Introduction
Hepatocellular carcinoma (HCC) is one of most common
causes of cancer-related mortality worldwide and the incidence of
this cancer has been increasing in recent years (1,2).
Improvements in survival are likely to depend on an improved
understanding of the molecular processes involved in tumorigenesis
and metastasis in hepatocellular cancer.
Human sulfatase 1 (hSulf-1), previously
characterized as a heparin-degrading endosulfatase, negatively
regulates growth factor and cytokine signaling and proteolysis by
desulfation of cell surface heparin sulfate proteoglycans (HSPGs),
major constituents of the extracellular matrix (3–6). This
biological effect requires sulfation of defined sites on
glycosaminoglycan chains. Previous studies have demonstrated that
hSulf-1 is downregulated in cancer cell lines originating from
various types of human cancer, including ovarian, breast, renal and
HCC, and that its expression is regulated by epigenetic mechanisms,
including DNA methylation and histone acetylation (7–10).
Additionally, previous studies have demonstrated
that re-expression of hSulf-1 in ovarian cells suppresses
fibroblast growth factor (FGF)-2 and heparin-binding epidermal
growth factor (HB-EGF) signaling and inhibits cell proliferation
and invasion in vitro (11–14). Further studies of the role
of hSulf-1 in tumorigenesis and progression have also found that
expression of hSulf-1 inhibits hepatocyte growth factor (HGF) and
vascular endothelial growth factor (VEGF) signaling (15–18).
While signal transducer and activator of transcription 3 (stat3)
signaling is known to be activated by several growth factor
receptors, including epidermal growth factor receptor (EGFR) and
platelet-derived growth factor receptor (PDGFR), which are
important in cell proliferation, migration, apoptosis and
angiogenesis (19–20), no previous studies have found a role
for hSulf-1 in regulating the stat3 signaling pathway.
Since the stat3 signaling pathway is one of the most
important signal transduction cascades characterized to date and is
known to be involved in the regulation of cytokine receptor
signaling in HCC (21–22), the potential link between hSulf-1
and stat3 in HCC must be investigated. Thus, in the current study,
to confirm the role of hSulf-1 in the proliferation, migration and
apoptosis of HCC, hSulf-1 was re-expressed in HCC cells and stat3
siRNA was constructed to manipulate the expression of this critical
signaling molecule in vitro. In addition, the present study
sought to determine whether the stat3 pathway is mediated by HGF, a
potent mitogen and key regulator of cell proliferation,
differentiation and motility in HCC (23–25),
in HCC cells exhibiting differential expression of hSulf-1. The
results indicated that hSulf-1 may inhibit HCC growth and migration
through suppression of the stat3 signaling pathway and that the
antiproliferative activity of hSulf-1 in HepG2 cells is due to cell
cycle arrest and apoptosis.
Materials and methods
Plasmids and siRNA
The plasmid containing whole-length hSulf-1
complementary DNA was purchased from Wuhan Genesil Biotechnology
Co., Ltd. (Wuhan, China) and stat3 siRNA was purchased from Origene
(Rockville, MD, USA). The stat3 siRNA target sequence used was
GGCGTCCAGTTC ACTACTA and the control siRNA target sequence used was
AATTCTCCGAACGTGTCACGT. Transfections were performed using
Lipofectamine 2000 (Invitrogen Life Technologies, Carlsbad, CA,
USA).
Cell culture
The HCC cell lines, HepG2, Hep3B, Huh-7 and
SMMC-7221, were purchased from Shanghai Cell Bank (Shanghai, China)
and were cultured in media according to the manufacturer’s
instructions. HepG2 cells stably expressing hSulf-1 were selected
with 600 μg/ml G418 (Invitrogen Life Technologies) and the
transfection results were detected by western blotting. Cells were
maintained at 37°C in an atmosphere of humidified air with 5%
CO2.
Treatment of cells with trichostatin A
(TSA) and 5-aza-dC
All drugs were added the day after the subculture of
HepG2 cells. The 5-aza-dC (Sigma-Aldrich, St. Louis, MO, USA) was
added at concentrations of 0, 0.5, 1.0, 2.5 and 5.0 μmol/l at 24,
48 and 72 h time points. For TSA (Sigma-Aldrich) treatment, HepG2
cells were treated with TSA concentrations of 0, 0.25 and 0.5
μmol/l and cells were incubated for 24 h. For combined treatment,
HepG2 cells were treated with 5.0 μmol/l 5-aza-dC at 24 and 48 h
time points and then with 0.5 μmol/l TSA for 24 h. When HepG2 cells
were treated with no drug, identical volumes of water were
added.
RNA extraction and semi-quantitative
reverse transcription-polymerase chain reaction (RT-PCR)
Total RNA was isolated from HCC cells using an
RNeasy kit (Qiagen, Valencia, CA, USA). Taq enzyme and PCR reagents
were purchased from Tiangen Corporation (Beijing, China). Primers
for amplifying hSulf-1 and GAPDH, which was used as
internal control in RT-PCR, were purchased from Sangon Corporation
(Shanghai, China). The forward and reverse primers used were as
follows: 5′-CTCACAGTCCGGAGCGGAAC-3′ (forward) and
5′-CACGGCGTTGCTGCTATCTGCCAGCATCC-3′ (reverse) for hSulf-1;
and 5′-AGTCAACGGATTTGGTCGT-3′ (forward) and
5′-TTGATTTTGGAGGGATCTG-3′ (reverse) for GAPDH. The primers
yielded amplicons of 371 and 238 bp, respectively. The PCR
conditions used were as follows: 94°C for 5 min, followed by 34
cycles of 15 sec at 94°C, 30 sec at 62°C and 30 sec at 72°C,
followed by a final extension at 72°C for 10 min. Semi-quantitative
RT-PCR products were analyzed on 1% agarose gels stained with
ethidium bromide.
