total of 24 h, at which cell
proliferation was inhibited by <10% (Fig. 1B). RMCCA-1 cells were pretreated
with RAD001 for 6 h prior to being allowed to invade through the
Matrigel-coated Transwell for an additional 18 h in the presence of
RAD001. Cell migration assay was performed in a similar manner to
the in vitro invasion assay, with the exception that
Matrigel was not applied to the upper surface of the Transwell
filters. Treatment with 0.1 and 0.5 μM RAD001 resulted in a
reduction of in vitro invasion to 62.6±9.6 and 25.7±3.7%,
respectively, compared with the negative control (DMSO-treated)
cells. Consistent with the in vitro invasion, the presence
of 0.1 and 0.5 μM RAD001 reduced the number of migrating cells to
85.9±9.0 and 53.9±7.4%, respectively, compared with the negative
control (Fig. 3A).
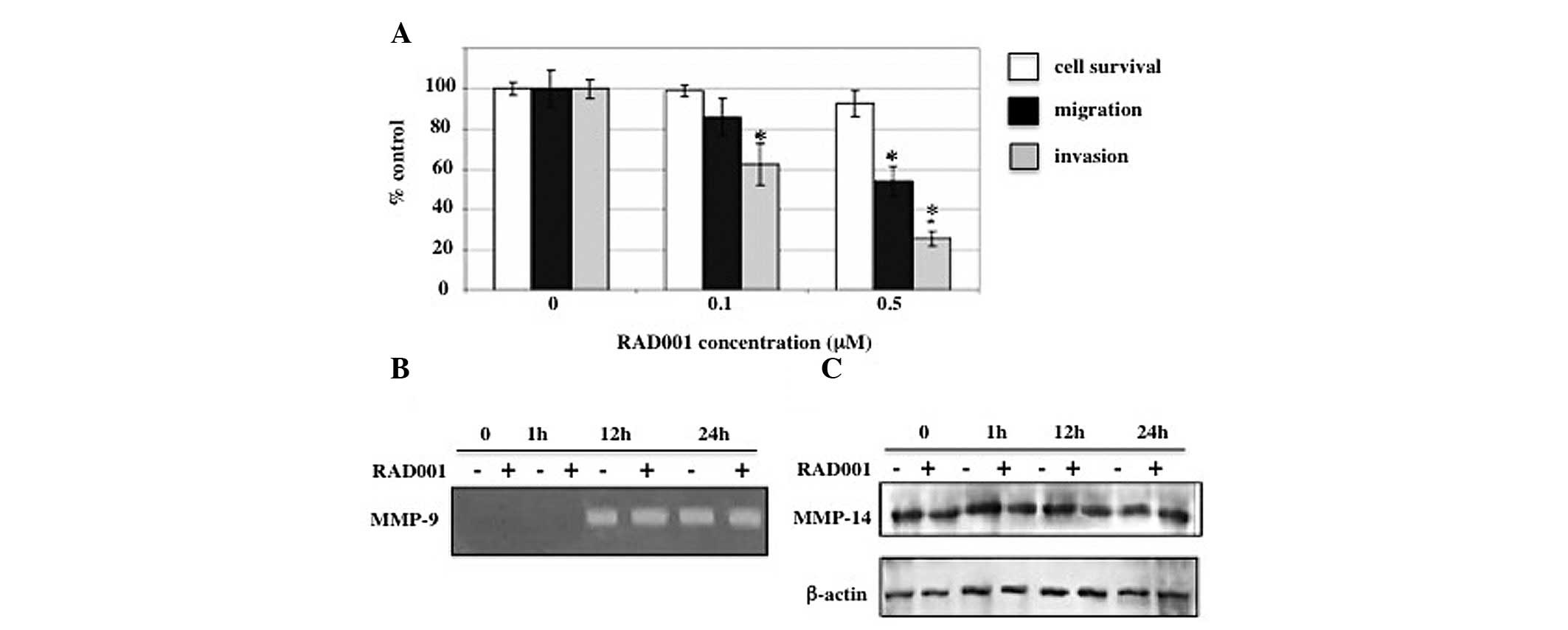 | Figure 3Effects of RAD001 on cell invasion,
migration and MMPs. (A) RMCCA-1 cells were exposed to 0, 0.1 and
0.5 μM RAD001 for a total of 24 h in each assay. For the in
vitro invasion and migration assay, RMCCA-1 cells were
pretreated with RAD001 for 6 h prior to being incubated in the
Transwell chamber for an additional 18 h. RAD001 was present in the
upper and lower chambers. Y-axis presents the mean ± SEM of results
expressed as a percentage of the control from three independent
experiments, each performed in duplicate. Controls were
DMSO-treated cells. X-axis presents the various RAD001
concentrations. (B) Effects of RAD001 on the secretion of MMP-9
into the conditioned medium from RMCCA-1 cells. RMCCA-1 cells were
treated with 0.5 μM RAD001 or DMSO (negative control) in serum-free
media for 0, 1, 12 and 24 h prior to determining the MMP-9 activity
by gelatin zymography. An intense band at 92 kDa corresponding to
