Introduction
Multiple myeloma (MM) is a malignant hematological
disease characterized by the accumulation of clonal plasma cells
and the presence of monoclonal immunoglobulin in blood, osteolytic
lesions, hypercalcemia and immunodeficiency. It accounts for
approximately 1% of all cancers and 10–15% of hematologic
malignancies (1,2). In recent years, the molecular and
clinical knowledge emerging from studies of MM pathogenesis and the
response to treatments has grown exponentially. This has
facilitated the generation of new drugs and clinical strategies
that have significantly improved the prognosis of certain patients
with MM. However, in the long term, MM remains an incurable
disease. Patients with acquired drug resistance invariably relapse,
and salvage therapy is not effective (3,4).
Methylation of DNA is one of the most important
modifications of the mammalian genome, DNA methylation is achieved
by a family of DNA methyltransferase enzymes (DNMTs) that transfer
the methyl group from the donor S-adenosyl methionine to the fifth
carbon of cytosine. Aberrant DNA methylation is a common epigenetic
mechanism implicated in the etiology of numerous human cancers.
Hypermethylation of a number of tumor suppressor genes occurs at
CpG islands in the promoter, leading to gene inactivation (5–8).
Previous Studies have shown that hypermethylation of genes encoding
cell cycle inhibitors p15 and p16, the apoptosis regulator
death-associated protein kinase, the tumor suppressor Ras
association domain-containing protein 1 and suppressor of cytokine
signaling 1 (SOCS1) occurs frequently in MM patients (9–14).
In the current study, RNA interference was employed
to knock down DNMT1 expression in human MM cells to
investigate the association between DNMT1 expression and the
proliferative activity, tumor suppressor gene expression and gene
methylation levels of myeloma cells.
Materials and methods
Cell culture and experimental
reagents
The RPMI-8226 human MM cell line was obtained from
the Cell Bank of the Chinese Academy of Sciences (Shanghai, China)
and was cultured in RPMI-1640 (Gibco-BRL, Grand Island, NY, USA)
supplemented with 10% fetal bovine serum (Gibco-BRL) in a 5%
CO2 atmosphere at 37°C. Lipofectamine 2000 was purchased
from Invitrogen Life Technologies (Carlsbad, CA, USA). RevertAid
First Strand cDNA synthesis kit and DreamTaq Green PCR master mix
were from Fermentas (Glen Burnie, MD, USA). QIAamp DNA mini kit and
EpiTect Bisulfite kit were from Qiagen (Hilden, Germany). Cell
Counting Kit-8 (CCK-8) and Cell Cycle Analysis kit were from
MultiSciences Biotech (Hangzhou, China).
siRNA transfection
Recombinant plasmids containing the green
fluorescent protein (GFP) gene which expresses GFP, and the
transfection efficiency may be directly observed under an inverted
fluorescence microscope (CKX41-F32FL, Olympus, Tokyo, Japan).
RPMI-8226 cells were seeded in six-well plates overnight and then
transfected with siRNA or negative control siRNA oligonucleotides
(containing the GFP gene which emits green light) that were
precomplexed with Lipofectamine 2000 (Invitrogen Life
Technologies). The medium was refreshed after 6 h with complete
growth medium, and the cells were incubated for an additional 48 h.
Following this, antibiotic selection (0.2 μg/ml puromycin;
Invitrogen Life Technolgies) was initiated and continued for 14–20
days prior to selection of stably transfected cells. The siRNA
sequences used to target DNMT1 were 5′-CACTGGTTCTGCGCTGGGA-3′
(sense) and 5′-AAGTCTTCTGACGCTGCTGCCTGGTCCAG-3′ (antisense), and
were designed based on GenBank accession no. NM_001379.1.
Quantification of proliferation using the
CCK-8 assay
Cells transfected with DNMT1 siRNA or siRNA control
were incubated for 1–5 days at a density of 1×103
cells/well in 96-well plates. CCK-8 (10 μl) was added in each well,
followed by an additional 3-h incubation prior to reading the
absorbance at 450 nm using a microplate reader (Bio-Rad 680;
Bio-Rad, Hercules, CA, USA). An average value from three wells was
obtained for each group of RPMI-8226 cells to plot the growth
curve.
