1. Introduction
Epigenetic mechanisms are important for human
carcinogenesis. Epigenetic abnormalities are involved in the early
stages of tumorigenesis and may trigger genetic events leading to
tumor development (1). Epigenetic
alterations result in aberrant gene expression profiles that do not
result from changes in the primary nucleic acid sequence, but
rather involve covalent modification of nucleotide bases in normal
DNA sequences. DNA methylation is the most commonly studied
epigenetic mechanism and is crucial in the development of nearly
all types of cancer (2). Aberrant
DNA methylation can result in altered patterns of gene expression
leading to cancerous features. During carcinogenesis, numerous
tumor suppressor genes are silenced by DNA methylation. DNA
methylation does not change genetic information, however, alters
the readability of the DNA and results in the inactivation of genes
by subsequent repression of transcription. Tumors often possess
decreased genomic DNA methylation levels and hypermethylated CpG
islands (3).
Hepatocellular carcinoma (HCC) is the primary
malignant tumor of the liver and the third leading cause of
cancer-related mortality worldwide (4,5) Rising
incidence and mortality rates from HCC have been observed in the
majority of countries, particularly in Asia (6). During human HCC development and
progression, DNA hypomethylation and regional CpG hypermethylation
are dominant events (7). DNA
methylation can occur as part of normal development, however, can
also occur as a result of age or exposure to risk factors,
potentially resulting in carcinogenesis in tissues with normal DNA
sequences. HCC typically occurs in the setting of chronic
inflammation that is secondary to the hepatitis B virus (HBV) or
hepatitis C virus (HCV) infection, or alcoholism; each increases
the risk for hepatocarcinogenesis. Furthermore, the HCV infection
has been found to accelerate the methylation process in HCC
(8).
Microarray analysis of HCC tissues has identified
novel genes with cancer-specific methylation and 221 novel DNA
methylation markers for HCC (9,10).
TNFRSF10C, HOXA9, NPY and IRF5 were found to be frequently
hypermethylated in HCC tissues and their methylation was identified
to be closely associated with the inactivation of gene expression
(9). In HCC cell lines, regional
DNA methylation in tumor suppressor genes has been reported
(11,12). The frequency of hypermethylation of
tumor suppressor genes is relatively high in HCC, indicating that
regional DNA hypermethylation is involved in hepatocarcinogenesis
(13). In the present review,
aberrant DNA methylation in tumor suppression of HCC is
discussed.
2. Methylation hot spots on different
chromosomes associated with early stage hepatocarcinogenesis
The epigenetic alteration of promoters by
methylation is an alternative mechanism for the inactivation of
tumor suppressor genes. Methylation hot spots on different
chromosomes have been reported during early stage
hepatocarcinogenesis. On chromosome 16, aberrant DNA methylation
participates in the precancerous stage of hepatocarcinogenesis by
preceding or causing loss of heterozygosity (14). At the D17S5 locus, aberrant DNA
hypermethylation may participate in hepatocarcinogenesis during the
early developmental stages and malignant progression of HCC
(15). On chromosome 3,
hypermethylation of multiple tumor suppressor genes, including
Ras-association domain family 1, isoform A (RASSF1A), BLU and
fragile histidine triad (FHIT), is a common and early event in
hepatocarcinogenesis, as observed in human HCC tissues.
Furthermore, CRBP1 methylation may be involved in later-stage
carcinogenesis (16).
3. Methylation status of CpG islands
A CpG island is an ~1-kb DNA sequence with a high
density of CpG dinucleotides and ~70% of human genes harbor CpG
islands in their promoters (17,18).
Promoter CpG island hypermethylation is an important mechanism for
inactivation of tumor suppressor genes or tumor-related genes in
human cancers and occurs in virtually all human cancer types
(19). In a HCC rat model, the
stages of multistage carcinogenesis following initiation are driven
primarily by carcinogen-induced epigenetic alterations, including
altered global histone lysine methylation patterns; increased
histone H3 lysine 9 and histone H3 lysine 27 trimethylation in the
promoter regions of the tumor suppressor genes RASSF1A, p16
(INK4a), suppressor of cytokine signaling (SOCS)1, E-cadherin 1
(CDH1)and Cx26, and early RASSF1A; and p16 (INK4a) promoter CpG
island hypermethylation. These changes are accompanied by
dysregulation of the balance between cell proliferation and
apoptosis, a fundamental protumorigenic event in
hepatocarcinogenesis (20).
Among the gene mutations mentioned above, p16
(INK4a) is important in regulating the cell cycle and mutations in
p16 (INK4a) increase the risk of developing a variety of cancers.
Adjacent to p16 (INK4a) is p15 (INK4b), which is also frequently
mutated and deleted in numerous types of tumor; thus, p16 (INK4a)
and p15 (INK4b) are candidates for putative tumor suppressor genes.
In tumors of HCC patients from Japan, p16 (INK4a) was identified to
be inactivated by extensive CpG methylation (21). However, tumors of HCC patients from
Taiwan showed no aberrant 5′-CpG island hypermethylation of p16
(INK4a) or p15 (INK4b) in any primary tumors (22). The findings from different
geographic regions vary. Environmental factors may affect the
frequency and concordance of the degree of hypermethylation in
multiple genes in HCC tumors, leading to the observed geographic
variations in CpG island methylation status (23). CpG island methylation phenotype may
be caused or facilitated by proliferative stimuli that are
associated with environmental exposures. The precise mechanism of
generating the CpG island phenotype requires investigation;
however, the phenotype may contribute to screening, prevention or
treatment of HCC in different geographic regions.
