Introduction
Hepatocellular carcinoma (HCC) is the third most
common cause of cancer-related mortality worldwide, with high
recurrence and a low five-year survival rate (1). Thus far, excision remains the most
significant method in the comprehensive treatment of HCC. However,
resection of the tumor using surgery is difficult in the majority
of cases of HCC, since >80% patients are suffering from advanced
or unresectable diseases at the final diagnosis (2,3). Even
following successful resection, the recurrence may be as high as
50% after two years (4).
Chemotherapy is used as an adjuvant post-operative treatment, with
the aim of reducing tumor recurrence. However, conventional
systemic chemotherapy has shown only minor effectiveness with
response rates of <10%, due to the intrinsic or acquired drug
resistance caused by multidrug resistance (MDR) (4,5). The
mechanisms of drug resistance are heterogeneous and include
increased anticancer agent efflux by adenosine triphosphate
(ATP)-binding cassette proteins, apoptotic inhibition, DNA repair
activation and detoxifying system enhancement (6). Although numerous MDR reversal agents
have been reported, the clinical application of these drugs is
limited due to side-effects or toxicity that are unacceptable at
the effective dose (7). Thus,
identifying MDR reversal agents with high reversal activity and low
toxicity is important.
Recently, evidence has accumulated demonstrating
that the activation of the mitogen-activated protein kinase (MAPK)
signaling pathway is associated with MDR in multiple types of
tumors and that this signaling pathway has a predominant role in
various cellular processes, including proliferation,
differentiation, apoptosis, angiogenesis and migration (8–13). The
MAPK group includes four distinct signaling cascades, which are
known by the corresponding MAPK tier component: Extracellular
signal-regulated kinase 1 and 2 (ERK1/2) (14–16);
c-Jun N-terminal kinase 1 to 3 (17,18);
p38 MAPK α, β, γ and δ (p38 α-δ) (19–22);
and ERK5, also known as Big MAPK (23,24).
The ERK1/2 cascade, which has been the most widely analyzed module
in the MAPK signaling pathways, transmits predominantly mitogenic
signals. The activation of the ERK1/2 signaling pathway is induced
by guanosine 5′-triphosphate loading of Ras at the plasma membrane,
which is followed by sequential activation of a series of protein
kinases, including a member of the Raf family (such as Raf-1), MAPK
or ERK kinase (MEK) 1/2, then ERK1 or ERK2 (25). Our previous study demonstrated that
ERK1/2 is highly expressed in several HCC cells with MDR (26). The aim of the present study was to
analyze whether the ERK/MAPK inhibitors reverse MDR in HCC cells.
Furthermore, one classical MDR reversal agent, cyclosporine A
(CsA), was selected to examine whether downregulation of ERK/MAPK
signaling pathway activity is involved in the reversal mechanism of
traditional methods, such as the inhibition of p-gp.
Materials and methods
Compounds
Sorafenib (Nexavar, BAY 43-9006), a multikinase
inhibitor [for vascular endothelial growth factor receptor,
platelet-derived growth factor receptor and rapidly accelerated
fibrosarcoma (RAF) kinases], was manufactured by Bayer
Pharmaceuticals (West Haven, CT, USA). PD98059, a MEK inhibitor,
was purchased from Cell Signaling Technology, Inc. (Beverly, MA,
USA). CsA was obtained from Enzo Life Sciences, Inc. (Farmingdale,
NY, USA). The compounds were dissolved in 100% dimethyl sulfoxide
(DMSO; Sigma-Aldrich, St. Louis, MO, USA) and diluted with RPMI
1640 to obtain a final DMSO concentration of 0.1% for the in
vitro experiments. DMSO was subsequently added to the cell
cultures at 0.1% (v/v) as a solvent control.
Cell lines and cell culture
The SMMC7721 and BEL7402 human HCC cell lines were
purchased from the Institute of Biochemistry and Cell Biology,
Shanghai Institutes for Biological Science, Chinese Academy of
Sciences (Shanghai, China). The SMMC7721 and BEL7402 cells were
cultured with RPMI-1640 (HyClone Laboratories, Inc., Logan, UT,
USA). The medium was supplemented with 10% calf serum, 100 IU/ml
penicillin and 100 μg/ml streptomycin (all HyClone), and maintained
at 37°C in a humidified atmosphere containing 50 ml/l
CO2 and 950 ml/l air. To establish SMMC7721/Adriamycin
(ADM) and BEL7402/ADM MDR cells, ADM (Shanghai Shenggong Biological
Engineering Co., Ltd., Shanghai, China) was added to SMMC7721 and
BEL7402 cells, respectively, at increasing stepwise concentrations
between 1 and 5 mg/l. Resistant cells were selected by removing the
non-resistant dead cells. MDR was maintained by culturing the cells
with 5 mg/l ADM; the MDR cells were termed the SMMC7721/ADM and
BEL7402/ADM cells. This study was approved by the ethics committee
of the Affiliated Hospital of Xiamen University (Xiamen,
China).