Cell cycle assays
Transfected cells were seeded at a density of
1×106 cells/well in six-well plates. After a 48-h
incubation, the cells were collected and washed twice with ice-cold
phosphate-buffered saline (PBS), fixed in 70% ethanol at room
temperature (RT) for at least 30 min, and stored at −20°C
overnight. For analysis, the cells were washed twice with PBS,
stained with propidium iodide (10 μg/ml; MP Biomedicals, Santa Ana,
CA, USA) and incubated at 37°C for 30 min. Fluorescence was
measured with a flow cytometer (BD Bioscience, San Jose, CA, USA),
and the data were analyzed using Cell ModFit software (BD
Bioscience). The experiments were performed three times in order to
derive a mean value.
Western blot analysis
RPMI-8226 cells were incubated at 4°C for 30 min in
lysis buffer. Protein concentration was determined with the Bio-Rad
protein assay system (Bio-Rad). Equal amounts of protein lysates
(50 μg) were analyzed by performing SDS-PAGE and electrotransfer of
proteins to polyvinylidene fluoride membranes. Membranes were
washed with 1× Tris-buffered saline with Tween-20 (TBST), blocked
for 1 h at RT in skimmed milk/TBST, and then immunoblotted with the
appropriate primary antibodies. The primary antibodies included
monoclonal mouse anti-human DNMT1, monoclonal rabbit anti-human
B-cell lymphoma 2 (BCL2), monoclonal rabbit anti-human nuclear
factor κB (NF-κB) (Abcam, Cambridge, UK), and monoclonal mouse
anti-human β-actin (Santa Cruz Biotechnology, Inc., Dallas, TX,
USA). The β-actin was used as a loading control. The next day, the
membranes were washed with 1× TBST and incubated with anti-rabbit
or anti-mouse IgG horseradish peroxidase-conjugated secondary
antibodies diluted to 1:3000 in skimmed milk/TBST for 1 h at RT.
Proteins were visualized using a Vazyme E411-01 enhanced
chemiluminescence detection kit [Vazyme Biotech (Nanjing) Co.,
Ltd., Nanjing, China].
Quantitative polymerase chain reaction
(qPCR) assays
Total RNA was isolated from transfected cells using
TRIzol reagent (Invitrogen Life Technologies). cDNA was synthesized
using the RevertAid First Strand cDNA synthesis kit (Fermentas)
according to the manufacturer’s instructions. The primers used for
amplifying DNMT1 were 5′-ACCATCACATCTCATTTTGC-3′ (sense) and
5′-GGTTTGACTTCGGAGTCTCT-3′ (antisense). The primers used for
amplifying β-actin were 5′-GTGGGGCGCCCCAGGCACCA-3′ (sense) and
5′-CTCCTTAATGTCACGCACGATTT-3′ (antisense). qPCR was carried out
with DreamTaq Green PCR master mix (Fermentas), and the analysis
was performed in an Eppendorf PCR device 00135 (Eppendorf, Hamburg,
Germany). The thermal cycling was as follows: 94°C for 3 min; 30
cycles of denaturation at 94°C for 30 sec, annealing at 55°C for 30
sec and extension at 72°C for 1 min; and a final 10-min extension
at 72°C. The relative quantity was analyzed with the
2−ΔΔCt method. β-actin mRNA was used as a control, and
each experiment was performed in triplicate.