MMP-9 activity was detected. Equal volumes of conditioned media
were loaded into each lane. (C) Effects of RAD001 on MMP-14
protein. RMCCA-1 cells were treated with 0.5 μM RAD001 in
serum-free media for 0, 15 min, 1 and 24 h, prior to the
preparation of the whole cell lysates, and subjected to 7.5%
SDS-PAGE. The blot was then probed with a monoclonal antibody
against MMP-14 and β-actin was used as a loading control.
*P<0.05, vs. controls. MMPs, matrix
metalloproteinases; DMSO, DMSO, dimethyl sulfoxide. |
Effect of RAD001 on MMPs
Degradation of the extracellular matrix is a key
factor which contributes to invasion and metastasis. Since the
results showed that RAD001 significantly reduced the in
vitro invasion and migration of RMCCA-1, the suppression of
these processes was analyzed to identify whether it is due to the
effect of RAD001 on the secretion of MMPs. Gelatin zymography
revealed that the intensity of the 92-kDa band representing the
MMP-9 activity was indifferent between RMCCA-1 cells treated with
0.5 μM RAD001 and untreated RMCCA-1 cells, following 12 or 24 h of
incubation (Fig. 3B).
In addition to secreted MMPs, membrane-bound MMPs
have been previously shown to be crucial in cancer invasion and
metastasis. Membrane-type 1 MMP (MT1-MMP or MMP-14), which is
expressed on the cell membrane, promotes the invasion of cancer
cells by directly degrading extracellular matrix components,
including fibronectin, vitronectin, laminin-1 and -5, fibrin,
proteoglycans and collagen types I, II and III (20–22).
The present study investigated the effect of RAD001 on MMP-14
protein expression using western blot analysis probed with a
monoclonal antibody against MMP-14. Consistent with the effect of
RAD001 on MMP-9, MMP-14 levels were not affected by the treatment
with RAD001 (Fig. 3C).