Western blotting
HepG2 cells were lysed in RIPA buffer (Beyotime
Institute of Biotechnology, Shanghai, China). Cell lysates (20 μg
protein/lane) were loaded and separated on gradient polyacrylamide
gels and then transferred to polyvinylidene difluoride membranes by
electroblotting (Millipore Corp., Boston, MA, USA). Following
blocking with 5% non-fat milk containing 0.3% Tween 20 for 1 h, the
membranes were incubated overnight with primary antibodies at 4°C,
including anti-hSulf-1 (1:250), -stat3 (1:500), -phospho-stat3
(1:500), -phospho-c-met (1:500), -bcl-2 (1:1000) and -cyclin D1
(1:500) (Santa Cruz Biotechnology, Inc., Santa Cruz, CA, USA). The
membranes were washed three times with Tris-buffered saline
containing Tween 20 and membranes were then incubated with
horseradish peroxidase-conjugated secondary antibodies (R&D
Systems China Co., Ltd., Shanghai, China) at 4°C for 1 h.
Subsequently, membranes were exposed to enhanced chemiluminescent
reagents for detection of protein bands. β-actin was used as an
internal control.
Cell proliferation analysis
Cell proliferation was measured using an MTT assay
(Sigma-Aldrich). Cells were harvested and plated in 96-well plates
at 4×103 cells/well in 100 ml culture medium and then
maintained at 37°C in an incubator containing 5% CO2 for
three days. In total, 20 μl MTT dye was added to each well (5
mg/ml). After 4 h of incubation, 100 μl dimethyl sulfoxide was
added for 10 min to dissolve the crystals. The absorbance was
measured by a microtiter plate reader at 490 nm (no. DG5033A,
Jinggong Industrial Co., Ltd., Shanghai, China). Cell viability was
expressed as an optical density value.
Transwell chamber assay
Migration was detected by the Transwell chamber
assay. A total of 5×105 cells per ml were starved
overnight in serum-free medium. In total, 100 μl of cells were then
added to each upper well in a 24-well Transwell plate (8.0-μm pore
size; Corning, Inc., Cambridge, MA, USA) and medium containing 10%
fetal bovine serum (600 μl) was added to the lower well. Cells were
incubated in the Transwell chambers for 24 h. Then, the Transwells
were extracted, the medium in the upper well was removed and the
Transwells were washed in phosphate-buffered saline (PBS) once. The
residual cells in the upper well were swabbed and stained with 0.5%
crystal violet for 20 min. Cells that had migrated through the
Transwell were dissolved in 10% acetic acid and the absorbance was
measured at 560 nm.
Cell cycle analysis
Cells were seeded at a density of ~6×105
cells/ml and treated with 5 μmol/l cisplatin to determine the
effects of hSulf-1 on cisplatin-induced cell cycle arrest for 24 h.
Following incubation, cells were washed with PBS and fixed with 70%
ethanol overnight at 4°C. Next, cells were stained with 1 ml
propidium iodide (PI, Sigma-Aldrich) synthetic dye solution (20
μg/ml PI, 20 μg/ml RNase, 0.5% Triton X-100 and 1 g/ml sodium
citrate) for 30 min at 37°C in the dark and then analyzed by flow
cytometry using an FC 500 MPL instrument (Beckman Coulter, Miami,
FL, USA). The cell number in each phase in every group was
calculated using ModFit software (Verity Software House Corp.,
Topsham, ME, USA).
Cellular apoptosis assay
Cells were plated at a density of 6×105
cells/ml. Following treatment with 5 μmol/l cisplatin, apoptotic
cells were quantified by Annexin V/PI double staining (Jingmei
Biotech Co., Ltd., Shenzhen, China). The double-staining technique
was performed as follows, according to the manufacturer’s
instructions. Cisplatin-treated cells were collected and then
washed twice in cold PBS. Cell pellets were resuspended in 250 μl
1X binding buffer (Jingmei Biotech Co., Ltd.) and resuspended cells
were gently vortexed and stained with 5 μl Annexin V-fluorescein
isothiocyanate and 10 μl PI for 15 min in the dark at room
temperature. The results were analyzed using flow cytometry (PC 500
MPL, Beckmann Coulter, Miami, FL, USA) according to the
manufacturer’s instructions.
Statistical analysis
All data obtained in triplicate independent
experiments were evaluated using GraphPad Prism 5.02 for Windows
(GraphPad Software, Inc., La Jolla, CA, USA). Data are expressed as
the mean ± standard error. The significance of differences between
groups was determined by two-sided t-tests. P<0.05 was
considered to indicate a statistically significant difference.