The HBV infection has a strong correlation with HCC
occurrence (24–27) and aberrant CpG island methylation of
genes has been recognized in hepatitis virus-related HCC. In
studies regarding hepatitis virus-related HCC, Kiran et al
(28) investigated promoter region
methylation of a panel of six tumor suppressor genes: p16 (INK4a),
p15 (INK4b), CDH1, glutathione S-transferase P (GSTP)1, SOCS1 and
adenomatous polyposis coli (APC). The authors identified that the
p15 (INK4b) methylation frequency and methylation allele density
were higher in HCC than that in hepatitis (28). Furthermore, in HBV-associated HCC,
the intensive hypermethylation of the CpG island of the tumor
suppressor gene RASSF1A may be pathologically important in this
tumor type, based on studies of human HBV-associated HCC tissues
and HCC cell lines (Hep3B, HepG2, SK-HEP-1 and Huh-7) (29). In two HCC cell lines (HepG2 and
Hep3B) RASSF1A can be inactivated and treatment of the cell lines
with a DNA methylation inhibitor reactivates RASSF1A transcription
(30).
A series of CpG island methylation alterations have
been observed in the HCC cell lines Hep3B, HepG2, PLC/RPF/5/RPF/5,
SMMC-7721, BEL-7402, MHCC97-H, MHCC97-L, HCCLM3 and HCCLM6. CpG
island hypermethylation of tumor suppressor genes leads to a
decrease in their expression (31,32).
4. DNA methyltransferases (DNMTs)
Aberrant DNA methylation on CpG islands is one of
the most consistent epigenetic changes in human cancers and the
methylation process is catalyzed by DNMTs. In mammals, five members
of the DNMT family have been reported, DNMT1, DNMT2, DNMT3a, DNMT3b
and DNMT3l. Among these proteins, only DNMT1, DNMT3a and DNMT3b
exhibit methyltransferase activity. DNMT3a and DNMT3b establish
methylation patterns at specific sequences, while DNMT1 maintains
DNA methylation during replication by copying the methylation
pattern of the parent DNA strand onto the newly synthesized strand
(33,34). Abnormal variations of DNMTs
participate in hepatocarcinogenesis.
In human hepatocarcinogenesis, DNMT1, DNMT3a and
DNMT3b show a progressively increasing expression from normal
liver, to chronic hepatitis/cirrhosis, to HCC (35). In the early and late stages of HCC
development, global DNA hypomethylation and aberrant expression of
DNMT1 and DNMT3b were identified in a glycine N-methyltransferase
gene knockout mouse model for HCC (36). In a human HCC cell line, the
depletion of DNMT3a suppressed cell proliferation and restored
phosphatase and tensin homolog (PTEN), which is a crucial tumor
suppressor in HCC. This indicated that PTEN may be the target of
DNMT3a (37). Fan et al
(38) observed a novel target of
DNMT3b, metastasis suppressor 1 (MTSS1), which acts as a tumor
suppressor in HCC. MTSS1 was repressed by DNMT3b via a DNA
methylation-independent mechanism (38).
Hepatitis-related HCC in the DNMT
mechanism
The hepatitis B virus X (HBx) protein is involved in
epigenetic modifications during hepatocarcinogenesis. Park et
al (39) found that HBx
repressed insulin-like growth factor-3 expression through de
novo methylation via DNMT3a1 and DNMT3a2. Furthermore, HBx
inhibited SP1 binding by recruiting methyl CpG binding protein 2 to
a newly methylated SP1 binding element. HBx also induced global
hypomethylation of satellite 2 repeat sequences by downregulating
DNMT3b (39). In addition, the
prevalence of these specific methylation abnormalities that are
induced by HBx was identified to be significantly correlated with
HBx expression in HBV-infected HCC patients (39). These findings indicated a potential
association between DNMTs and HBV-infected HCC.
MicroRNAs (miRs) have also been identified to
participate in the regulation of abnormal DNA methylation status in
HBV-related HCC. By combining with the 3′-noncoding region of
corresponding target mRNAs, miRs act as potent negative regulators
of protein translation by disrupting mRNA stability, which affects
the post-transcriptional regulation of genetic expression and is
physiologically important (40).
Zhang et al (41) identified
that the expression of miR-152 was downregulated in the livers of
HBx transgenic mice compared with the livers of wild-type mice. The
authors also investigated the function of miR-152 as a tumor
suppressor in epigenetic aberrations of HBV-related HCC (42). In HCC cell lines, the forced
expression of miR-152 resulted in a marked reduction in the
expression of DNMT1 by directly targeting the 3′-untranslated
regions of DNMT1, which in turn led to a decrease in global DNA
methylation. Inhibition of miR-152 resulted in global DNA
hypermethylation and increased the methylation levels of two tumor
suppressor genes, GSTP1 and CDH1 (42). miR-101 was also reported to be
downregulated by HBx and to induce aberrant DNA methylation by
targeting DNMT3a (43). Thus, miRs
may participate in hepatocarcinogenesis by directly targeting
DNMTs, during which HBx may act as a regulator (Fig. 1). miRs with key roles in regulation
may be potential targets for inhibiting the development of
HBV-related HCC.