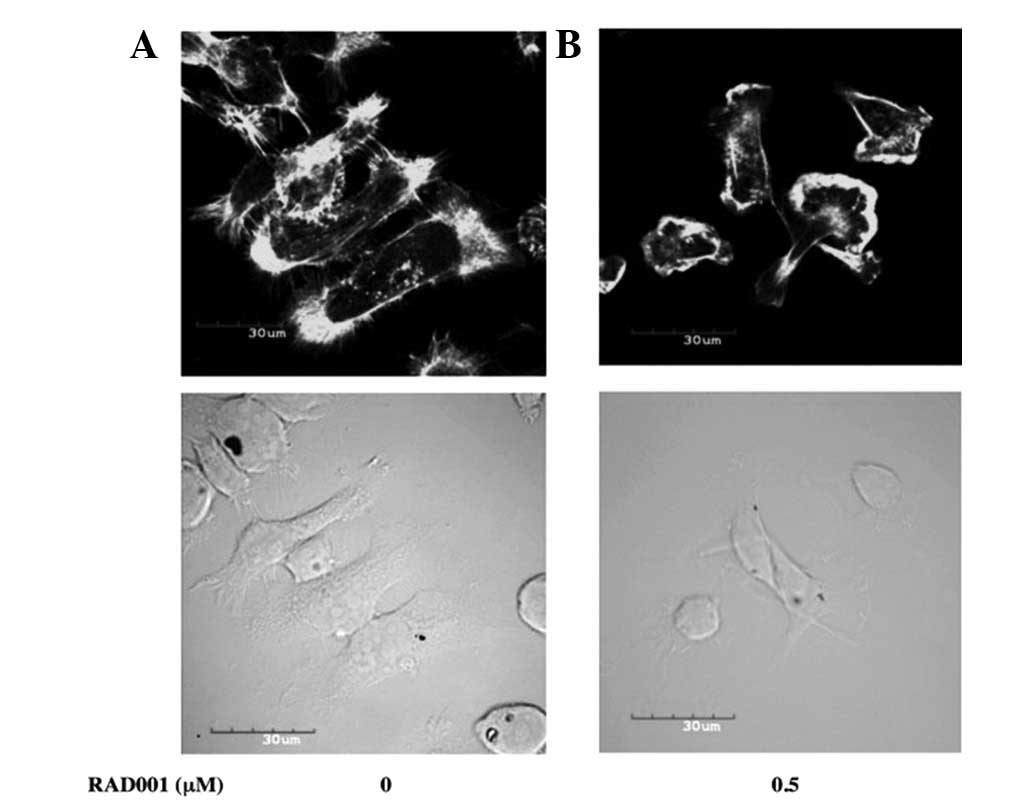
Effect of RAD001 on the actin
cytoskeleton
The results showed that RAD001 significantly reduced
the in vitro invasion and migration of the RMCCA-1 cells,
suggesting that the actin cytoskeleton may have been affected by
RAD001. RAD001 was examined to identify whether it alters the actin
cytoskeleton of RMCCA1 by staining actin with Alexa Fluor
488-conjugated phalloidin and by confocal microscopy. RMCCA-1 cells
treated with DMSO (negative control) exhibited a well-spread cell
morphology with high levels of actin polymerization in the
periphery of the cells. Distinct filopodia and lamellipodia were
evident. Treatment with 0.5 μM RAD001 for 24 h caused the cells to
round up and appear smaller. The lamellipodia and filopodia
formation was also diminished (Fig.
4).
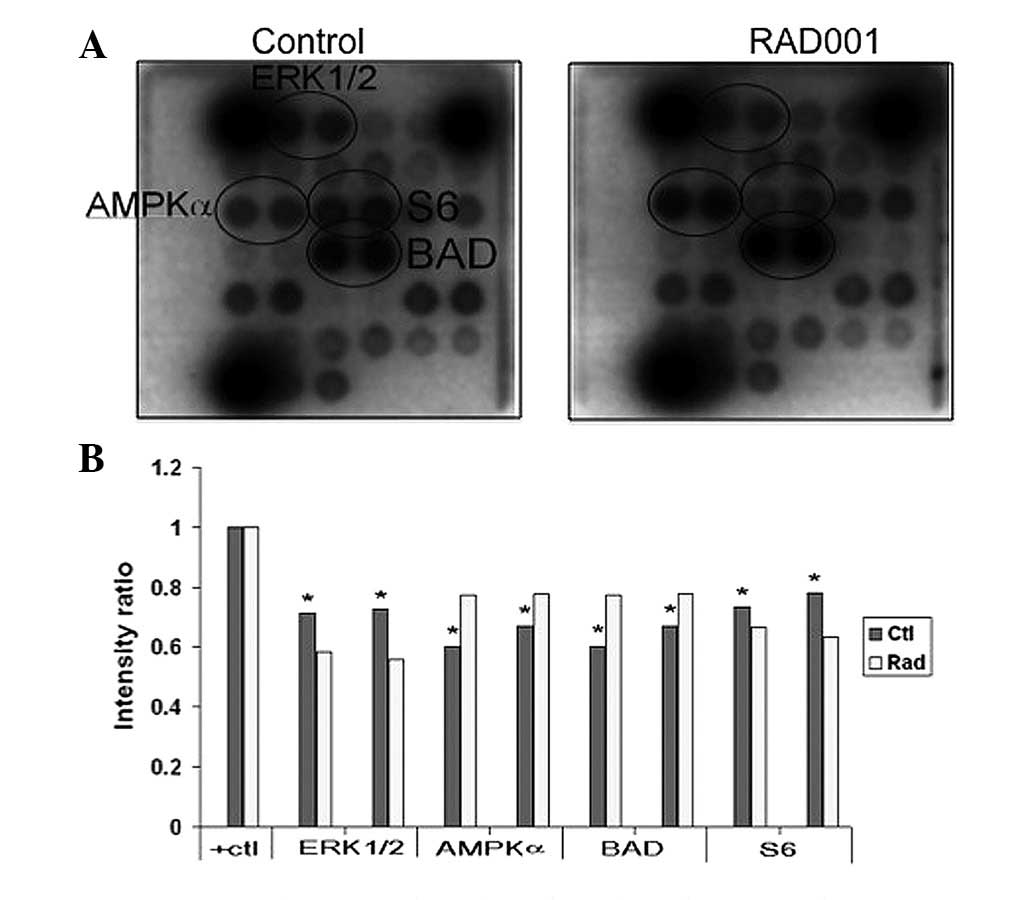
Effects of RAD001 on CCA cell
signaling
To elucidate the signal mediated by RAD001 in CCA
cells, the phosphorylation of 18 significant and well-characterized
signaling molecules were examined simultaneously using the PathScan
intracellular signaling array kit. The results showed that
RAD001-treated cells demonstrate a lower extent of phosphorylation
of multiple signaling molecules, including ERK1/2, AMP-activated
protein kinase α (AMPKα), proline-rich Akt substrate of 40 kDa,
Bcl-2-associated death promoter (BAD) and p38, than the control
cells (Fig. 5).
Discussion
To date, despite an improved understanding of CCA
pathophysiology, only marginal improvements in the treatment of
this disease have been suggested. The PI3K/Akt/mTOR pathway has
been shown to be upregulated in CCA cells and this pathway may be a
suitable target for the effective treatment of this cancer. RAD001
has been previously shown to inhibit mTOR activity, thereby halting