CellTiter-Glo® luminescent
cell viability assay
The cells were plated at 3,000 cells per well in
96-well microtiter plates and incubated overnight at 37°C in a
humidified incubator containing 5% CO2. On the following
days, the corresponding compounds were added to the wells and the
cultures were incubated for an additional 48 h. To investigate the
drug resistance of MDR cells, the parental cells and MDR cells were
exposed to various concentrations of ADM (0, 0.25, 1, 4, 16 or 64
μg/ml). A combination of various concentrations of ADM (0, 0.25, 1,
4, 16 or 64 μg/ml) and sorafenib (2.5 μM), PD98059 (5 μM) or CsA (4
μg/ml) were added to the experimental groups. Cell viability was
determined using the CellTiter-Glo luminescent cell viability kit
from Promega Corporation (Madison, WI, USA) according to the
manufacturer’s instructions. This method was based on the
measurement of ATP production in the cells, proportional to the
number of viable cells, detected by luciferin-luciferase reaction.
The cell proliferation inhibition rate was calculated by the
following formula: Cell proliferation inhibition rate = (1 −
relative luminescence of the experimental group/relative
luminescence of the control group) × 100. All experiments were
repeated at least three times and the average values were used as
the final results. The half maximal inhibitory concentration
(IC50) value, which signifies 50% cell growth inhibition
compared with the control, was calculated by non-linear regression
analysis with GraphPad Prism version 5.0 software (GraphPad
Software, Inc., San Diego, CA, USA), according to the results of at
least three independent experiments, with four replicates of each
cell line per experiment. The resistance index (RI) and reversal
fold were calculated according to the following formulae: RI =
(IC50 of MDR cells)/(IC50 of parental cells);
and reversal fold = (IC50 of MDR cells)/(IC50
of MDR cells following reversal).
Western blot analysis
The cells were cultured in culture medium until
60–70% confluence was reached. DMSO and PD98059 (2.5, 5, 10 or 20
μM) were added to the control and experimental groups, which were
incubated for 1 h. Sorafenib (2.5, 5 or 10 μM) or CsA (0.25, 1, 4,
or 16 μg/ml) were then added to the experimental groups, and DMSO
was added to the control groups. The cells were then incubated for
24 h. Adherent cells were washed with cold phosphate-buffered
saline and lysed directly in the dish for 20 min on ice with cell
lysis buffer [containing 150 mmol/l NaCL, 50 mmol/l Tris-HCL (pH
7.4), 2 mmol/l EDTA, 1% NP-40, protease inhibitor cocktail and
phosphatase inhibitor cocktail; Applygen Technologies Inc.,
Beijing, China]. The lysates were then incubated at 4°C for 20 min
and centrifuged for 10 min at 12,000 × g. The protein levels in the
extracts were quantified using a bicinchoninic acid assay (Pierce
Biotechnology, Inc., Rockford, IL, USA). Subsequently, the protein
was denatured in a lithium dodecyl sulfate sample buffer for 5 min
at 105°C. Equal quantities of total protein (20 μg per lane) were
resolved on 12% polyacrylamide gels using standard sodium dodecyl
sulfate polyacrylamide gel electrophoresis and then transferred to
a polyvinylidene difluoride membrane (0.45 μm, Millipore,
Billerica, MA, USA). The membranes were blocked with 5% dry milk in
Tris-buffered saline (TBS) containing 0.05% Tween-20 (TBST) for 1 h
at room temperature and incubated overnight at 4°C with the
following primary antibodies: monoclonal rabbit anti-human, -mouse,
-rat, -hamster, -monkey, -mink, -D. melanogaster, -zebrafish,
-bovine, -dog, -pig, -S. cerevisiae, phosphorylated (p)ERK1/2
(1:2,000; Thr202/Tyr204; Cell Signaling Technology, Inc.),
polyclonal rabbit anti-human, -mouse, -rat, -equine, -canine,
-bovine, -porcine and -avian, ERK1/2 (1:200; Santa Cruz
Biotechnology, Inc., Santa Cruz, CA, USA), polyclonal rabbit
anti-human, -mouse, -rat and -baboon, GAPDH (1:1,000; Epitomics,
Inc., Burlingame, CA, USA). Following incubation with the
respective primary antibodies, the membranes were washed three
times for 5 min in TBST. The memebranes were then exposed to
horseradish peroxidase-conjugated monoclonal goat anti-rabbit
immunoglobulin G (1:1,000; Multi Sciences (Lianke) Biotech Co.,
Ltd., Hangzhou, China) for 1 h at room temperature. Following
incubation with the secondary antibodies, the membranes were washed
three times for 5 min in TBST. The signal was detected with an
Enhanced Chemiluminesence Western Blotting Detection kit (Applygen
Technologies Inc.). The results are presented as the ratio of the
density of the target protein to that of GAPDH. Each experiment was
repeated at least three times and the final results are shown as
the mean values.
Statistical analysis
Statistical analysis was performed using SPSS
version 13.0 (SPSS, Inc., Chicago, IL, USA) and data are shown as
the mean ± standard deviation. Student’s t-test and a one-way
analysis of variance were used for the statistical analyses.
P<0.05 was considered to indicate a statistically significant
difference.
Results
SMMC7721/ADM and BEL7402/ADM cells
exhibit stable drug resistance
The ADM IC50 values of the SMMC7721 and
SMMC7721/ADM cells were 0.089±0.026 and 1.463±0.168 μg/ml,
respectively, and the RI of the SMMC7721/ADM cells was 16.44. The
ADM IC50 values of the BEL7402 and BEL7402/ADM cells
were 0.161±0.039 μg/ml and 3.266±0.271 μg/ml, respectively, and the
RI of the BEL7402/ADM cells was 20.34. The results are shown in
Fig. 1 and Table I. The data show that the ADM
sensitivities of the SMMC7721/ADM and BEL7402/ADM cells were
significantly lower than those of the corresponding non-resistant
parent cells (P=0.000), which indicates that the SMMC7721/ADM and
BEL7402/ADM cells exhibited stable chemoresistance.