Results
Expression of hSulf-1 mRNA decreases in
HCC cell lines and reactivates with 5-aza-dC and/or TSA in HepG2
cells
Firstly, the expression of hSulf-1 mRNA was
evaluated in four human HCC cell lines (HepG2, Hep3B, Huh-7 and
SMMC-7221) by semi-quantitative RT-PCR. All the HCC cell lines
tested were found to express low or undetectable levels of hSulf-1
mRNA (Fig. 1A and B). The
expression of hSulf-1 increased when treated with 5-aza-dC or TSA
in HepG2 cells. In addition, hSulf-1 may be reactivated
significantly with the appropriate concentration of 5.0 μmol/l
5-aza-dC and 0.5 μmol/l TSA. The expression of hSulf-1 also
increased due to a synergistic effect of 5.0 μmol/l 5-aza-dC and
0.5 μmol/l TSA combined treatment (Fig.
1C–E).
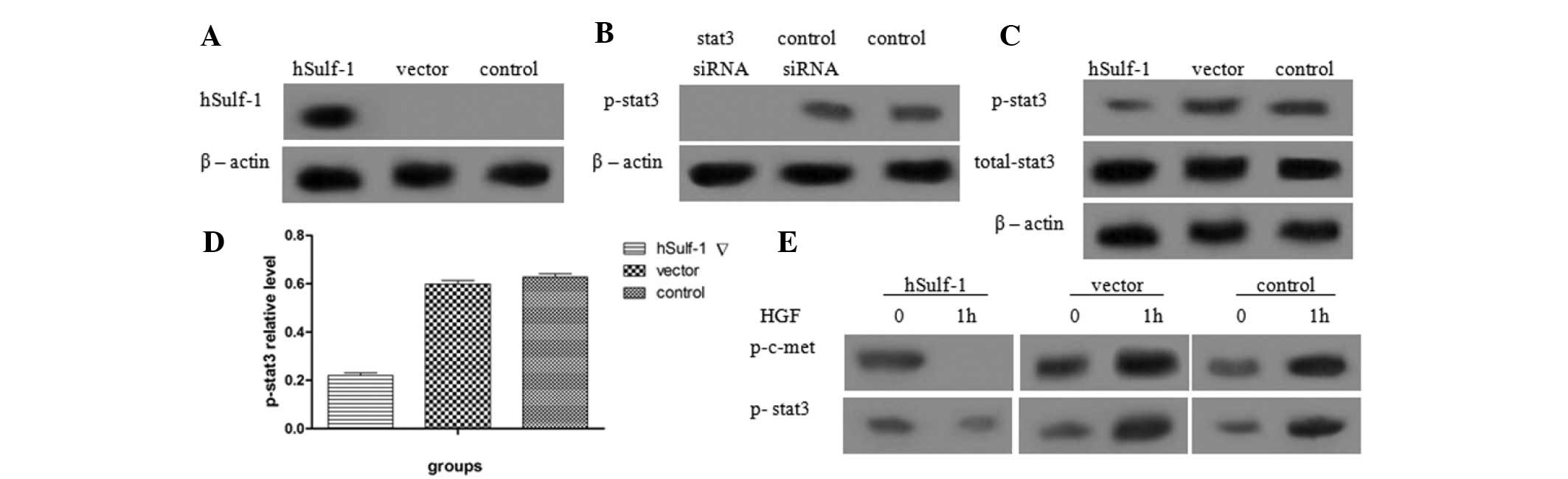
hSulf-1 inhibits the phosphorylation of
stat3 in HepG2 cells
Previous studies have shown that dysregulation of
the stat3 signaling pathway is involved with HCC development and
metastasis. Therefore, the effects of hSulf-1 on the stat3
signaling pathway were investigated in HepG2 cells. The expression
of hSulf-1 was detected in hSulf-1-transfected HepG2 cells and
HepG2 cells transfected with empty vector. Additionally, the
phosphorylation of stat3 in hSulf-1-transfected HepG2 cells and
control siRNA-transfected HepG2 cells was examined (Fig. 2A and B). The results showed that
overexpression of hSulf-1 reduced the phosphorylation of stat3 in
HepG2 cells, but did not affect the expression of total stat3
(Fig. 2C and D). Furthermore, when
hSulf-1-negative HepG2 cells were treated with 5 ng/ml HGF, the
phosphorylation of c-met and stat3 increased. By contrast, in
hSulf-1-transfected HepG2 cells, the phosphorylation of c-met and
stat3 decreased (Fig. 2E).
hSulf-1 inhibits the proliferation of
HepG2 cells through stat3 signaling
The effects of the stable transfection of hSulf-1
into hSulf-1-negative HepG2 cells on cell proliferation was
examined and measured by MTT assay. The viability of
hSulf-1-transfected cells was significantly decreased compared with
that of vector-transfected cells (Fig.
3C). To confirm whether the stat3 signaling pathway is involved
in mediating these inhibitory effects on cell proliferation, cells
were also transfected with stat3 siRNA to knockdown the expression
of stat3. The results showed that the inhibitory effects of hSulf-1
on cell proliferation were decreased following the knockdown of
stat3 (Fig. 3C). Cyclin D1 protein,
a protein involved in proliferation downstream of stat3, was also
expressed at extremely low levels following transfection with
hSulf-1 (Fig. 3A and B). In
addition, following the knockdown of stat3 in hSulf-1-transfected
cells, cell viability was further decreased (Fig. 3C). These results suggested that
hSulf-1 inhibits HepG2 cell growth by suppressing stat3
signaling.
 | Figure 3Effects of hSulf-1 on HepG2 cell
proliferation and migration. (A and B) Cyclin D1 expression was
examined by western blotting and then analyzed by densitometry.