Nested methylation-specific PCR
assays
Genomic DNA was extracted from transfected cells
with a QIAamp DNA mini kit (Qiagen) and was subjected to bisulfite
modification with EpiTect Bisulfite kit (Qiagen) according to the
manufacturer’s instructions. The stage-1 PCR products were diluted
50-fold, and 5 μl of the product was subjected to a stage-2 PCR, in
which primers specific to methylated or unmethylated template were
used. Primer sequences used in the stage-1 amplification of the
SOCS1 and p16 genes are as follows: SOCS1 sense,
5′-AACTGCTTTTTCGCCCTTAGC-3′ and SOCS1 antisense,
5′-CAGCTCGAAGAGGCAGTCG-3′; p16 sense, 5′-GAAGAAAGAGGAGGGGTTGG-3′
and p16 antisense 5′-CTACAAACCCTCTACCCACC-3′. The PCR amplification
protocol for stage 1 was as follows: 95°C for 3 min; 40 cycles of
denaturation at 95°C for 30 sec, annealing at 60°C for 30 sec and
extension at 72°C for 30 sec; and a final 10-min extension at 72°C.
In the stage-2 PCR, annealing temperatures were increased to 65°C
for p16, and annealing times were increased to 45 sec for
SOCS1. All assays were conducted in triplicate.
Statistical analysis
Data are presented as the mean ± standard deviation.
Statistical analysis was performed using Student’s t-test or
analysis of variance. P<0.05 was considered to indicate a
statistically significant difference. GraphPad Prism 5.0 software
(GraphPad Software Inc., La Jolla, CA, USA) was used for
statistical analysis.
Results
Effective downregulation of DNMT1
expression in human RPMI-8226 cells by DNMT1 siRNA
The recombinant plasmid of siRNA targeted against
DNMT1 was constructed and successfully transfected into RPMI-8226
cells. The transfected RPMI-8226 cells were visible (emitted green
light) under an inverted fluorescence microscope (CKX41-F32FL;
Olympus Corporation). Compared with the two control groups, the
DNMT1 expression was decreased significantly, both at the mRNA
(Fig. 1A) and protein (Fig. 1B) levels (P<0.001), as determined
by qPCR and western blot analysis, respectively. This confirmed the
transfection of siRNA to the genome and its stable expression.