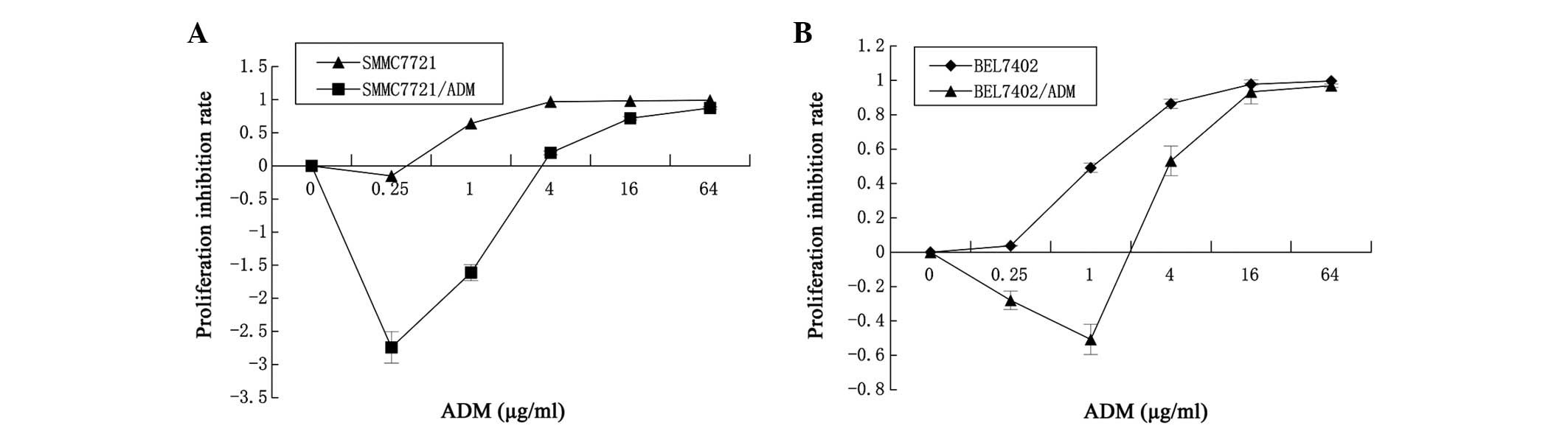 | Figure 1Increased drug resistance of
SMMC7721/Adriamycin (ADM) and BEL7402/ADM cells to the cytotoxic
drug, ADM, compared with that of SMMC7721 and BEL7402 cells. (A)
The SMMC7721 and SMMC7721/ADM cells were incubated with 0, 0.25, 1,
4, 16 or 64 μg/ml ADM for 48 h. (B) The BEL7402 and BEL7402/ADM
cells were also incubated with 0, 0.25, 1, 4, 16 or 64 μg/ml ADM
for 48 h. At the end of incubation, the cell survival rates were
determined by CellTiter-Glo® luminescent cell viability
assay and the proliferation inhibition rate was calculated. Results
are reported as the mean ± standard deviation of three independent
experiments performed in five replicates. |
 | Table IADM IC50 in SMMC7721,
SMMC7721/ADM, BEL7402 and BEL7402/ADM cells. |
Table I
ADM IC50 in SMMC7721,
SMMC7721/ADM, BEL7402 and BEL7402/ADM cells.
| Cell line | ADM IC50,
μg/ml | Resistance index |
|---|
| SMMC7721 | 0.089±0.006 | |
| SMMC7721/ADM | 1.463±0.068 | 16.44a |
| BEL7402 | 0.161±0.03 | |
| BEL7402/ADM | 3.266±0.072 | 20.34b |
PD98059 and sorafenib inhibit ERK/MAPK
signaling pathway activity in SMMC7721/ADM and BEL7402/ADM
cells
Subsequent to 1 h of treatment with PD98059, the
pERK1/2 expression rates (% of control) in the SMMC7721/ADM
(Fig. 2A) and BEL7402/ADM (Fig. 2B) cells were downregulated in a
dose-dependent manner. At concentrations of 2.5, 5, 10 and 20 μM,
the rates declined to 97.43±1.51, 70.53±4.23, 62.33±3.34 and
25.79±5.33%, respectively, in the SMMC7721/ADM cells, and to
91.01±2.27, 86.31±6.54, 84.54±4.98 and 55.53±3.75%, respectively,
in the BEL7402/ADM cells (Fig. 2E).
Following 24 h of treatment with sorafenib at these same
concentrations, pERK expression was again inhibited in a
concentration-dependent manner (Fig. 2C
and D). At concentrations of 2.5, 5 and 10 μM sorafenib, the
pERK1/2 expression rates were reduced to 91.71±3, 50.41±2.3 and
42.76±2.6%, respectively, in the SMMC7721/ADM cells, and to
88.45±3.1, 68.79±2.9 and 31.28±3.3%, respectively, in the
BEL7402/ADM cells (Fig. 2F).