Bands were normalized with the corresponding β-actin density. (C)
Cell viability was measured by MTT assay. The absorbance at 490 nm
was measured using a microtiter plate reader and the viable cell
number was calculated as a percentage. (D) Parental HepG2 cells,
HepG2 cells transfected with the hSulf-1 expression vector, HepG2
cells transfected with the empty vector or control siRNA, HepG2
cells transfected with stat3 siRNA and the hSulf-1 expression
vector, and HepG2 cells transfected with stat3 siRNA and the empty
vector were seeded in transwell plates at a density of
5×105 cells/ml. Cells that migrated through the
Transwell were quantified by Transwell migration assay.
▽P<0.05, vs. control or vector groups;
▽▽P<0.05, vs. control, vector or stat3 siRNA groups
and ▽▽▽P<0.05, vs. remaning groups. hSulf-1, human
sulfatase-1; stat3, signal transducer and activator of
transcription 3. |
hSulf-1 inhibits the migration of HepG2
cells
To further elucidate the relevance of hSulf-1
function in HCC, the effects of hSulf-1 on migration were
investigated by Transwell chamber assay. The results showed that
hSulf-1 transfection inhibits cell migration ability compared with
vector transfection alone. Additionally, stat3 siRNA transfection
decreased the migration of HepG2 cells and cells doubly transfected
with stat3 siRNA, and the hSulf-1 expression vector exhibited
further reductions in migration (Fig.
3D). These results suggested that hSulf-1 may affect
stat3-mediated migration.
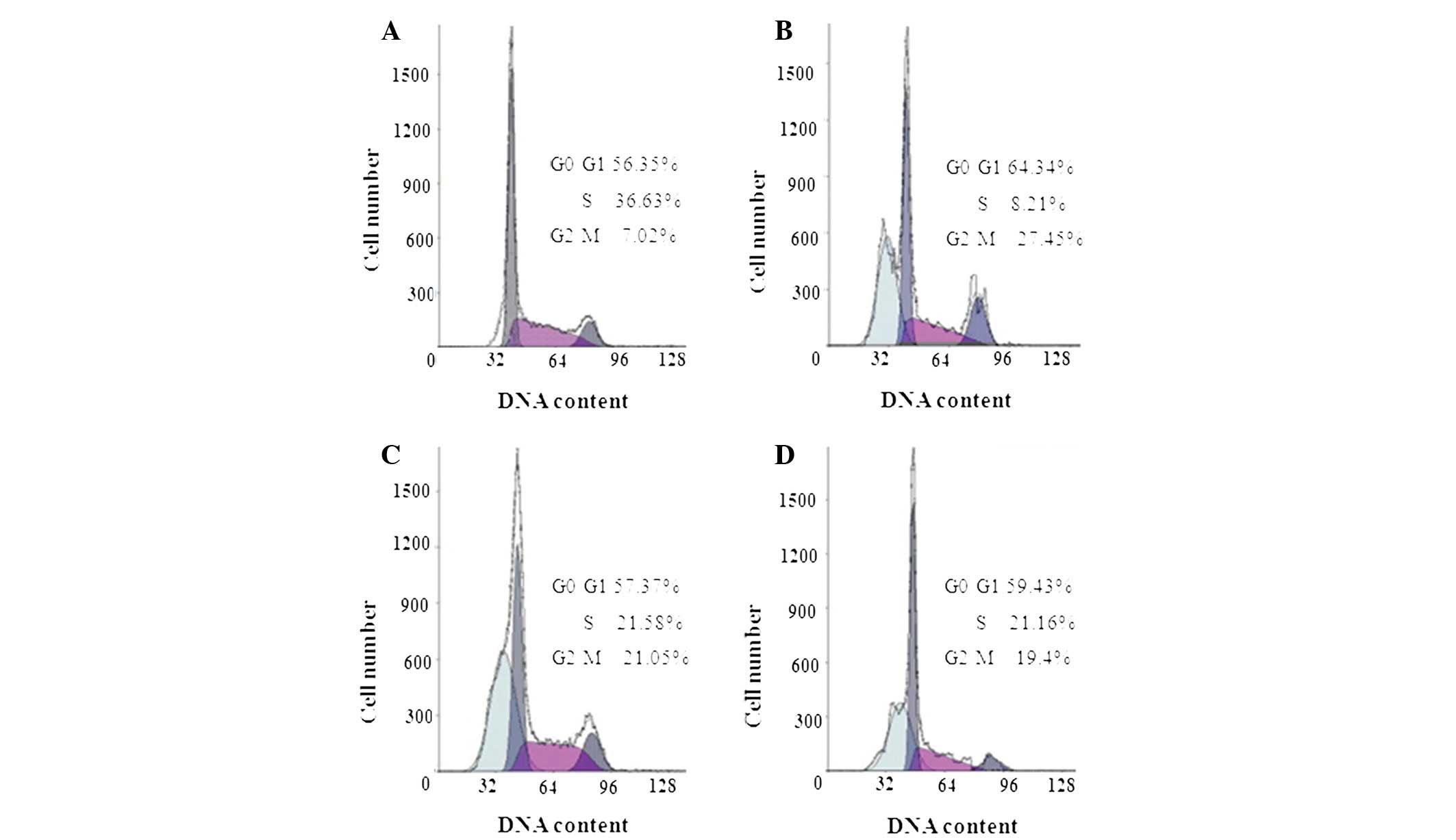
hSulf-1 induces
G0/G1 phase cell cycle arrest through stat3
signaling and promotes G2/M phase cell cycle arrest in
HepG2 cells
Next, it was investigated whether the
antiproliferative activity of hSulf-1 in HepG2 cells correlates
with cell cycle arrest. As demonstrated in Fig. 4A–D, cell cycle analysis revealed
that, compared with the vector and control siRNA groups, stat3
siRNA-transfected cells exhibited an increased number of cells in
the G0/G1 phase, while the number of cells in
the G2/M phase did not change. By contrast, when hSulf-1
was transfected into HepG2 cells, the number of cells in the
G0/G1 and G2/M phases was
increased. Therefore, it was assumed that hSulf-1 induces
G0/G1 phase arrest in HepG2 cells through the
stat3 signal pathway and also promotes G2/M phase
arrest.
