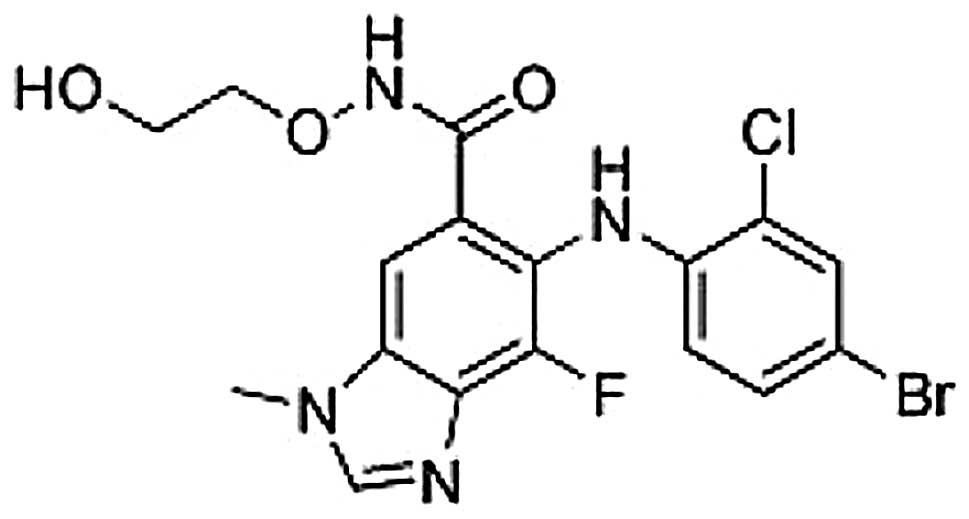
Introduction
Non-small cell lung cancer (NSCLC) remains the
cancer type with the highest mortality in the United States, with
>150,000 fatalities anticipated in 2013 (1). The majority of NSCLC patients are
diagnosed with the advanced stage of the disease, leading to a poor
prognosis, with a median survival time of only 10–12 months
(2,3).
Advances in the molecular characterization of NSCLC, have resulted
in the use of novel drugs that are able to target oncogenic
mutations. The efficacy of NSCLC therapy has increased following
the development of epidermal growth factor receptor tyrosine kinase
inhibitors (EGFR-TKIs), such as gefitinib and erlotinib (1,4). EGFR-TKIs
attenuate EGFR autophosphorylation and the subsequent activation of
downstream pathways that regulate cell survival and growth,
including the phosphoinositide 3-kinase (PI3K)-Akt-mammalian target
of rapamycin signal pathway and the mitogen-activated protein
kinase (MAPK) cascade [Ras-Raf-mitogen-activated protein kinase
kinase (MEK)-extracellular signal-regulated kinases (ERK)]
(5).
Despite EGFR-TKIs being an effective clinical
therapy in patients with advanced NSCLC and EGFR-activated
mutations, all patients will eventually develop acquired resistance
to these drugs. Following continuous use of TKIs for 9–11 months,
the majority of patients who initially respond to the treatment
subsequently experience progression of the disease (6); ~50% of those with an initial response to
EGFR-TKIs acquire resistance to these drugs. The challenge of
overcoming the resistance to TKIs in clinical practice requires a
novel method.
A range of mechanisms have been proposed to underlie
the acquired resistance to EGFR-TKIs. For example, a second
mutation in the EGFR kinase domain, particularly a T790M mutation
in exon 20, accounts for 50% of this acquired resistance (7); the T790M gate keeper mutation decreases
the binding of EGFR-TKI to the mutant EGFR (8). Another 20% of patients develop TKI
acquired resistance results from MET amplification (9); MET amplification-induced persistent
activation of the PI3K-Akt pathway via a HER3-dependent pathway is
believed to be the common downstream pathway of the acquired
resistance of NSCLC.
Moreover, the presence of the K-Ras mutation is
associated with the primary resistance to EGFR-TKIs. In total,
15–30% of patients with NSCLC exhibit a gain of function mutation
in the K-Ras gene, resulting in a failure to respond to EGFR-TKI
treatment. In NSCLC, the activation of K-Ras leads to the
overexpression of ERK1/2 via the Raf/MEK/ERK signaling pathway.