PD98059 and sorafenib increase the
sensitivity of SMMC7721/ADM and BEL7402/ADM cells to ADM
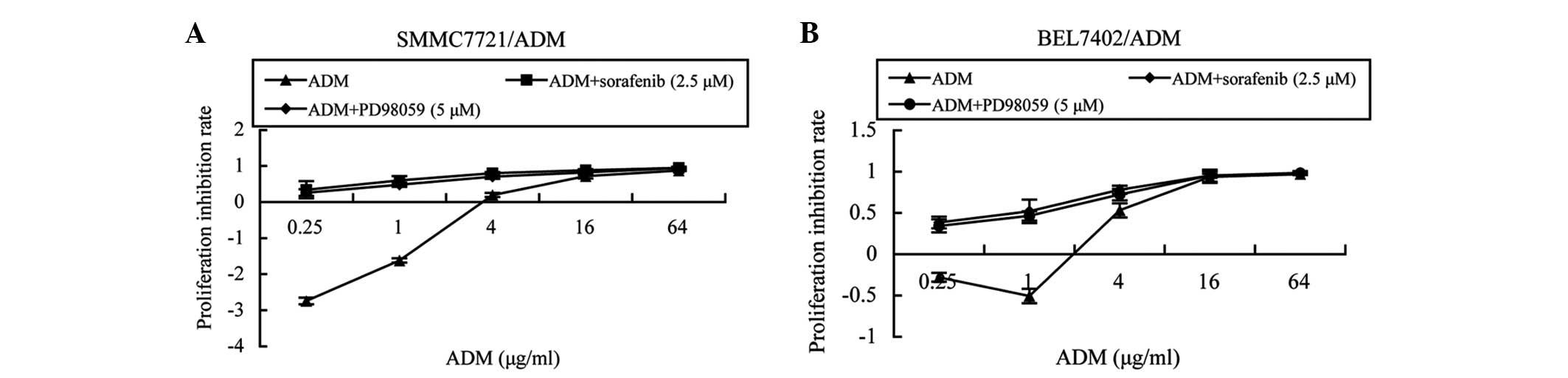
Subsequent to 72 h of treatment, sorafenib inhibited
MDR cell proliferation in a dose-dependent manner. When the drug
concentration was 2.5 μM, the cell inhibition rates were <5%, at
4.35 and 1.84% for the SMMC7721/ADM and BEL7402/ADM cells,
respectively. Thus, 2.5 μM was selected to be the concentration of
sorafenib used for MDR reversal. Similarly, when the concentration
of PD98059 was 5 μM, the cell inhibition rates for SMMC7721/ADM and
BEL7402/ADM cells were 3.78 and 1.21%, respectively; therefore, 5
μM was selected as the reversal concentration of PD98059. The cell
proliferation inhibition rates in the cells treated with a
combination of ADM plus PD98059 or sorafenib were higher than those
treated with ADM only (Fig. 3A and
B). When 5 μM PD98059 was added, the ADM IC50 values
of the SMMC7721/ADM and BEL7402/ADM cells were 0.8±0.056 and
1.583±0.284 μg/ml, respectively. Furthermore, the reverse fold
values were 1.83 in the SMMC7721/ADM cells and 2.06 in the
BEL7402/ADM cells. When treated with 2.5 μM sorafenib, the ADM
IC50 values of the SMMC7721/ADM and BEL7402/ADM cells
were 0.264±0.049 and 1.099±0.135 μg/ml, respectively. The reversal
fold ADM resistance levels of the SMMC7721/ADM and BEL7402/ADM
cells were 5.54-fold and 2.97-fold, respectively (Table II).
| Table IIADM IC50 in hepatocellular
carcinoma multidrug-resistant cells treated with PD98059, sorafenib
and CsA. |
Table II
ADM IC50 in hepatocellular
carcinoma multidrug-resistant cells treated with PD98059, sorafenib
and CsA.