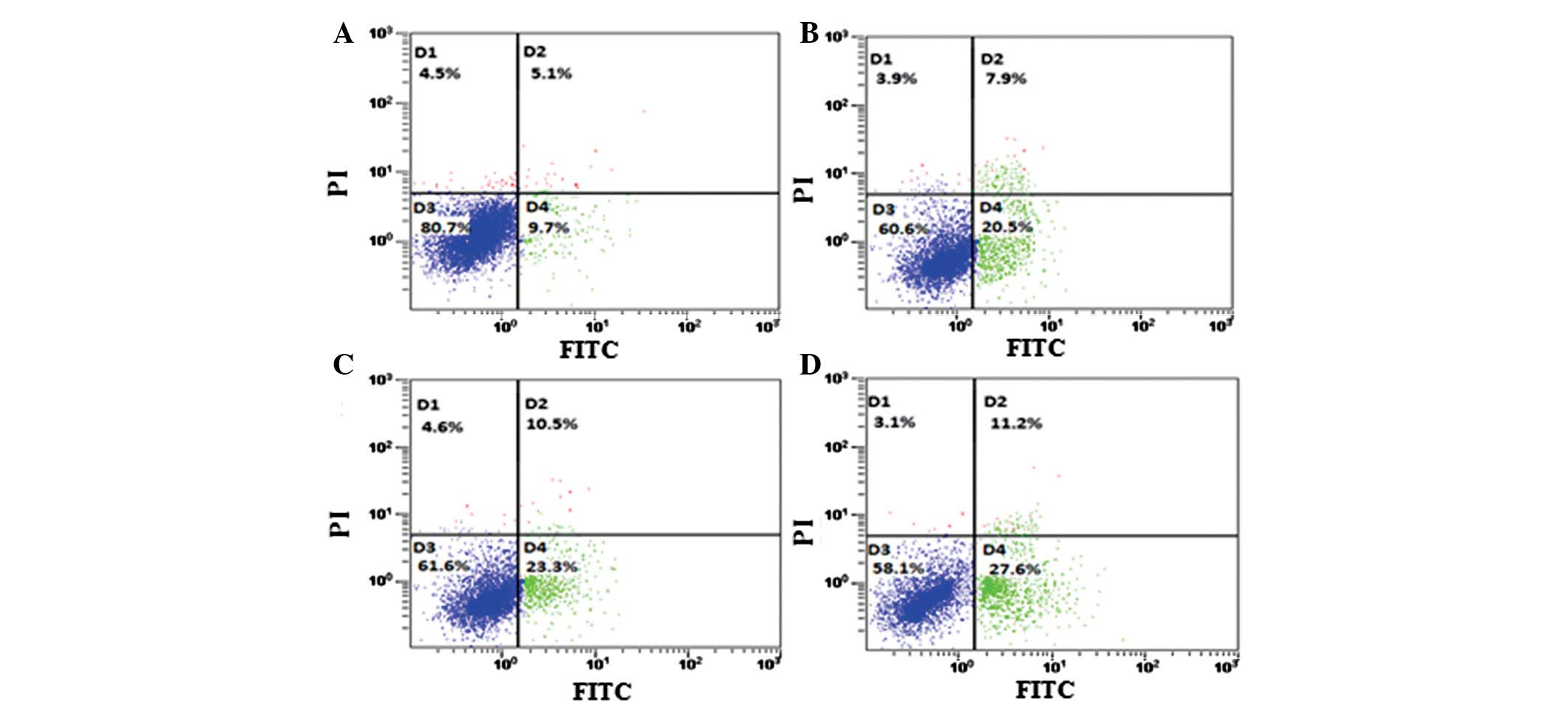
hSulf-1 promotes cisplatin-induced
apoptosis in HepG2 cells
Dysregulation of cell growth and apoptosis is
considered to lead to carcinogenesis. Therefore, the effects of
hSulf-1 on cisplatin-induced apoptosis were examined in HCC cells
using Annexin V/PI double-staining. hSulf-1-transfected HepG2 cells
showed a higher sensitivity to cisplatin-induced apoptosis compared
with that in control cells. Furthermore, the apoptosis rate in
HepG2 cells was decreased following transfection with stat3 siRNA
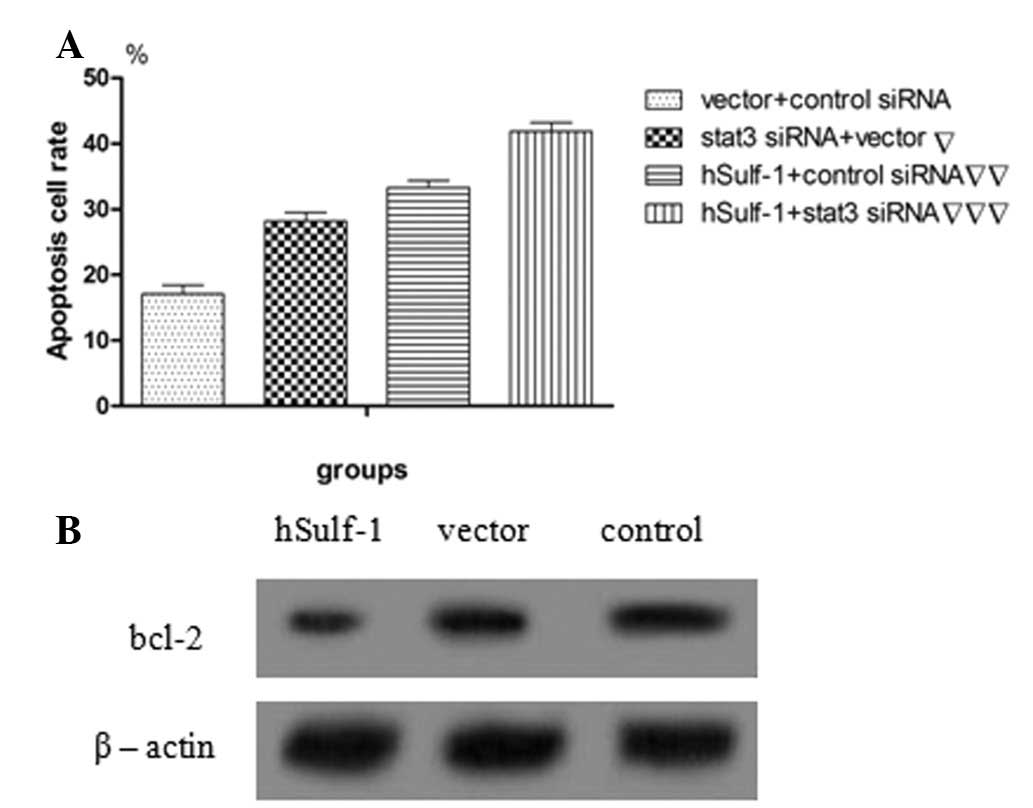
(Figs. 5A–D and 6A). Consistent with these observations,
the expression of the antiapoptotic protein, bcl-2, which signals
downstream of stat3, was found to increase in vector-transfected
HepG2 cells compared with that in hSulf-1-transfected HepG2 cells
(Fig. 6B). The results showed that
the hSulf-1 expression promotes cisplatin-induced apoptosis in
HepG2 cells and correlates with stat3 signaling to a certain
degree.
Discussion
Previous studies have reported that the hSulf-1
protein is an arylsulfatase that negatively regulates the sulfation
of HSPGs (5,26–27).
Notably, hSulf-1 desulfates cell surface HSPGs and subsequently
downregulates receptor tyrosine kinase signaling. Therefore,
hSulf-1 may be considered a tumor suppressor gene (8,12,28).
hSulf-1 also affects the binding of heparin-binding factors to
their receptors in several signaling pathways and suppresses the
phosphorylation and activation of receptor tyrosine kinases.