the proliferation of cancer cells, in vitro and in
vivo (23,24). The results of the present study
showed that treatment of CCA cells with a high concentration of
RAD001 (2 μM) significantly reduced cell proliferation, whereas a
low concentration of RAD001 (0.5 μM) impaired cell invasion and the
organization of the actin cytoskeleton. These observations are
consistent with a previous study, where low concentrations of
RAD001 imparted significant effects on cell invasion and migration,
but not cell proliferation (25,26).
This suggested that high concentrations of RAD001 or combination of
RAD001 with other chemotherapeutic drugs is required for effective
inhibition of cancer growth. Pretreatment with RAD001 was found to
increase the sensitivity of CCA cells to oxaliplatin (27).
The mTOR pathway regulates multiple cellular
signals, mediated by the mTORC1 and mTORC2 complexes. Previously,
mTORC1 has been shown to regulate cellular processes, including
cell proliferation, differentiation, cell cycle and protein
translation; whereas, mTORC2 has been shown to regulate the actin
cytoskeleton (5–7). RAD001, a derivative of rapamycin, has
been considered to confer inhibition primarily via the inhibition
of mTORC1 (28). However, the
results of the present study demonstrated that RAD001 inhibits CCA
cell invasion. This suggested that these events involve the
activation of the ERK cascade, a central pathway that transmits
signals from a number of extracellular agents to regulate cellular
processes. The assertion is based on the observation that low doses
of RAD001 suppress the phosphorylation of ERK1/2 and also inhibit
the invasive property of CCA cells. These observations are
consistent with our previous study showing that ERK1/2 activation
is required for CCA cell invasion (2).