Reactive oxygen species (ROS) and the
DNMT mechanism
In addition to being involved in inflammatory
stimuli and associated proliferative changes associated with HBV,
oxidative damage, which is associated with chronic inflammation
directly affects the methylation status of DNA via DNMTs in HCC.
ROS increase Snail expression, which recruits histone deacetylase 1
and DNMT1, and induces hypermethylation of the CDH1 promoter
(44). Since CDH1 is a regulator of
the epithelial-to-mesenchymal transition, this result is
potentially relevant to understanding the activity of ROS in
silencing tumor suppressor genes, and in subsequent tumor
progression and metastasis. ROS accumulation mediates signal
transduction cascades, and the activation of stress kinases and
phosphorylation of substrates (45–47).
Various studies have shown reduced histone deacetylases (HDAC)
activity during oxidative stress (47–49)
and in HCC, the expression of HDACs is associated with the HCC
grade (50). In HCC cell lines,
deacetylase inhibitors exert a dual effect on DNMT activity and
expression, with rapid inhibition of enzyme activity from
interference with post-translational acetylation and a delayed
effect on transcriptional control of DNMT genes by HDACs or miR
mechanisms (51).
5. DNA methylation in the cell cycle
DNA methylation contributes to HCC tumorigenesis by
regulating cell proliferation. For example, DNA methylation of the
promoter region of a candidate SRY box-containing gene 17 is found
in 82% of HCC tissues and is associated with nuclear accumulation
of β-catenin (52). β-catenin is an
indispensable component of the canonical WNT signaling pathway
(53) and is involved in cell
differentiation, migration and proliferation during embryonic
development and adult homeostasis (5). In HCC cell lines, the expression of
family with sequence similarity 43, a novel tumor suppressor gene,
reduced cell growth and colony formation in vitro, delayed
the cell cycle and regulated DNA methylation (54). These findings indicated the
involvement of DNA methylation in HCC by regulating cell
proliferation.
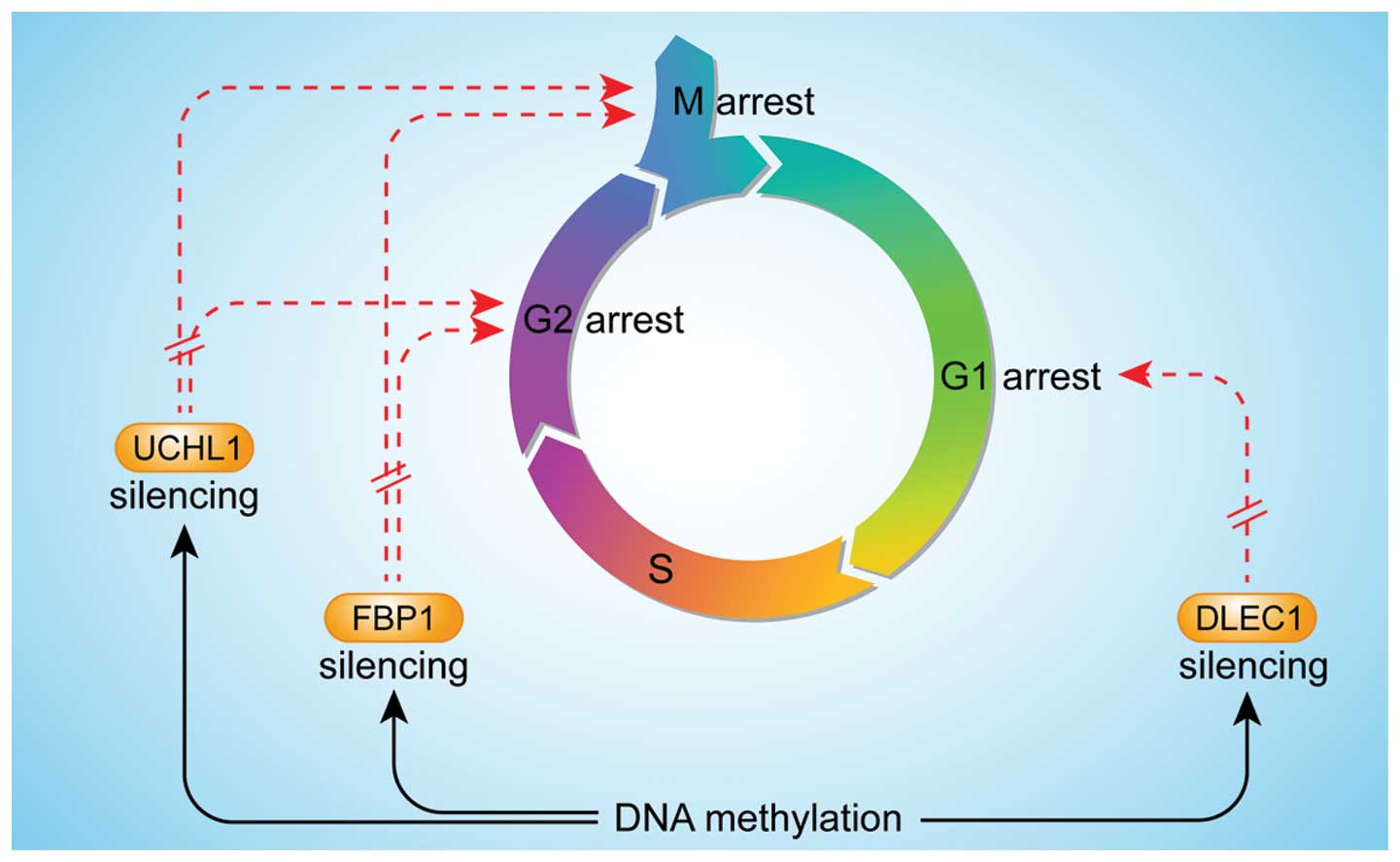
The cell cycle consists of four distinct phases, G1,
S, G2 and M. In HCC cell lines, the tumor suppressor gene, deleted
in lung and esophageal cancer 1 (DLEC1) decreases cell growth and
cell size, and induces G1 arrest in the cell cycle, whereas DNA
methylation silences DLEC1 (55). A
comparable effect of DNA methylation on ubiquitin carboxyl-terminal
hydrolase L1 (UCHL1) and fructose-1,6-bisphosphatase-1 (FBP1)
indicates that they are tumor suppressors. UCHL1 silencing is
reversed by genetic demethylation of the promoter, indicating
direct epigenetic silencing. Restoring UCHL1 expression in silenced
cell lines significantly inhibits their growth and colony formation
ability, by inhibiting cell proliferation through cell cycle arrest
in the G2/M phase and inducing apoptosis through the intrinsic
caspase-dependent pathway (56). In
addition, promoter hypermethylation mediates downregulation of FBP1
in human HCC, whereas restoring the FBP1 expression in the cells in
which FBP1 expression is low significantly inhibits cell growth
through the induction of G2-M cell cycle arrest (57) (Fig.
2).
In addition to the tumor suppressor genes mentioned
above, DNA methylation is involved in silencing the expression and
function of mac25/insulin-like growth factor binding protein-7,
methylthioadenosine phosphorylase and TMEM7 in HCC (58–60).
6. Clinical applications
Patient prognosis
The accumulating evidence for DNA methylation of