DNMT1 silencing inhibits the
proliferation capacity of human RPMI-8226 cells
The effect of DNMT1 silencing on cell
proliferation was determined by a CCK-8 assay. As shown in Fig. 2, it was found that the in
vitro cell growth rate of the DNMT1 siRNA group was
significantly lower than that of the other two groups 5 days after
siRNA transfection (P<0.01), while no significant difference was
found between the negative control group and the non-transfection
group (P>0.05).
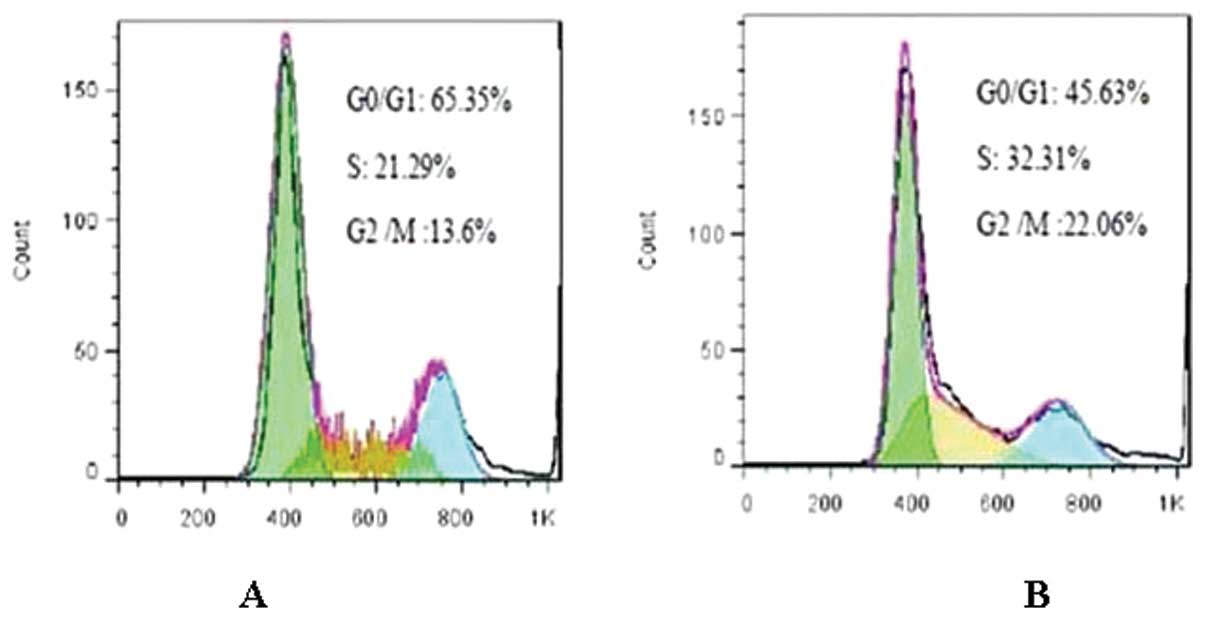
Effect of DNMT1 siRNA on the cell cycle
of RPMI-8226 cells
Cell cycle analysis showed that compared with
untransfected cells, treatment with DNMT1 siRNA increased
the number of cells in the G0/G1 phase (65.35±0.08 vs. 45.63±1.10%,
P<0.05), while reducing the number of cells in the S and G2/M
phases (S stage: 21.29±1.54 vs. 32.31±0.72%, P<0.05; G2/M stage:
13.6±1.03 vs. 22.06±0.66%, P<0.05) (Fig. 3). The percentage of each phase of
cells in the siRNA-control group was similar to that of the
RPMI-8226 group (P>0.05). The percentage of each phase of cells
was not considered to be statistically significant between the
negative control group and the RPMI-8226 group (P>0.05).
DNMT1 siRNA reduces NF-κB and Bcl-2
protein expression in human RPMI-8226 cells
RPMI-8226 cells were grown and transfected with
DNMT1 siRNA or negative control siRNA oligonucleotides.
Protein expression was detected by western blot analysis. The
results suggested that compared with the negative control group and
the untransfected group, both NF-κB and Bcl-2 protein expression
was significantly reduced in the DNMT1 siRNA group
(P<0.05, Fig. 4).
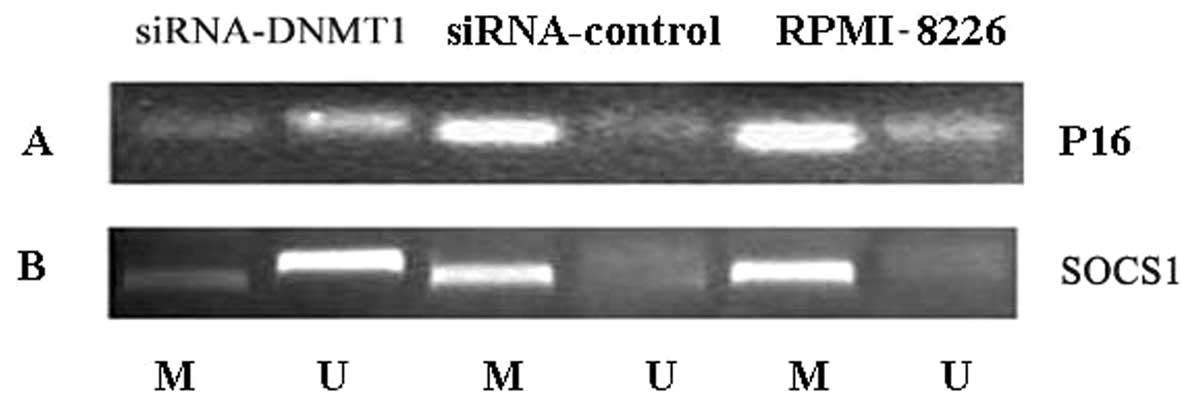
DNMT1 siRNA induces demethylation of the
tumor suppressor genes SOCS1 and p16
In human RPMI-8226 cells, the tumor suppressor genes
SOCS1 and p16 were identified to be highly methylated
by nested methylation-specific PCR (Fig. 5). Following transfection of
RPMI-8226 cells with DNMT1 siRNA, SOCS1 and
p16 genes lost methylation marks, showing that the
high-level methylation at these loci can in part be reversed
(Fig. 5).