The results from the PathScan intracellular
signaling array performed in the current study demonstrated that
the level of phosphorylated S6 protein was decreased by RAD001
treatment. Ribosomal protein S6 is a component of a 40S ribosomal
subunit which has been previously shown to be phosphorylated at
Ser235/236 by S6K1, a downstream effector of mTOR (29). This phosphorylation is required for
the translation of a group of mRNAs possessing a 5′-terminal
oligopyrimidine tract. As predicted, S6 phosphorylation levels were
decreased by RAD001 treatment, confirming that the mTOR signaling
pathway is inhibited by RAD001.
In the current study, RAD001-treated cells also
exhibited higher levels of AMPK phosphorylation compared with the
control cells. AMPK is a highly conserved sensor of cellular energy
status. In addition, a role for AMPK in the regulation of cancer
cell invasion has recently been demonstrated and the
pharmacological activation of AMPK reduces cancer cell invasion
(30). Therefore, we suggested that
the activation of AMPK by RAD001 inhibits the invasive property of
CCA cells. Future studies must be performed to demonstrate the
exact roles of AMPK in CCA cells.
BAD is a proapoptotic member of the Bcl-2 gene
family and the proapoptotic activity of BAD is regulated through
its phosphorylation. Only non-phosphorylated BAD induces apoptosis
by forming a heterodimer with Bcl-2 and Bcl-xL, inactivating them
and allowing Bax/Bak-triggered apoptosis (31). However, phosphorylation suppresses
apoptotic activity due to sequestration of p-BAD in the cytoplasm
by 14-3-3 proteins, preventing neutralization of the antiapoptotic
BCL-2 proteins. Notably, the results from the PathScan
intracellular signaling array kit performed in the current study
showed that, under the condition where RAD001 induces apoptosis and
reduces ERK1/2 phosphorylation, the level of p-Ser112 was
significantly increased. This observation was unexpected since
Ser112 is a known substrate of p90 ribosomal S6K (p90RSK), a
downstream target of the mitogen-activated protein kinase pathway,
thus, a reduction of ERK1/2 is predicted to parallel a reduction of
the p-Ser112 of BAD. However, Ser112 is a substrate for multiple
kinases in addition to p90RSK, including protein kinase A, PIM
kinases and p21-activated kinases (32). It is possible that the reduction of
p-Ser112 caused by the suppression of ERK1/2 activity is shadowed
by an increase of phosphorylation by the other kinases. Although
the present study showed that RAD001 treatment caused an
enhancement of p-Ser112, apoptosis was induced. This may be
explained by the evidence that the phosphorylation at Ser112 alone
is not sufficient to suppress apoptosis. Previously, a tiered
phosphorylation model was proposed, where the phosphorylation of at
least two serine residues is required to fully neutralize the
proapoptotic activity of BAD (33,34).
In addition, the other proapoptotic proteins, including BH3
interacting-domain death agonist, Bcl-2 interacting protein, p53
upregulated modulator of apoptosis and NADPH oxidase activator 1
(35,36), may substitute for BAD in inducing
apoptosis under this condition.
The results of the present study suggest a mechanism
that controls CCA cell proliferation and invasion by activation of
the mTOR signaling pathway. In addition, results showed that RAD001
may be a suitable new molecular target for CCA therapy.
Acknowledgements
The current study was supported by the Royal Golden
Jubilee Ph.D program and the Thailand Research Fund.
References
|
1
|
Sano T, Shimada K, Sakamoto Y, Yamamoto J,
Yamasaki S and Kosuge T: One hundred two consecutive hepatobiliary
resections for perihilar cholangiocarcinoma with zero mortality.
Ann Surg. 244:240–247. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Leelawat K, Leelawat S, Narong S and
Hongeng S: Roles of the MEK1/2 and AKT pathways in CXCL12/CXCR4
induced cholangiocarcinoma cell invasion. World J Gastroenterol.