tumor suppressor genes in HCC presents a potential clinical
benefit. First, DNA methylation of tumor suppressor genes may aid
in predicting the individual patient risk of tumor development. In
chronic HCV patients, the methylation frequency of tumor suppressor
genes, such as HIC1, GSTP1, SOCS1, RASSF1, CDKN2A, APC, RUNX3 and
PRDM2 are associated with shorter time-to-HCC, and the number of
methylated genes is an independent risk factor for HCC (61). These results indicate that
characteristic patterns of altered DNA methylation are critical for
the earliest steps of hepatocarcinogenesis and may predict the
emergence of human HCC in HCV patients (61). Second, DNA methylation of tumor
suppressor genes is associated with tumor biological features. For
example, DLEC1 methylation is associated with the American Joint
Committee on Cancer tumor staging (55).
Third, the DNA methylation status of tumor
suppressor genes is valuable as a prognostic indicator in HCC
patients. Calvisi et al (7)
analyzed the global levels of DNA methylation and the methylation
status of 105 putative tumor suppressor genes and identified that
the extent of genome-wide hypomethylation and CpG hypermethylation
correlated with the clinical outcome of HCC patients. The promoter
DNA methylation of the Klotho gene was a predictive factor for poor
HCC prognosis (62). Univariate and
multivariate survival analysis revealed that HIST1H2AE methylation
status is closely correlated with overall survival (10). The increased expression of DNMT3a
and DNMT3b is suggested to be a predictor of poor HCC survival
(35). MGMT methylation is
considered to predict a shorter disease-free survival time
(63), however, the sample sizes of
these studies were small. Further studies with larger samples may
aid in selecting the predictors of HCC survival.
The retinoblastoma protein-interacting zinc finger
(RIZ1) gene performs tumor suppressor activity and is frequently
silenced in numerous human cancers, including HCC (64–67).
Promoter methylation of RIZ1 and H3K9 modifications act together in
HCC to silence the RIZ1 gene, which is involved in HCC
tumorigenesis, particularly in the early stage of the disease
(68,69). Comparative analysis of promoter
methylation and gene expression endpoints between tumor and
non-tumor tissues from HCV-positive patients with HCC showed that
RIZ1 methylation and increased levels of LINE-1 hypomethylation in
non-tumor tissues are associated with time to recurrence. This
underscores the importance of assessing the epigenetic state of
liver remnants (70). In addition
to RIZ1, methylation of CpG sites of the potential tumor
suppressors, CFH and MYRIP, is associated with HCC recurrence
(71).
Clinical testing
Methods that are based upon DNA methylation patterns
are useful in clinical testing for HCC. Iyer et al (72) compared tumor methylation profiles
for the tumor suppressor genes APC, FHIT, p15, p16 and CDH1 in
tumor tissues and plasma, and found that plasma DNA can be used for
the reliable assessment of methylation profiles in HCC patients in
an Egyptian population (72). To
assess the medical applicability of CpG methylation as a molecular
marker for cancer diagnosis, Kimura et al (73) established a novel system to
determine DNA methylation based on TaqMan polymerase chain reaction
combined with a methyl-binding-domain polypeptide 2. The
availability of DNA methylation profiles for cancer diagnosis
enable clinical predictions to be made from pre-therapy biopsies,
paraffin-embedded samples or plasma DNA.