However, its molecular mechanisms are not well known. The stat3
signaling pathway, which may be activated by several growth factor
receptors, such as EGFR and PDGFR (19–20),
is known to be associated with the progression of HCC; thus, the
effects of hSulf-1 on stat3 signaling must also be explored in HCC
cells. The current study demonstrated that hSulf-1 expression is
downregulated in HCC cell lines, including HepG2, Hep3B, Huh-7 and
SMMC-7221. In various types of cancer, DNA methylation and histone
modification are involved in gene regulation. The present study
demonstrated that DNA methylation and histone modification regulate
hSulf-1 expression that synergistically effects the demethylating
agent and histone deacytelase inhibitor, resulting in the
expression of hSulf-1. This indicated that epigenetic modifications
of DNA and histones are a mechanism of hSulf-1 inactivation and
other mechanisms involved in the interaction between DNA
methylation and histone modification in HCC. In addition, the link
between hSulf-1 and stat3 signaling was further investigated. The
results revealed that hSulf-1 inhibits cell proliferation and
migration, induces G2/M phase cell cycle arrest and
promotes apoptosis through the suppression of stat3 signaling in
HepG2 cells. To verify the negative effects of hSulf-1 on cancer
angiogenesis, hSulf-1-expression vector and stat3 siRNA were
constructed. hSulf-1 expression was found to downregulate the
phosphorylation of stat3, but had no effect on total stat3
expression, indicating that hSulf-1 regulates the activity of
stat3.