13:1561–1568. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Alvarez M, Roman E, Santos ES and Raez LE:
New targets for non-small-cell lung cancer therapy. Expert Rev
Anticancer Ther. 7:1423–1437. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Yuan ZQ, Sun M, Feldman RI, et al:
Frequent activation of AKT2 and induction of apoptosis by
inhibition of phosphoinositide-3-OH kinase/Akt pathway in human
ovarian cancer. Oncogene. 19:2324–2330. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Burnett PE, Barrow RK, Cohen NA, Snyder SH
and Sabatini DM: RAFT1 phosphorylation of the translational
regulators p70 S6 kinase and 4E-BP1. Proc Natl Acad Sci USA.
95:1432–1437. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Beugnet A, Wang X and Proud CG: Target of
rapamycin (TOR)-signaling and RAIP motifs play distinct roles in
the mammalian TOR-dependent phosphorylation of initiation factor
4E-binding protein 1. J Biol Chem. 278:40717–40722. 2003.
View Article : Google Scholar
|
|
7
|
Sehgal SN, Baker H and Vézina C: Rapamycin
(AY-22, 989), a new antifungal antibiotic. II Fermentation,
isolation and characterization. J Antibiot (Tokyo). 28:727–732.
1975. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Moreno A, Akcakanat A, Munsell MF, Soni A,
Yao JC and Meric-Bernstam F: Antitumor activity of rapamycin and
octreotide as single agents or in combination in neuroendocrine
tumors. Endocr Relat Cancer. 15:257–266. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Motzer RJ, Escudier B, Oudard S, et al:
Efficacy of everolimus in advanced renal cell carcinoma: a double
blind, randomised, placebo-controlled phase III trial. Lancet.
372:449–456. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Di Paolo S, Teutonico A, Ranieri E,
Gesualdo L and Schena PF: Monitoring antitumor efficacy of
rapamycin in Kaposi sarcoma. IS J Kidney Dis. 49:462–470.
2007.PubMed/NCBI
|
|
11
|
Oshiro N, Yoshino K, Hidayat S, et al:
Dissociation of raptor from mTOR is a mechanism of
rapamycin-induced inhibition of mTOR function. Genes Cells.
9:359–366. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Schöniger-Hekele M and Müller C: Pilot
study: rapamycin in advanced hepatocellular carcinoma. Aliment
Pharmacol Ther. 32:763–768. 2010.PubMed/NCBI
|
|
13
|
Okada T, Sawada T and Kubota K: Rapamycin
inhibits growth of cholangiocarcinoma cells.
Hepatogastroenterology. 56:6–10. 2009.
|
|
14
|
Agarwala SS and Case S: Everolimus
(RAD001) in the treatment of advanced renal cell carcinoma: a
review. Oncologist. 15:236–245. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Rattanasinganchan P, Leelawat K,
Treepongkaruna SA, et al: Establishment and characterization of a
cholangiocarcinoma cell line (RMCCA-1) from a Thai patient. World J
Gastroenterol. 12:6500–6506. 2006.PubMed/NCBI
|
|
16
|
Cybulski N and Hall MN: TOR complex 2: a
signaling pathway of its own. Trends Biochem Sci. 34:620–627. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Vousden KH and Lu X: Live or let die: the
cell’s response to p53. Nat Rev Cancer. 2:594–604. 2002.
|
|
18
|
Delhalle S, Duvoix A, Schnekenburger M,
Morceau F, Dicato M and Diederich M: An introduction to the
molecular mechanisms of apoptosis. Ann N Y Acad Sci. 1010:1–8.
2003. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Gao CF, Xie Q, Su YL, et al: Proliferation
and invasion: plasticity in tumor cells. Proc Natl Acad Sci USA.