7. Conclusion
Aberrant DNA methylation results in altered gene
expression, leading to cancerous features. During
hepatocarcinogenesis, numerous tumor suppressor genes are silenced
by DNA methylation. Hypermethylation of promoter CpG islands is an
important mechanism for inactivating tumor suppressor genes in HCC.
Although promoter CpG island hypermethylation of p16 (INK4a) and
p15 (INK4b) may increase the risk of developing HCC, individuals
from different geographic regions exhibit different methylation
statuses for CpG islands. Aberrant DNA methylation of CpG islands
are catalyzed by DNMTs, thus, abnormal variations of DNMTs may
participate in hepatocarcinogenesis. Aberrant CpG island
methylation of genes and DNMTs is involved in hepatitis-related
HCC. miRs and ROS may participate in hepatocarcinogenesis by
directly targeting DNMTs. Furthermore, DNA methylation may
contribute to HCC tumorigenesis by regulating the cell cycle. Based
on the importance of DNA methylation in tumor suppression in HCC,
particularly the patterns of DNA methylation, it may predict the
risk of tumor development, tumor staging, patient survival and HCC
recurrence.
Abbreviations:
|
HCC
|
hepatocellular carcinoma
|
|
HBV
|
hepatitis B virus
|
|
DNMTs
|
DNA methyltransferases
|
|
HCV
|
hepatitis C virus
|
|
HBx
|
hepatitis B virus X
|
|
miR
|
microRNA
|
|
GSTP
|
glutathione S-transferase P
|
|
CDH1
|
E-cadherin 1
|
|
ROS
|
reactive oxygen species
|
|
UCHL1
|
ubiquitin carboxyl-terminal hydrolase
L1
|
|
FBP1
|
fructose-1,6-bisphosphatase-1
|
References
|
1
|
Dumitrescu RG: Epigenetic targets in
cancer epidemiology. Methods Mol Biol. 471:457–467. 2009.
|
|
2
|
Jaenisch R and Bird A: Epigenetic
regulation of gene expression: how the genome integrates intrinsic
and environmental signals. Nat Genet. 33(Suppl): S245–S254.
2003.
|
|
3
|
Liu WR, Shi YH, Peng YF and Fan J:
Epigenetics of hepatocellular carcinoma, a new horizon. Chin Med J
(Engl). 125:2349–2360. 2012.
|
|
4
|
Logan CY and Nusse R: The Wnt signaling
pathway in development and disease. Annu Rev Cell Dev Biol.
20:781–810. 2004.
|
|
5
|
Kim M, Lee HC, Tsedensodnom O, et al:
Functional interaction between Wnt3 and Frizzled-7 leads to
activation of the Wnt/betacatenin signaling pathway in
hepatocellular carcinoma cells. J Hepatol. 48:780–791. 2008.
|
|
6
|
Parkin DM, Bray F, Ferlay J and Pisani P:
Global cancer statistics 2002. CA Cancer J Clin. 55:74–108.
2005.
|
|
7
|
Calvisi DF, Ladu S, Gorden A, et al:
Mechanistic and prognostic significance of aberrant methylation in
the molecular pathogenesis of human hepatocellular carcinoma. J
Clin Invest. 117:2713–2722. 2007.
|
|
8
|
Nishida N, Nagasaka T, Nishimura T, Ikai
I, Boland CR and Goel A: Aberrant methylation of multiple tumor
suppressor genes in aging liver, chronic hepatitis, and
hepatocellular carcinoma. Hepatology. 47:908–918. 2008.
|
|
9
|
Shin SH, Kim BH, Jang JJ, Suh KS and Kang
GH: Identification of novel methylation markers in hepatocellular
carcinoma using a methylation array. J Korean Med Sci.
25:1152–1159. 2010.
|
|
10
|
Jung N, Won JK, Kim BH, et al:
Pharmacological unmasking microarray approach-based discovery of
novel DNA methylation markers for hepatocellular carcinoma. J
Korean Med Sci. 27:594–604. 2012.
|
|
11
|
Huang J, Zhang YL, Teng XM, et al:
Down-regulation of SFRP1 as a putative tumor suppressor gene can
contribute to human hepatocellular carcinoma. BMC Cancer.
7:1262007.
|
|
12
|
Zhang C, Li H, Zhou G, et al:
Transcriptional silencing of the TMS1/ASC tumour suppressor gene by
an epigenetic mechanism in hepatocellular carcinoma cells. J
Pathol. 212:134–142. 2007.
|
|
13
|
Park HJ, Yu E and Shim YH: DNA
methyltransferase expression and DNA hypermethylation in human
hepatocellular carcinoma. Cancer Lett. 233:271–278. 2006.
|
|
14
|
Kanai Y, Ushijima S, Tsuda H, Sakamoto M
and Hirohashi S: Aberrant DNA methylation precedes loss of
heterozygosity on chromosome 16 in chronic hepatitis and liver
cirrhosis. Cancer Lett. 148:73–80. 2000.
|
|
15
|
Kanai Y, Hui AM, Sun L, et al: DNA
hypermethylation at the D17S5 locus and reduced HIC-1 mRNA
expression are associated with hepatocarcinogenesis. Hepatology.