Discussion
DNA methyltransferases, including DNMT1, DNMT3A and
DNMT3B, catalyze the methylation of human genomic DNA. Methylation
of DNA at C-5 of cytosine plays a key role in the regulation of
human genes and can result in X-chromosome inactivation, genomic
imprinting and silencing of proviral elements and retrotransposons
(15). Aberrant methylation,
particularly in the promoter regions of tumor suppressor genes,
alters gene expression and can facilitate human tumorigenesis.
DNMT1, the major DNMT in adult cells, preferentially acts on
hemimethylated CpG substrates and is involved in the maintenance of
genomic DNA methylation during DNA replication (16,17).
Aberrant methylation in the promoter regions of
tumor suppressor genes is implicated in the pathogenesis of
numerous types of malignant tumors. Thus, epigenetic therapies
incorporating DNMT inhibitors are expected to induced
demethylation, re-expression, and functional recovery of silenced
tumor suppressor genes (18–20).
Both SOCS1 and p16 are commonly silenced in malignant
tumors, and are also silenced by methylation in familial MM
(21–23).
In the present study, it was found that DNMT1
expression was significantly downregulated at both the mRNA and
protein level in RPMI-8226 myeloma cells following treatment with
DNMT1 siRNA. The downregulation of DNMT1 expression
inhibited cell growth, most likely by inducing arrest at the G0/G1
phase. Our results also showed that upon DNMT1 silencing,
the methylation of the SOCS1 and p16 promoters was
reduced, and the genes were re-expressed. These observations
suggest that demethylation of tumor suppressor gene promoters
should be further evaluated as a therapeutic strategy in
myeloma.
Tumor suppressor gene inactivation has previously
been correlated with DNMT1 overexpression in various types of
cancer, including hematological malignancies (24–27).
Robert et al (28) reported
that siRNA-mediated DNMT1 silencing can trigger reactivation
of tumor suppressor gene expression and function in HCT116 colon
cancer cells. This is consistent with the findings of the present
study that inactivation of SOCS1 and p16 may be
associated with DNMT1 overexpression. By contrast, Ting et
al reported that single DNMT1 gene knockout was
insufficient to trigger demethylation of tumor suppressor gene
promoters and restore their function (29). This highlights the fact that
mechanisms that control the expression and inactivation of tumor
suppressor genes vary across different tumor cells.
NF-κB is an important transcription factor involved
in transcriptional regulation of various genes that in turn
modulate immune cell activation, apoptosis and differentiation
processes. Sustained activation of NF-κB plays an important role in
the pathogenesis of MM. For example, NF-κB activation can enhance
the expression of adhesion molecules, promoting homing of myeloma
precursors and the production of tumor cell growth factors. NF-κB
can also promote the secretion of interleukin 6 by adhesion with
extracellular matrix proteins and bone marrow stromal cells,
initiating various signal transduction pathways and promoting tumor
cell proliferation and drug resistance (30,31).
Therefore, inhibition of NF-κB overexpression is an effective way
to induce apoptosis and overcome the drug resistance associated
with certain MM cells. Indeed, NF-κB is now one of the key
therapeutic targets in MM (32,33).
BCL2 is a critical pro-survival member of the BH
domain-containing superfamily. BCL2 inhibits apoptosis and is
involved in the pathogenesis of a variety of hematological tumors,
including MM (34). Specifically,
overexpression of BCL2 protein is associated with the survival and
drug resistance of MM cells (35).
In the present study, compared with the
untransfected and negative control groups, the expression of NF-κB
and BCL2 proteins was significantly reduced upon DNMT1
knockdown. Although the precise mechanism for this remains unclear,
these data suggest that targeting DNA methylases may lead to
downregulation of critical tumor survival factors, thereby inducing
tumor cell death. In summary, the results of the present study
provide key evidence that targeting methylases, either by
RNA-mediated knockdown approaches or through the use of small
molecules, may be an effective means of inducing tumor regression.
The challenge will be to selectively de-repress only those gene
targets responsible for tumor survival and to avoid de-repression
of genes in neighboring normal cells, as this could have
deleterious effects.