102:10528–10533. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Pei D and Weiss SJ: Transmembrane-deletion
mutants of the membrane-type matrix metalloproteinase-1 process
progelatinase A and express intrinsic matrix-degrading activity. J
Biol Chem. 271:9135–9140. 1996. View Article : Google Scholar
|
|
21
|
Ohuchi E, Imai K, Fujii Y, Sato H, Seiki M
and Okada Y: Membrane type 1 matrix metalloproteinase digests
interstitial collagens and other extracellular matrix
macromolecules. J Biol Chem. 272:2446–2451. 1997. View Article : Google Scholar
|
|
22
|
Koshikawa N, Giannelli G, Cirulli V,
Miyazaki K and Quaranta V: Role of cell surface metalloprotease
MT1-MMP in epithelial cell migration over laminin-5. J Cell Biol.
148:615–624. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Mabuchi S, Altomare DA, Cheung M, et al:
RAD001 inhibits human ovarian cancer cell proliferation, enhances
cisplatin-induced apoptosis, and prolongs survival in an ovarian
cancer model. Clin Cancer Res. 13:4261–4270. 2007. View Article : Google Scholar
|
|
24
|
Brown VI, Fang JJ, Barr R, Alcorn K, Kim J
and Grupp SA: The mTOR inhibitors rapamycin and RAD001 are active
in experimental models of ALL and are antagonized by interleukin-7.
Blood. 100:761a2002.
|
|
25
|
Hirashima K, Baba Y, Watanabe M, et al:
Aberrant activation of the mTOR pathway and anti-tumour effect of
everolimus on oesophageal squamous cell carcinoma. Br J Cancer.
106:876–882. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Ida S, Miki Y, Ono K, et al: Synergistic
anti-tumor effects of RAD001 with MEK inhibitors in neuroendocrine
tumors: a potential mechanism of therapeutic limitation of mTOR
inhibitor. Mol Cell Endocrinol. 350:99–106. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Leelawat K, Narong S, Udomchaiprasertkul
W, Leelawat S and Tungpradubkul S: Inhibition of PI3K increases
oxaliplatin sensitivity in cholangiocarcinoma cells. Cancer Cell
Int. 9:32009. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Gorshtein A, Rubinfeld H, Kendler E, et
al: Mammalian target of rapamycin inhibitors rapamycin and RAD001
(everolimus) induce anti-proliferative effects in GH-secreting
pituitary tumor cells in vitro. Endocr Relat Cancer. 16:1017–1027.
2009. View Article : Google Scholar
|
|
29
|
Thomas G, Siegmann M and Gordon J:
Multiple phosphorylation of ribosomal protein S6 during transition
of quiescent 3T3 cells into early G1, and cellular
compartmentalization of the phosphate donor. Proc Natl Acad Sci
USA. 76:3952–3956. 1979. View Article : Google Scholar
|
|
30
|
Fitzgerald JP, Nayak B, Shanmugasundaram
K, et al: Nox4 mediates renal cell carcinoma cell invasion through
hypoxia-induced interleukin 6- and 8- production. PLoS One.
7:e307122012. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Bergmann A: Survival signaling goes BAD.
Dev Cell. 3:607–608. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Schürmann A, Mooney AF, Sanders LC, et al:
p21-activated kinase 1 phosphorylates the death agonist bad and
protects cells from apoptosis. Mol Cell Biol. 20:453–461.
2000.PubMed/NCBI
|
|
33
|
Datta SR, Dudek H, Tao X, Masters S, Fu H,
Gotoh Y, et al: Akt phosphorylation of BAD couples survival signals
to the cellintrinsic death machinery. Cell. 91:231–241. 1997.
View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Datta SR, Katsov A, Hu L, Petros A, Fesik
SW, Yaffe MB, et al: 14-3-3 proteins and survival kinases cooperate
to inactivate BAD by BH3 domain phosphorylation. Mol Cell. 6:41–51.
2000. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Cheng EH, Wei MC, Weiler S, et al: BCL-2,
BCL-X(L) sequester BH3 domain-only molecules preventing BAX- and
BAK-mediated mitochondrial apoptosis. Mol Cell. 8:705–711. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Chen L, Willis SN, Wei A, et al:
Differential targeting of prosurvival Bcl-2 proteins by their
BH3-only ligands allows complementary apoptotic function. Mol Cell.
17:393–403. 2005. View Article : Google Scholar
|