29:703–709. 1999.
|
|
16
|
Zhang X, Li HM, Liu Z, et al: Loss of
heterozygosity and methylation of multiple tumor suppressor genes
on chromosome 3 in hepatocellular carcinoma. J Gastroenterol.
48:132–143. 2012.
|
|
17
|
Saxonov S, Berg P and Brutlag DL: A
genome-wide analysis of CpG dinucleotides in the human genome
distinguishes two distinct classes of promoters. Proc Natl Acad Sci
USA. 103:1412–1417. 2006.
|
|
18
|
Weber M, Hellmann I, Stadler MB, et al:
Distribution, silencing potential and evolutionary impact of
promoter DNA methylation in the human genome. Nat Genet.
39:457–466. 2007.
|
|
19
|
Baylin SB and Chen WY: Aberrant gene
silencing in tumor progression, implications for control of cancer.
Cold Spring Harb Symp Quant Biol. 70:427–433. 2005.
|
|
20
|
Pogribny IP, Muskhelishvili L, Tryndyak VP
and Beland FA: The role of epigenetic events in genotoxic
hepatocarcinogenesis induced by 2-acetylaminofluorene. Mutat Res.
722:106–113. 2011.
|
|
21
|
Matsuda Y, Ichida T, Matsuzawa J, Sugimura
K and Asakura H: p16 (INK4) is inactivated by extensive CpG
methylation in human hepatocellular carcinoma. Gastroenterology.
116:394–400. 1999.
|
|
22
|
Lin YW, Chen CH, Huang GT, et al:
Infrequent mutations and no methylation of CDKN2A (P16/MTS1) and
CDKN2B (p15/MTS2) in hepatocellular carcinoma in Taiwan. Eur J
Cancer. 34:1789–1795. 1998.
|
|
23
|
Shen L, Ahuja N, Shen Y, et al: DNA
methylation and environmental exposures in human hepatocellular
carcinoma. J Natl Cancer Inst. 94:755–761. 2002.
|
|
24
|
Marotta F, Vangieri B, Cecere A and
Gattoni A: The pathogenesis of hepatocellular carcinoma is
multifactorial event. Novel immunological treatment in prospect.
Clin Ter. 155:187–199. 2004.
|
|
25
|
Cougot D, Neuveut C and Buendia MA: HBV
induced carcinogenesis. J Clin Virol. 34(Suppl 1): S75–S78.
2005.
|
|
26
|
Anzola M: Hepatocellular carcinoma, role
of hepatitis B and hepatitis C viruses proteins in
hepatocarcinogenesis. J Viral Hepat. 11:383–393. 2004.
|
|
27
|
Barazani Y, Hiatt JR, Tong MJ and Busuttil
RW: Chronic viral hepatitis and hepatocellular carcinoma. World J
Surg. 31:1243–1248. 2007.
|
|
28
|
Kiran M, Chawla YK and Kaur J: Methylation
profiling of tumor suppressor genes and oncogenes in hepatitis
virus-related hepatocellular carcinoma in northern India. Cancer
Genet Cytogenet. 195:112–119. 2009.
|
|
29
|
Zhong S, Yeo W, Tang MW, et al: Intensive
hypermethylation of the CpG island of Ras association domain family
1A in hepatitis B virus-associated hepatocellular carcinomas. Clin
Cancer Res. 9:3376–3382. 2003.
|
|
30
|
Schagdarsurengin U, Wilkens L, Steinemann
D, Flemming P, et al: Frequent epigenetic inactivation of the
RASSF1A gene in hepatocellular carcinoma. Oncogene. 22:1866–1871.
2003.
|
|
31
|
Zheng D, Liu BB, Liu YK, et al: Screening
for differential methylation status by CpG island microarray in the
hepatocellular carcinoma cell lines. Zhonghua Zhong Liu Za Zhi.
30:891–896. 2008.(In Chinese).
|
|
32
|
Liu BB, Zheng D, Liu YK, et al:
Array-based profiling of the differential methylation status of CpG
islands in hepatocellular carcinoma cell lines. Oncol Lett.
1:815–820. 2010.
|
|
33
|
Jurkowska RZ and Jeltsch A: Silencing of
gene expression by targeted DNA methylation: concepts and
approaches. Methods Mol Biol. 649:149–161. 2010.
|
|
34
|
Kim JK, Samaranayake M and Pradhan S:
Epigenetic mechanisms in mammals. Cell Mol Life Sci. 66:596–612.
2009.
|
|
35
|
Oh BK, Kim H, Park HJ, et al: DNA
methyltransferase expression and DNA methylation in human
hepatocellular carcinoma and their clinicopathological correlation.
Int J Mol Med. 20:65–73. 2007.
|
|
36
|
Liao YJ, Liu SP, Lee CM, et al:
Characterization of a glycine N-methyltransferase gene knockout
mouse model for hepatocellular carcinoma: Implications of the
gender disparity in liver cancer susceptibility. Int J Cancer.