HGF is a key regulating factor in cell
proliferation, motility and differentiation. We first hypothesized
that the stat3 signaling pathway may be activated by HGF, similar
to other growth factors. Subsequently, it was determined whether
hSulf-1 may mediate the HGF-dependent stat3 signal pathway.
Following HGF treatment, the phosphorylation of stat3 and the
expression of c-met decreased in hSulf-1-transfected HepG2 cells,
indicating that hSulf-1 suppresses the phosphorylation of stat3,
which is mediated by HGF. This effect may correlate with the
activity of receptor molecules since c-met activates the
phosphorylation of stat3. However, when hSulf-1 was re-expressed
following transfection with the hSulf-1 expression vector, c-met
expression was also inhibited, thereby further influencing the
phosphorylation of stat3.
In addition, the current study investigated the
correlation between hSulf-1 and stat3 signaling on the effects of
cell proliferation, migration and apoptosis. hSulf-1 has been
previously shown to inhibit cell growth and invasion through HGF,
FGF, HB-EGF, VEGF and wnt signaling (6,12,29–30).
In the present study, when HepG3 cells were transfected with stat3
siRNA to silence the expression of stat3, cell viability and
motility were decreased to a certain extent. This effect was more
apparent following the additional transfection of the HepG2 cells
with hSulf-1, suggesting that hSulf-1 is involved in the regulation
of cancer cell proliferation and migration, partly due to the
inhibition of stat3 signaling. Consistent with these observations,
the expression of cyclin D1, a downstream effector of stat3
signaling in the regulation of cell proliferation (31), decreased in hSulf-1-transfected
HepG2 cells. These results demonstrated that stat3 activation in
cancer cell proliferation and migration is mediated by the effects
of hSulf-1.
Next, the present study investigated whether the
antiproliferative activity of hSulf-1 in HepG2 cells was due to
cell cycle arrest and apoptosis. hSulf-1 was found to induce
G0/G1 arrest and apoptosis partly through the
stat3 signaling, and to also promote G2/M phase arrest.
The results of the Annexin V/PI double staining demonstrated that
hSulf-1 transfection increases the number of apoptotic HepG2 cells.
Previous studies have revealed that stat3 signaling is pivotal in
the antiapoptotic process, mediated through downstream proteins,
including bcl-2 and bcl-XL, which allow cells to resist apoptosis
(32–33). Additionally, stat3 signaling has
been shown to promote G0/G1 phase cell cycle
arrest (34–35). In the current study, cisplatin
treatment also induced G0/G1 phase arrest and
downregulation of stat3 signaling in HepG2 cells. Therefore, the
effects of hSulf-1 on apoptosis and G0/G1
phase cell cycle arrest may involve downregulation of the stat3
pathway. Furthermore, it was found that hSulf-1 expression alone
induces G2/M phase arrest in HepG2 cells; elucidation of
the mechanisms involved requires further investigation.
In conclusion, the results of the present study
demonstrated that hSulf-1 re-expression attenuates the
phosphorylation of stat3, suppresses cell proliferation and
motility and promotes cancer cell apoptosis. These effects
correlate with the downregulation of stat3 signaling in HepG2
cells. Thus, the study provides a novel insight into the molecular
mechanisms of hSulf-1 in HCC cells. These observations strengthen
the theory that hSulf-1 is a promising target for therapeutic
intervention through the stat3 signaling pathway in HCC.
References
|
1
|
el-Serag HB: Epidemiology of
hepatocellular carcinoma. Clin Liver Dis. 5:87–107. 2001.
View Article : Google Scholar
|
|
2
|
He J, Gu D, Wu X, et al: Major causes of
death among men and women in China. N Engl J Med. 353:1124–1134.
2005. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Lin X: Functions of heparan sulfate
proteoglycans in cell signaling during development. Development.
131:6009–6021. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Park PW, Reizes O and Bernfield M: Cell
surface heparan sulfate proteo-glycans: selective regulators of
ligand-receptor encounters. J Biol Chem. 275:29923–29926. 2000.
View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Kirkpatrick CA and Selleck SB: Heparan
sulfate proteoglycans at a glance. J Cell Sci. 120:1829–1832. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Kikuchi A and Yamamoto H: Regulation of
Wnt signalling by receptor-mediated endocytosis. J Biochem.
141:443–451. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Staub J, Chien J, Pan Y, et al: Epigenetic
silencing of HSulf-1 in ovarian cancer: implications
inchemoresistance. Oncogene. 26:4969–4978. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Lai J, Chien J, Staub J, et al: Loss of
HSulf-1 up-regulates heparin-binding growth factor signaling in
cancer. J Biol Chem. 278:23107–23117. 2003. View Article : Google Scholar
|
|
9
|
Chen Z, Fan JQ, Li J, et al: Promoter
hypermethylation correlates with the HSulf-1 silencing in human
breast and gastric cancer. Int J Cancer. 124:739–744. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Monneret C: Histone deacetylase
inhibitors. Eur J Med Chem. 40:1–13. 2005. View Article : Google Scholar
|
|
11
|
Pye DA, Vives RR, Turnbull JE, Hyde P and
Gallagher JT: Heparan sulfate oligosaccharides require
6-O-sulfation for promotion of basic fibroblast growth factor
mitogenic activity. J Biol Chem. 273:22936–22942. 1998. View Article : Google Scholar
|
|
12
|
Lai JP, Chien J, Strome SE, et al: HSulf-1
modulates HGF-mediated tumor cell invasion and signaling in head
and neck squamous carcinoma. Oncogene. 23:1439–1447. 2004.