Acknowledgements
This study was supported by the National Nature
Science Fund (grant no. 81172246).
References
|
1
|
Benjamin M, Reddy S and Brawley OW:
Myeloma and race: a review of the literature. Cancer Metastasis
Rev. 22:87–93. 2003.
|
|
2
|
Siegel R, Ward E, Brawley O, et al: Cancer
statistics, 2011: the impact of eliminating socioeconomic and
racial disparities on premature cancer deaths. CA Cancer J Clin.
61:212–236. 2011.
|
|
3
|
Richardson PG, Barlogie B, Berenson J, et
al: Clinical factors predictive of outcome with bortezomib in
patients with relapsed, refractory multiple myeloma. Blood.
106:2977–2981. 2005.
|
|
4
|
Richardson PG, Weller E, Lonial S, et al:
Lenalidomide, bortezomib, and dexamethasone combination therapy in
patients with newly diagnosed multiple myeloma. Blood. 116:679–686.
2010.
|
|
5
|
Chen T and Li E: Structure and function of
eukaryotic DNA methyltransferases. Curr Top Dev Biol. 60:55–89.
2004.
|
|
6
|
Robertson KD: DNA methylation and human
disease. Nat Rev Genet. 6:597–610. 2005.
|
|
7
|
Belinsky SA, Nikula KJ, Baylin SB, et al:
A microassay for measuring cytosine DNA methyltransferase activity
during tumor progression. Toxicol Lett. 82–83:335–340. 1995.
|
|
8
|
Vertino PM, Yen RW, Gao J and Baylin SB:
De novo methylation of CpG island sequences in human fibroblasts
overexpressing DNA (cytosine-5-)-methyltransferase. Mol Cell Biol.
16:4555–4565. 1996.
|
|
9
|
Ng MH, Chung YF, Lo KW, et al: Frequent
hypermethylation of p16 and p15 genes in multiple myeloma. Blood.
89:2500–2506. 1997.
|
|
10
|
Mateos MV, García-Sanz R, Lopez-Perez R,
et al: Methylation is an inactivating mechanism of the p16 gene in
multiple myeloma associated with high plasma cell proliferation and
short survival. Br J Haematol. 118:1034–1040. 2002.
|
|
11
|
Chim CS, Liang R, Fung TK, et al:
Epigenetic dysregulation of the death-associated protein
kinase/p14/HDM2/p53/Apaf-1 apoptosis pathway in multiple myeloma. J
Clin Pathol. 60:664–669. 2007.
|
|
12
|
Ng MH, Lau KM, Wong WS, et al: Alterations
of RAS signalling in Chinese multiple myeloma patients: absent BRAF
and rare RAS mutations, but frequent inactivation of RASSF1A by
transcriptional silencing or expression of a non-functional variant
transcript. Br J Haematol. 123:637–645. 2003.
|
|
13
|
Galm O, Yoshikawa H, Esteller M, et al:
SOCS-1, a negative regulator of cytokine signaling, is frequently
silenced by methylation in multiple myeloma. Blood. 101:2784–2788.
2003.
|
|
14
|
Galm O, Wilop S, Reichelt J, et al: DNA
methylation changes in multiple myeloma. Leukemia. 18:1687–1692.
2004.
|
|
15
|
Baylin SB and Ohm JE: Epigenetic gene
silencing in cancer - a mechanism for early oncogenic pathway
addiction? Nat Rev Cancer. 6:107–116. 2006.
|
|
16
|
Bestor TH: The DNA methyltransferases of
mammals. Hum Mol Genet. 9:2395–2402. 2000.
|
|
17
|
Szyf M, Pakneshan P and Rabbani SA: DNA
methylation and breast cancer. Biochem Pharmacol. 68:1187–1197.
2004.
|
|
18
|
Xu M, Gao J, Du YQ, et al: Reduction of
pancreatic cancer cell viability and induction of apoptosis
mediated by siRNA targeting DNMT1 through suppression of total DNA
methyltransferase activity. Mol Med Rep. 3:699–704. 2010.
|
|
19
|
Milutinovic S, Knox JD and Szyf M: DNA
methyltransferase inhibition induces the transcription of the tumor
suppressor p21 (WAF1/CIP1/sdi1). J Biol Chem. 275:6353–6359.