124:816–826. 2009.
|
|
37
|
Zhao Z, Wu Q, Cheng J, et al: Depletion of
DNMT3A suppressed cell proliferation and restored PTEN in
hepatocellular carcinoma cell. J Biomed Biotechnol.
2010:7375352010.
|
|
38
|
Fan H, Chen L, Zhang F, et al: MTSS1, a
novel target of DNA methyltransferase 3B, functions as a tumor
suppressor in hepatocellular carcinoma. Oncogene. 31:2298–2308.
2012.
|
|
39
|
Park IY, Sohn BH, Yu E, et al: Aberrant
epigenetic modifications in hepatocarcinogenesis induced by
hepatitis B virus X protein. Gastroenterology. 132:1476–1494.
2007.
|
|
40
|
Bartel DP: MicroRNAs, genomics,
biogenesis, mechanism, and function. Cell. 116:281–297. 2004.
|
|
41
|
Zhang X, Liu S, Hu T, Liu S, He Y and Sun
S: Up-regulated microRNA-143 transcribed by nuclear factor kappa B
enhances hepatocarcinoma metastasis by repressing fibronectin
expression. Hepatology. 50:490–499. 2009.
|
|
42
|
Huang J, Wang Y, Guo Y and Sun S:
Down-regulated microRNA-152 induces aberrant DNA methylation in
hepatitis B virus-related hepatocellular carcinoma by targeting DNA
methyltransferase 1. Hepatology. 52:60–70. 2010.
|
|
43
|
Wei X, Xiang T, Ren G, et al: miR-101 is
down-regulated by the hepatitis B virus x protein and induces
aberrant DNA methylation by targeting DNA methyltransferase 3A.
Cell Signal. 25:439–446. 2013.
|
|
44
|
Lim SO, Gu JM, Kim MS, et al: Epigenetic
changes induced by reactive oxygen species in hepatocellular
carcinoma: methylation of the E-cadherin promoter.
Gastroenterology. 135:2128–2140. 2008.
|
|
45
|
Blackburn RV, Spitz DR, Liu X, et al:
Metabolic oxidative stress activates signal transduction and gene
expression during glucose deprivation in human tumor cells. Free
Radic Biol Med. 26:419–430. 1999.
|
|
46
|
Lee YJ, Galoforo SS, Berns CM, et al:
Glucose deprivation-induced cytotoxicity and alterations in
mitogen-activated protein kinase activation are mediated by
oxidative stress in multidrug-resistant human breast carcinoma
cells. J Biol Chem. 273:5294–5299. 1998.
|
|
47
|
Rahman I, Marwick J and Kirkham P: Redox
modulation of chromatin remodeling: impact on histone acetylation
and deacetylation, NF-kappaB and pro-inflammatory gene expression.
Biochem Pharmacol. 68:1255–1267. 2004.
|
|
48
|
Adenuga D, Yao H, March TH, Seagrave J and
Rahman I: Histone deacetylase 2 is phosphorylated, ubiquitinated,
and degraded by cigarette smoke. Am J Respir Cell Mol Biol.
40:464–473. 2009.
|
|
49
|
Ito K, Lim S, Caramori G, Chung KF, Barnes
PJ and Adcock IM: Cigarette smoking reduces histone deacetylase 2
expression, enhances cytokine expression, and inhibits
glucocorticoid actions in alveolar macrophages. FASEB J.
15:1110–1112. 2001.
|
|
50
|
Quint K, Agaimy A, Di Fazio P, et al:
Clinical significance of histone deacetylases 1, 2, 3, and 7, HDAC2
is an independent predictor of survival in HCC. Virchows Arch.
459:129–139. 2011.
|
|
51
|
Zopf S, Ocker M, Neureiter D, et al:
Inhibition of DNA methyltransferase activity and expression by
treatment with the pan-deacetylase inhibitor panobinostat in
hepatocellular carcinoma cell lines. BMC Cancer. 12:3862012.
|
|
52
|
Jia Y, Yang Y, Liu S, Herman JG, Lu F and
Guo M: SOX17 antagonizes WNT/β-catenin signaling pathway in
hepatocellular carcinoma. Epigenetics. 5:743–749. 2010.
|
|
53
|
Grigoryan T, Wend P, Klaus A and
Birchmeier W: Deciphering the function of canonical Wnt signals in
development and disease: conditional loss- and gain-of-function
mutations of beta-catenin in mice. Genes Dev. 22:2308–2341.
2008.
|
|
54
|
Xu X, Liu RF, Wan BB, et al: Expression of
a novel gene FAM43B repressing cell proliferation is regulated by
DNA methylation in hepatocellular carcinoma cell lines. Mol Cell
Biochem. 354:11–20. 2011.
|
|
55
|
Qiu GH, Salto-Tellez M, Ross JA, et al:
The tumor suppressor gene DLEC1 is frequently silenced by DNA
methylation in hepatocellular carcinoma and induces G1 arrest in
cell cycle. J Hepatol. 48:433–441. 2008.
|
|
56
|
Yu J, Tao Q, Cheung KF, et al: Epigenetic
identification of ubiquitin carboxyl-terminal hydrolase L1 as a
functional tumor suppressor and biomarker for hepatocellular
carcinoma and other digestive tumors. Hepatology. 48:508–518.