| Cell
line/treatment | ADM IC50,
μg/ml | Reverse-fold |
|---|
| SMMC7721/ADM | 1.463±0.168 | |
|
SMMC7721/ADM+PD98059 | 0.800±0.056 | 1.83a |
|
SMMC7721/ADM+sorafenib | 0.264±0.049 | 5.54a |
| SMMC7721/ADM+CsA | 0.349±0.023 | 4.19a |
| BEL7402/ADM | 3.266±0.271 | |
|
BEL7402/ADM+PD98059 | 1.583±0.284 | 2.06b |
|
BEL7402/ADM+sorafenib | 1.099±0.135 | 2.97b |
| BEL7402/ADM+CsA | 0.427±0.039 | 7.65b |
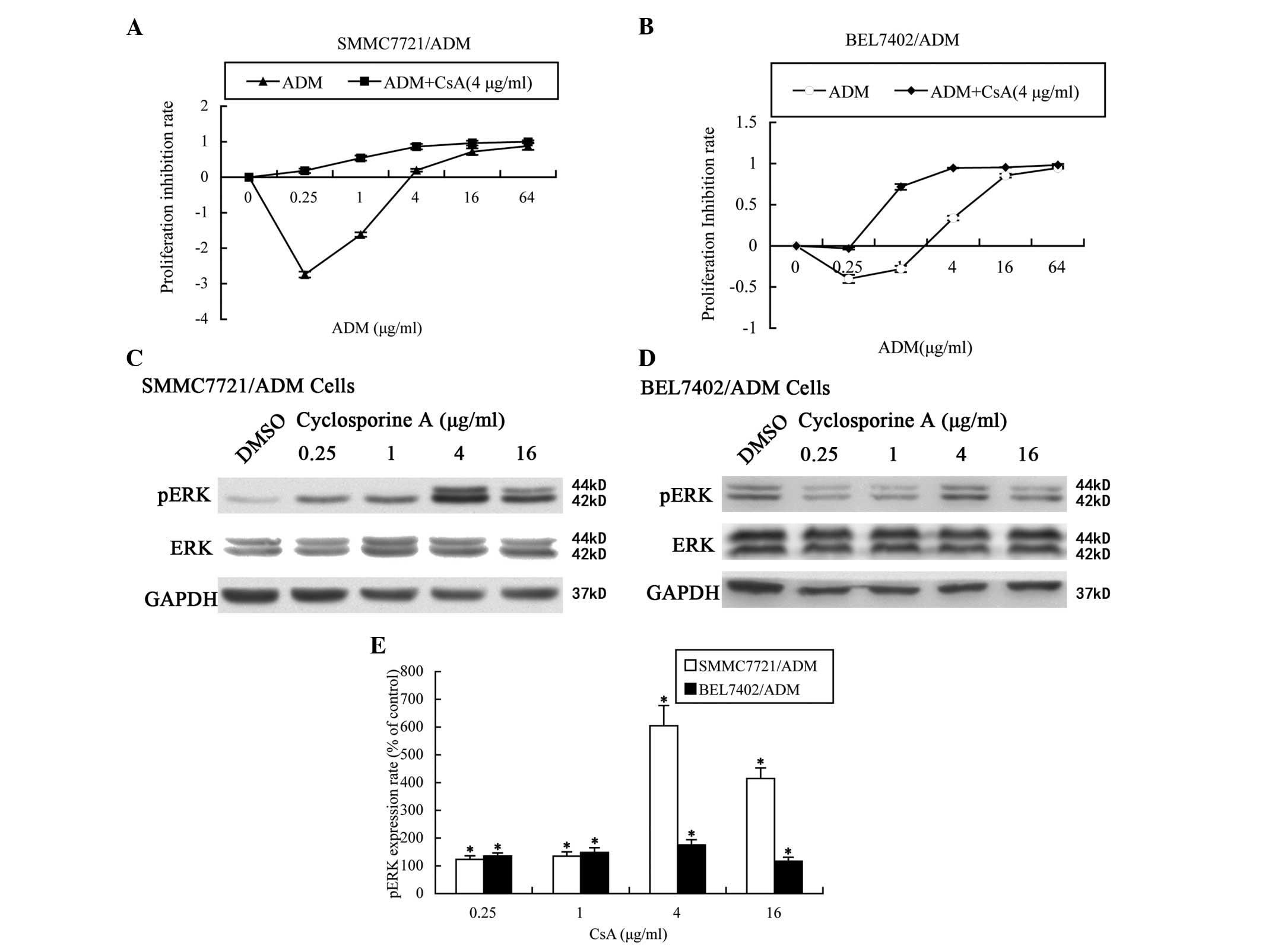
CsA enhances ADM sensitivity and
upregulates ERK 1/2 phosphorylation in MDR HCC cells
As shown in Fig. 4A and
B, the cell proliferation inhibition rate of the cells cultured
with ADM plus CsA was significantly increased compared with that of
the cells cultured with ADM only (P=0.000). When combined with 4
μg/ml CsA, the ADM IC50 values of the SMMC7721/ADM and
BEL7402/ADM cells were 0.349±0.023 and 0.427±0.039 μg/ml,
respectively. The ADM resistance reversal levels of the
SMMC7721/ADM and BEL7402/ADM cells were 4.19-fold and 7.65-fold,
respectively (Table II). Following
CsA treatment for 24 h, the pERK1/2 levels increased in a
dose-dependent manner up to 4 μg/ml CsA and then declined at higher
concentrations, but remained above the basal level. The total
ERK1/2 levels were unchanged (Fig.
4C–E).
Discussion
HCC is the third most common cause of cancer
mortality, resulting in more than half a million fatalities
annually worldwide (27,28). In addition to surgical intervention,
systemic chemotherapy also has a significant role in HCC treatment,
particularly for patients with advanced HCC (29). However, since traditional systemic
chemotherapy has limited benefits in advanced-stage HCC patients
due to MDR, novel approaches to overcome this resistance and offer
patients tailored treatment strategies are urgently required
(2,3). A number of MDR reversal agents have
been developed, using the possible mechanisms being reported
(30,31). However, toxicity has become the
predominant obstacle in the wide application of these agents in
clinical treatment (32). For
instance, verapamil treatment induces cardiac toxicity and CsA
exerts significant immunosuppressive effects and has renal
toxicity. Therefore, identifying safe and efficient reversal agents
is of vital importance for treating advanced-stage HCC
patients.
The MAPK signaling pathway is known to mediate a
number of cellular processes, including cell growth,
differentiation, survival and apoptosis. Aside from these
fundamental functions, MAPK has also been reported to be involved
in the development of MDR, conferring imatinib resistance in acute
lymphoblastic leukemia cells (33),
vincristine resistance in gastric cancer cells (34) and anthracycline resistance in breast
cancer cells (35).