View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Lai JP, Chien JR, Moser DR, et al: hSulf-1
sulfatase promotes apoptosis of hepatocellular cancer cells by
decreasing heparin-binding growth factor signaling.
Gastroenterology. 126:231–248. 2004. View Article : Google Scholar
|
|
14
|
Li J, Kleeff J, Abiatari I, et al:
Enhanced levels of Hsulf-1 interfere with heparin-binding growth
factor signaling in pancreatic cancer. Mol Cancer. 4:142005.
View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Dai Y, Yang Y, MacLeod V, et al: HSulf-1
and HSulf-2 are potent inhibitors of myeloma tumor growth in vivo.
J Biol Chem. 280:40066–40073. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Lai JP, Sandhu DS, Shire AM and Roberts
LR: The tumor suppressor function of human sulfatase 1 (SULF1) in
carcinogenesis. J Gastrointest Cancer. 39:149–158. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Narita K, Staub J, Chien J, et al: HSulf-1
inhibits angiogenesis and tumorigenesis in vivo. Cancer Res.
66:6025–6032. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Ji W, Yang J, Wang D, et al: hSulf-1 gene
exhibits anticancer efficacy through negatively regulating VEGFR-2
signaling in human cancers. PLoS ONE. 6:e232742011. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Bowman T, Garcia R, Turkson J and Jove R:
STATs in oncogenesis. Oncogene. 19:2474–2788. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Rahaman SO and Harbor PC: Inhibition of
constitutively active Stat3 suppresses proliferation and induces
apoptosis in glioblastoma multiforme cells. Oncogene. 21:8404–8413.
2002. View Article : Google Scholar
|
|
21
|
Iwamaru A, Szymanski S, Iwado E, et al: A
novel inhibitor of the stat3 pathway induces apoptosis in malignant
glioma cells both in vitro and in vivo. Oncogene. 26:2435–2444.
2007. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Jacoby JJ, Kalinowski A, Liu MG, et al:
Cardiomyocyte restricted knockout of stat3 results in higher
sensitivity to inflammation, cardiac fibrosis, and heart failure
with advanced age. Proc Natl Acad Sci USA. 100:12929–12934. 2003.
View Article : Google Scholar
|
|
23
|
Kurdi M and Booz GW: Can the protective
actions of JAK-stat in the heart be exploited therapeutically?
Parsing the regulation of interleukin-6-type cytokine signaling. J
Cardiovasc Pharmacol. 50:126–141. 2007. View Article : Google Scholar
|
|
24
|
Bores P and Miller CM: Hepatocyte growth
factor: a multifunctional cytokine. Lancet. 345:293–295. 1995.
View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Lee KH, Choi EY, Hyun MS, et al: Role of
hepatocyte growth factor/c-Met signaling in regulating urokinase
plasminogen activator on invasiveness in human hepatocellular
carcinoma: a potential therapeutic target. Clin Exp Metastasis.
25:89–96. 2008. View Article : Google Scholar
|
|
26
|
Moore AE, Greenhough A, Roberts HR, et al:
HGF/Met signaling promotes PGE2 biogenesis via regulation of COX-2
and 15-PGDH expression in colorectal cancer cells. Carcinogenesis.
30:1796–1804. 2009. View Article : Google Scholar
|
|
27
|
Ai X, Do AT, Kusche-Gullberg M, Lindahl U,
Lu K and Emerson CP Jr: Substrate specificity and domain functions
of extracellular heparan sulfate 6-O-endosulfatases, QSulf1 and
QSulf2. J Biol Chem. 281:4969–4976. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Diez-Roux G and Ballabio A: Sulfatases and
human disease. Annu Rev Genomics Hum Genet. 6:355–379. 2005.
View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Forsten-Williams K, Chu CL, Fannon M,
Buczek-Thomas JA and Nugent MA: Control of growth factor networks
by heparan sulfate proteoglycans. Ann Biomed Eng. 36:2134–2148.
2008. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Dhoot GK, Gustafsson MK, Ai X, Sun W,
Standiford DM and Emerson CP Jr: Regulation of Wnt signaling and
embryo patterning by an extracellular sulfatase. Science.
293:1663–1666. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Ornitz DM: FGFs, heparan sulfate and
FGFRs: complex interactions essential for development. Bioessays.
22:108–112. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Fletcher S, Turkson J and Gunning PT:
Molecular approaches towards the inhibition of the signal
transducer and activator of transcription 3 (Stat3) protein.
ChemMedChem. 3:1159–1168. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Chen Z and Han ZC: STAT3: a critical
transcription activator in angiogenesis. Med Res Rev. 28:185–200.
2008. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Frank DA: STAT3 as a central mediator of
neoplastic cellular transformation. Cancer Lett. 251:199–210. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Yu ZY, Huang R, Xiao H, et al:
Fluacrypyrim, a novel stat3 activation inhibitor, induces cell
cycle arrest and apoptosis in cancer cells harboring
constitutively-active stat3. Int J Cancer. 127:1259–1270. 2010.
View Article : Google Scholar : PubMed/NCBI
|