2008.
|
|
57
|
Chen M, Zhang J, Li N, Qian Z, Zhu M, Li
Q, Zheng J, Wang X and Shi G: Promoter hypermethylation mediated
downregulation of FBP1 in human hepatocellular carcinoma and colon
cancer. PLoS One. 6:e255642011.
|
|
58
|
Komatsu S, Okazaki Y, Tateno M, et al:
Methylation and downregulated expression of mac25/insulin-like
growth factor binding protein-7 is associated with liver
tumorigenesis in SV40T/t antigen transgenic mice, screened by
restriction landmark genomic scanning for methylation (RLGS-M).
Biochem Biophys Res Commun. 267:109–117. 2000.
|
|
59
|
Berasain C, Hevia H, Fernández-Irigoyen J,
Larrea E, Caballería J, Mato JM, Prieto J, Corrales FJ,
García-Trevijano ER and Avila MA: Methylthioadenosine phosphorylase
gene expression is impaired in human liver cirrhosis and
hepatocarcinoma. Biochim Biophys Acta. 1690:276–284. 2004.
|
|
60
|
Zhou X, Popescu NC, Klein G and Imreh S:
The interferon-alpha responsive gene TMEM7 suppresses cell
proliferation and is downregulated in human hepatocellular
carcinoma. Cancer Genet Cytogenet. 177:6–15. 2007.
|
|
61
|
Nishida N, Kudo M, Nagasaka T, Ikai I and
Goel A: Characteristic patterns of altered DNA methylation predict
emergence of human hepatocellular carcinoma. Hepatology.
56:994–1003. 2012.
|
|
62
|
Xie B, Zhou J, Yuan L, Ren F, Liu DC, Li Q
and Shu G: Epigenetic silencing of Klotho expression correlates
with poor prognosis of human hepatocellular carcinoma. Hum Pathol.
44:795–801. 2012.
|
|
63
|
Lou C, Yang B, Gao YT, Wang YJ, Nie FH,
Yuan Q, Zhang CL and Du Z: Aberrant methylation of multiple genes
and its clinical implication in hepatocellular carcinoma. Zhonghua
Zhong Liu Za Zhi. 30:831–836. 2008.(In Chinese).
|
|
64
|
Nishida N, Nagasaka T, Nishimura T, Ikai
I, Boland CR and Goel A: Aberrant methylation of multiple tumor
suppressor genes in aging liver, chronic hepatitis, and
hepatocellular carcinoma. Hepatology. 47:908–918. 2008.
|
|
65
|
Piao GH, Piao WH, He Y, Zhang HH, Wang GQ
and Piao Z: Hyper-methylation of RIZ1 tumor suppressor gene is
involved in the early tumorigenesis of hepatocellular carcinoma.
Histol Histopathol. 23:1171–1175. 2008.
|
|
66
|
Lal G, Padmanabha L, Smith BJ, Nicholson
RM, Howe JR, O’Dorisio MS and Domann FE Jr: RIZ1 is epigenetically
inactivated by promoter hypermethylation in thyroid carcinoma.
Cancer. 107:2752–2759. 2006.
|
|
67
|
Chadwick RB, Jiang GL, Bennington GA, Yuan
B, Johnson CK, Stevens MW, Niemann TH, Peltomaki P, Huang S and de
la Chapelle A: Candidate tumor suppressor RIZ is frequently
involved in colorectal carcinogenesis. Proc Natl Acad Sci USA.
97:2662–2667. 2000.
|
|
68
|
Piao GH, Piao WH, He Y, et al:
Hyper-methylation of RIZ1 tumor suppressor gene is involved in the
early tumorigenesis of hepatocellular carcinoma. Histol
Histopathol. 23:1171–1175. 2008.
|
|
69
|
Zhang C, Li H, Wang Y, et al: Epigenetic
inactivation of the tumor suppressor gene RIZ1 in hepatocellular
carcinoma involves both DNA methylation and histone modifications.
J Hepatol. 53:889–895. 2010.
|
|
70
|
Formeister EJ, Tsuchiya M, Fujii H, et al:
Comparative analysis of promoter methylation and gene expression
endpoints between tumorous and non-tumorous tissues from
HCV-positive patients with hepatocellular carcinoma. Mutat Res.
692:26–33. 2010.
|
|
71
|
Yang JD, Seol SY, Leem SH, Kim YH, Sun Z,
Lee JS, Thorgeirsson SS, Chu IS, Roberts LR and Kang KJ: Genes
associated with recurrence of hepatocellular carcinoma: integrated
analysis by gene expression and methylation profiling. J Korean Med
Sci. 26:1428–1438. 2011.
|
|
72
|
Iyer P, Zekri AR, Hung CW, Schiefelbein E,
Ismail K, Hablas A, Seifeldin IA and Soliman AS: Concordance of DNA
methylation pattern in plasma and tumor DNA of Egyptian
hepatocellular carcinoma patients. Exp Mol Pathol. 88:107–111.
2010.
|
|
73
|
Kimura N, Moribe T, Iizuka N, Miura T,
Tamatsukuri S, Ishitsuka H, Hamamoto Y and Oka M: Rapid and
quantitative detection of CpG-methylation status using TaqMan PCR
combined with methyl-binding-domain polypeptide. Clin Biochem.
42:1113–1122. 2009.
|