Among the four MAPK signaling pathways, the ERK1/2
cascade is the most widely investigated. Thus far, the majority of
studies have reported a positive correlation between the
overactivation of ERK and the development of chemoresistance in
numerous types of cancer cells (36–38).
Certain studies have suggested that modulation of ERK activation
may be a novel method in reversing MDR (40,41).
Our previous study also demonstrated that ERK1/2 activity is
upregulated in MDR HCC cells (26).
As determined by these findings, inhibitors of the ERK/MAPK
signaling pathway have been suggested to reverse MDR in HCC cells.
In the present study, the upstream proteins, RAF and MEK, key
proteins in the RAS/RAF/MEK/ERK cascade, were selected as the
targets of inhibition. The results revealed that sorafenib and
PD98059, which inhibit RAF and MEK kinases, respectively,
downregulated the pERK1/2 levels without affecting the levels of
total ERK1/2. The results of the cell viability assays demonstrated
that sorafenib and PD98059 reduced the ADM IC50 values
in the SMMC7721/ADM and BEL7402/ADM cells, indicating that the two
inhibitors reverse the resistance of HCC cells to ADM. Together,
these results suggest the possibility of using the inhibitors of
the ERK/MAPK signaling pathway as MDR reversal agents, thus
providing evidence for the use of these inhibitors in combination
with traditional chemotherapeutic drugs in treating HCC
patients.
CsA, an inhibitor first used to analyze MDR
reversal, enhances the apoptosis in tumor cells induced by
chemotherapeutics through increasing the intracellular drug
concentration. The MDR reversal mechanism of CsA possibly occurs
through inhibiting the pump function of p-gp (42,43).
In the present study, CsA upregulated, but did not downregulate,
the expression of pERK1/2 in the HCC MDR cells. These results
indicate that the downregulation of pERK1/2 was not involved in the
reversal function of CsA, suggesting that the inhibition of the
ERK/MAPK signaling pathway is not the only method to reverse MDR in
HCC cells. In addition to leukocytes, CsA exerts potent effects on
a number of distinct types of cells and thus, regulates disparate
biological functions (44–48). Furthermore, evidence is emerging
that CsA regulates cell proliferation and invasion through ERK
(44,49–51).
Therefore, the increased pERK1/2 levels observed may be involved in
certain other CsA effects in HCC cells. CsA treatment may therefore
render cells in a state of stress, thus resulting in the
upregulation of pERK1/2 through negative feedback.
The combined application of various
chemotherapeutants is one method for mitigating drug resistance in
classic chemotherapy (52). If the
signaling pathway that exerts a predominant role in the growth
tumor of a particular tumor becomes clear prior to the patient
accepting therapy, the inhibitors of the corresponding signaling
pathway may be pointedly selected to raise the effectiveness of
chemotherapy. All results from the present study indicate that
inhibition of ERK/MAPK signaling pathway activity may indeed
reverse MDR in HCC cells, thus providing evidence for the use of
ERK/MAPK signaling pathway inhibitors combined with traditional
drugs in treating HCC. In addition, downregulation of the ERK/MAPK
signaling pathway activity was not found to be involved in the CsA
reversal function, which indicates that inhibiting ERK/MAPK
signaling pathway activity is not a unique method to reverse MDR in
HCC cells.
References
|
1
|
Tang S, Bao H, Zhang Y, et al:
14-3-3epsilon mediates the cell fate decision-making pathways in
response of hepatocellular carcinoma to Bleomycin-induced DNA
damage. PLoS One. 8:e552682013.
|
|
2
|
Zhong X, Xiong M, Meng X and Gong R:
Comparison of the multi-drug resistant human hepatocellular
carcinoma cell line Bel-7402/ADM model established by three
methods. J Exp Clin Cancer Res. 29:1152010.
|
|
3
|
Zhang Z, Zhou X, Shen H, Wang D and Wang
Y: Phosphorylated ERK is a potential predictor of sensitivity to
sorafenib when treating hepatocellular carcinoma: evidence from an
in vitro study. BMC Med. 7:412009.
|
|
4
|
Zhu AX: Systemic therapy of advanced
hepatocellular carcinoma: how hopeful should we be? Oncologist.
11:790–800. 2006.
|
|
5
|
Wakamatsu T, Nakahashi Y, Hachimine D,
Seki T and Okazaki K: The combination of glycyrrhizin and
lamivudine can reverse the cisplatin resistance in hepatocellular
carcinoma cells through inhibition of multidrug
resistance-associated proteins. Int J Oncol. 31:1465–1472.
2007.
|
|
6
|
Gottesman MM, Fojo T and Bates SE:
Multidrug resistance in cancer: role of ATP-dependent transporters.
Nat Rev Cancer. 2:48–58. 2002.
|
|
7
|
List AF, Kopecky KJ, Willman CL, et al:
Benefit of cyclosporine modulation of drug resistance in patients
with poor-risk acute myeloid leukemia: a Southwest Oncology Group
study. Blood. 98:3212–3220. 2001.
|
|
8
|
Yamamoto K, Kikuchi Y, Kudoh K and Nagata
I: Modulation of cisplatin sensitivity by taxol in
cisplatin-sensitive and -resistant human ovarian carcinoma cell
lines. J Cancer Res Clin Oncol. 126:168–172. 2000.
|
|
9
|
Holmström TH, Tran SE, Johnson VL, et al:
Inhibition of mitogen-activated kinase signaling sensitizes HeLa
cells to Fas receptor-mediated apoptosis. Mol Cell Biol.