2000.
|
|
20
|
Ghoshal K and Bai S: DNA
methyltransferases as targets for cancer therapy. Drugs Today
(Barc). 43:395–422. 2007.
|
|
21
|
Auerkari EI: Methylation of tumor
suppressor genes p16(INK4a), p27(Kip1) and E-cadherin in
carcinogenesis. Oral Oncol. 42:5–13. 2006.
|
|
22
|
Chim CS, Fung TK and Liang R: Disruption
of INK4/CDK/Rb cell cycle pathway by gene hypermethylation in
multiple myeloma and MGUS. Leukemia. 17:2533–2535. 2003.
|
|
23
|
Komazaki T, Nagai H, Emi M, et al:
Hypermethylation-associated inactivation of the SOCS-1 gene, a
JAK/STAT inhibitor, in human pancreatic cancers. Jpn J Clin Oncol.
34:191–194. 2004.
|
|
24
|
Jost E, Gezer D, Wilop S, et al:
Epigenetic dysregulation of secreted Frizzled-related proteins in
multiple myeloma. Cancer Lett. 281:24–31. 2009.
|
|
25
|
Tshuikina M, Jernberg-Wiklund H, Nilsson
K, et al: Epigenetic silencing of the interferon regulatory factor
ICSBP/IRF8 in human multiple myeloma. Exp Hematol. 36:24–31.
2008.
|
|
26
|
Lin RK, Hsu HS, Chang JW, et al:
Alteration of DNA methyltransferases contributes to 5′CpG
methylation and poor prognosis in lung cancer. Lung Cancer.
55:205–213. 2007.
|
|
27
|
Ahluwalia A, Hurteau JA, Bigsby RM, et al:
DNA methylation in ovarian cancer. II. Expression of DNA
methyltransferases in ovarian cancer cell lines and normal ovarian
epithelial cells. Gynecol Oncol. 82:299–304. 2001.
|
|
28
|
Robert MF, Morin S, Beaulieu N, et al:
DNMT1 is required to maintain CpG methylation and aberrant gene
silencing in human cancer cells. Nat Genet. 33:61–65. 2003.
|
|
29
|
Ting AH, Schuebel KE, Herman JG, et al:
Short double-stranded RNA induces transcriptional gene silencing in
human cancer cells in the absence of DNA methylation. Nat Genet.
37:906–910. 2005.
|
|
30
|
Giuliani N, Colla S, Sala R, et al: Human
myeloma cells stimulate the receptor activator of nuclear
factor-kappa B ligand (RANKL) in T lymphocytes: a potential role in
multiple myeloma bone disease. Blood. 100:4615–4621. 2002.
|
|
31
|
Xiao W, Hodge DR, Wang L, et al: NF-kappaB
activates IL-6 expression through cooperation with c-Jun and
IL6-AP1 site, but is independent of its IL6-NFkappaB regulatory
site in autocrine human multiple myeloma cells. Cancer Biol Ther.
3:1007–1017. 2004.
|
|
32
|
Conticello C, Giuffrida R, Adamo L, et al:
NF-κB localization in multiple myeloma plasma cells and mesenchymal
cells. Leuk Res. 35:52–60. 2004.
|
|
33
|
Kannaiyan R, Hay HS, Rajendran P, et al:
Celastrol inhibits proliferation and induces chemosensitization
through down-regulation of NF-κB and STAT3 regulated gene products
in multiple myeloma cells. Br J Pharmacol. 164:1506–1521. 2011.
|
|
34
|
Baliga BC and Kumar S: Role of Bcl-2
family of proteins in malignancy. Hematol Oncol. 20:63–74.
2002.
|
|
35
|
Chen Q, Ray S, Hussein MA, et al: Role of
Apo2L/TRAIL and Bcl-2-family proteins in apoptosis of multiple
myeloma. Leuk Lymphoma. 44:1209–1214. 2003.
|