19:5991–6002. 1999.
|
|
10
|
Cowley GP and Smith ME: Modulation of
E-cadherin expression and morphological phenotype in the
intravascular component of adenocarcinomas. Int J Cancer.
60:325–329. 1995.
|
|
11
|
Huang C, Jacobson K and Schaller MD: MAP
kinases and cell migration. J Cell Sci. 117:4619–4628. 2004.
|
|
12
|
Mansour SJ, Matten WT, Hermann AS, et al:
Transformation of mammalian cells by constitutively active MAP
kinase kinase. Science. 265:966–970. 1994.
|
|
13
|
Mansour SJ, Resing KA, Candi JM, et al:
Mitogen-activated protein (MAP) kinase phosphorylation of MAP
kinase kinase: determination of phosphorylation sites by mass
spectrometry and site-directed mutagenesis. J Biochem. 116:304–314.
1994.
|
|
14
|
Sturgill TW, Ray LB, Erikson E and Maller
JL: Insulin-stimulated MAP-2 kinase phosphorylates and activates
ribosomal protein S6 kinase II. Nature. 334:715–718. 1988.
|
|
15
|
Boulton TG, Nye SH, Robbins DJ, et al:
ERKs: a family of protein-serine/threonine kinases that are
activated and tyrosine phosphorylated in response to insulin and
NGF. Cell. 65:663–675. 1991.
|
|
16
|
Seger R and Krebs EG: The MAPK signaling
cascade. FASEB J. 9:726–735. 1995.
|
|
17
|
Hibi M, Lin A, Smeal T, Minden A and Karin
M: Identification of an oncoprotein- and UV-responsive protein
kinase that binds and potentiates the c-Jun activation domain.
Genes Dev. 7:2135–2148. 1993.
|
|
18
|
Kyriakis JM, Banerjee P, Nikolakaki E, et
al: The stress-activated protein kinase subfamily of c-Jun kinases.
Nature. 369:156–160. 1994.
|
|
19
|
Han J, Lee JD, Bibbs L and Ulevitch RJ: A
MAP kinase targeted by endotoxin and hyperosmolarity in mammalian
cells. Science. 265:808–811. 1994.
|
|
20
|
Freshney NW, Rawlinson L, Guesdon F, et
al: Interleukin-1 activates a novel protein kinase cascade that
results in the phosphorylation of Hsp27. Cell. 78:1039–1049.
1994.
|
|
21
|
Rouse J, Cohen P, Trigon S, et al: A novel
kinase cascade triggered by stress and heat shock that stimulates
MAPKAP kinase-2 and phosphorylation of the small heat shock
proteins. Cell. 78:1027–1037. 1994.
|
|
22
|
Lee JC, Laydon JT, McDonnell PC, et al: A
protein kinase involved in the regulation of inflammatory cytokine
biosynthesis. Nature. 372:739–746. 1994.
|
|
23
|
Zhou G, Bao ZQ and Dixon JE: Components of
a new human protein kinase signal transduction pathway. J Biol
Chem. 270:12665–12669. 1995.
|
|
24
|
Lee JD, Ulevitch RJ and Han J: Primary
structure of BMK1: a new mammalian map kinase. Biochem Biophys Res
Commun. 213:715–724. 1995.
|
|
25
|
Kohno M, Tanimura S and Ozaki K: Targeting
the extracellular signal-regulated kinase pathway in cancer
therapy. Biol Pharm Bull. 34:1781–1784. 2011.
|
|
26
|
Yan X, Ruan W, Wang X, et al: The effects
of multidrug resistance on cell proliferation, apoptosis and
invasion activity and the expression of mitogen-activated protein
kinase in human hepatocellular cancer cells. Zhong Liu. 32:507–515.
2012.(In Chinese).
|
|
27
|
Philip PA, Mahoney MR, Allmer C, et al:
Phase II study of Erlotinib (OSI-774) in patients with advanced
hepatocellular cancer. J Clin Oncol. 23:6657–6663. 2005.
|
|
28
|
El-Serag HB and Rudolph KL: Hepatocellular
carcinoma: epidemiology and molecular carcinogenesis.
Gastroenterology. 132:2557–2576. 2007.
|
|
29
|
Geng W, Ng KT, Sun CK, et al: The role of
proline rich tyrosine kinase 2 (Pyk2) on cisplatin resistance in
hepatocellular carcinoma. PLoS One. 6:e273622011.
|
|
30
|
Long Sheng and Maoming Xiong: Progress in
primary liver cancer resistant MDR reversal of Hepatobiliary
Surgery. 20:236–238. 2012.
|
|
31
|
Dai CL and Fu LW: The development of the
reversal of the tumor multidrug resistance. Chin Pharm Bulletin.
21:513–8. 2005.
|
|
32
|
Pétriz J, Sánchez J, Bertrán J and
García-López J: Comparative effect of verapamil, cyclosporin A and
SDZ PSC 833 on rhodamine 123 transport and cycle in
vinblastine-resistant Chinese hamster ovary cells overexpressing
P-glycoprotion. Anticancer Drugs. 8:869–875. 1997.
|
|
33
|
Dreuw A, Hermanns HM, Heise R, et al:
Interleukin-6-type cytokines upregulate expression of multidrug
resistance-associated proteins in NHEK and dermal fibroblasts. J
Invest Dermatol. 124:28–37. 2005.
|
|
34
|
Imai Y, Ohmori K, Yasuda S, et al: Breast
cancer resistance protein/ABCG2 is differentially regulated
downstream of extracellular signal-regulated kinase. Cancer Sci.
100:1118–1127. 2009.
|
|
35
|
Gómez-Martínez A, García-Morales P,
Carrato A, et al: Post-transcriptional regulation of P-glycoprotein
expression in cancer cell lines. Mol Cancer Res. 5:641–653.
2007.
|
|
36
|
Guan J, Chen XP, Zhu H, et al: Involvement
of extracellular signal-regulated kinase/mitogen-activated protein
kinase pathway in multidrug resistance induced by HBx in hepatoma
cell line. World J Gastroenterol. 10:3522–3527. 2004.
|
|
37
|
Zhu H, Chen XP, Luo SF, et al: The role of
extracellular signal-regulated kinase/mitogen-activated protein
kinase pathway in multidrug resistance of hepatocellular carcinoma.
Zhonghua Wai Ke Za Zhi. 45:917–920. 2007.(In Chinese).
|
|
38
|
Kisucká J, Barancík M, Bohácová V and
Breier A: Reversal effect of specific inhibitors of
extracellular-signal regulated protein kinase pathway on
P-glycoprotein mediated vincristine resistance of L1210 cells. Gen
Physiol Biophys. 20:439–444. 2001.
|
|
39
|
Chen B, Jin F, Lu P, et al: Effect of
mitogen-activated protein kinase signal transduction pathway on
multidrug resistance induced by vincristine in gastric cancer cell
line MGC803. World J Gastroenterol. 10:795–799. 2004.
|
|
40
|
Li Y, Li S, Han Y, et al: Calebin-A
induces apoptosis and modulates MAPK family activity in drug
resistant human gastric cancer cells. Eur J Pharmacol. 591:252–258.
2008.
|
|
41
|
Hu Y, Bally M, Dragowska WH and Mayer L:
Inhibition of mitogen-activated protein kinase/extracellular
signal-regulated kinase kinase enhances chemotherapeutic effects on
H460 human non-small cell lung cancer cells through activation of
apoptosis. Mol Cancer Ther. 2:641–649. 2003.
|
|
42
|
Anglicheau D, Pallet N, Rabant M, et al:
Role of P-glycoprotein in cyclosporine cytotoxicity in the
cyclosporine-sirolimus interaction. Kidney Int. 70:1019–1025.
2006.
|
|
43
|
Tanaka S, Hirano T, Saito T, Wakata N and
Oka K: P-glycoprotein function in peripheral blood mononuclear
cells of myasthenia gravis patients treated with tacrolimus. Biol
Pharm Bull. 30:291–296. 2007.
|
|
44
|
Alvarez-Arroyo MV, Yagüe S, Wenger RM, et
al: Cyclophilin-mediated pathways in the effect of cyclosporin A on
endothelial cells: role of vascular endothelial growth factor. Circ
Res. 91:202–209. 2002.
|
|
45
|
Robida AM, Xu K, Ellington ML and Murphy
TJ: Cyclosporin A selectively inhibits mitogen-induced
cyclooxygenase-2 gene transcription in vascular smooth muscle
cells. Mol Pharmacol. 58:701–708. 2000.
|
|
46
|
Chen HW, Chien CT, Yu SL, Lee YT and Chen
WJ: Cyclosporine A regulate oxidative stress-induced apoptosis in
cardiomyocytes: mechanisms via ROS generation, iNOS and Hsp70. Br J
Pharmacol. 137:771–781. 2002.
|
|
47
|
Kiely B, Feldman G and Ryan MP: Modulation
of renal epithelial barrier function by mitogen-activated protein
kinases (MAPKs): mechanism of cyclosporine A-induced increase in
transepithelial resistance. Kidney Int. 63:908–916. 2003.
|
|
48
|
Hojo M, Morimoto T, Maluccio M, et al:
Cyclosporine induces cancer progression by a cell-autonomous
mechanism. Nature. 397:530–534. 1999.
|
|
49
|
Paslaru L, Trigon S, Kuhlmann M and
Morange M: MAP kinase activation by cyclosporine A. Biochem Biophys
Res Commun. 236:599–603. 1997.
|
|
50
|
Du MR, Zhou WH, Yan FT, et al:
Cyclosporine A induces titin expression via MAPK/ERK signalling and
improves proliferative and invasive potential of human trophoblast
cells. Hum Reprod. 22:2528–2537. 2007.
|
|
51
|
Zhou WH, Du MR, Dong L, et al: Cyclosporin
A increases expression of matrix metalloproteinase 9 and 2 and
invasiveness in vitro of the first-trimester human trophoblast
cells via the mitogen-activated protein kinase pathway. Hum Reprod.
22:2743–2750. 2007.
|
|
52
|
Cen J and Li YM: Combination therapy on
multidrug-resistance tumor. World Notes Antibiotics. 5:224–228.
2009.
|