Therefore, inhibiting the Raf/MEK/ERK signaling pathway would
result in an improvement in the antitumor effects following
TKI-acquired resistance, however, the function of the Raf/MEK/ERK
pathway in acquired resistance in NSCLC has not been extensively
examined. In the present study, the antitumor effects of gefitinib
alone compared with gefitinib in combination with MEK inhibitor,
AZD6244, was investigated in EGFR-TKI-resistant NSCLC cells.
Materials and methods
Drugs and cell lines
Gefitinib and AZD6244 (selumetinib) were obtained
from Astrazeneca (Cheshire, UK). Gefitinib and AZD6244 were
prepared in dimethyl sulfoxide to obtain a stock solution of 10 mM.
The EGFR-TKI-sensitive PC-9 (mutant EGFR/wild-type K-Ras) and
EGFR-TKI-resistant A549 (wild-type EGFR/mutant K-Ras) human NSCLC
cell lines were purchased from the American Type Culture Collection
(Manassas, VA, USA) and maintained in RPMI-1640 medium (Hyclone,
Logan, UT, USA), supplemented with 10% heat-inactivated fetal
bovine serum (Hyclone), penicillin (100 U/ml), streptomycin (100
µg/ml) and L-glutamine (2 mM) at 37°C in a 5% CO2
atmosphere, and then harvested with trypsin-EDTA when the cells
reached exponential growth.
Anti-proliferative effects of
agents
The anti-proliferative effects of gefitinib and
AZD6244 as single agents on A549 and PC-9 cells were evaluated by
MTT assay. The cells were cultured in 96-well plates, with 4,000
A549 cells per well or 6,000 PC-9 cells per well. Gefitinib was
added at concentrations of 10, 20, 30, 40 and 50 µmol/l after 24 h.
AZD6244 was added at concentrations of 0.001, 0.01, 0.1, 1 and 10
µmol/l after 24 h. The half maximal inhibitory concentration
(IC50) was determined following 72 h of exposure to the
drug compared with unexposed control cells. Once the cells had been
exposed to each drug for 72 h in the 96-well plates, 20 ml MTT
solution was added to each well. The optical density (OD) of each
well was measured at 490 nm following incubation for 4 h. The
percentage of cell growth inhibition resulting from each drug was
calculated as: [(OD490 of control cells -
OD490 of treated cells) / OD490 of control
cells] × 100. This assay was repeated for >3 independent
experiments.
Western blotting
The cells were treated with gefitinib plus AZD6244
at various concentrations. To evaluate downstream signaling,
treated cells were harvested, washed with phosphate-buffered saline
and lysed. Western blotting was performed as described previously
(10). The primary antibodies were as
follows: β-actin (rabbit anti-human monoclonal; cat no. 8457;
1:1,000; Cell Signaling Technology, Beverly, MA, USA), EGFR (rabbit
anti-human monoclonal; cat no. ab32562; 1:5,000) and its
phosphorylated (p) components pY1173 (rabbit anti-human monoclonal;
cat no. ab32578; 1:1,000), Akt (mouse anti-human monoclonal; cat
no. ab124341; 1:1,000) and pAkt (rabbit anti-human monoclonal; cat
no. 4060; 1:2,000), ERK (rabbit anti-human polyclonal; cat no.
9102; 1:1,000) and pERK (rabbit anti-human monoclonal; cat no.
4370; 1:2,000) (Cell Signaling Technology Inc., Danvers, MA, USA).
Proteins were visualized with a horseradish peroxidase-coupled
secondary antibody (anti-rabbit IgG; cat no. 7071; 1:5,000)
obtained from Cell Signaling Technology Inc. Membranes were then
washed again three times for 10 min each with Tris-buffered saline
with Tween 20. Target protein bands were visualized using the
enhanced chemiluminescence method. The intensity of the bands was
quantified using the Tanon GIS system (Tanon, Shanghai, China) and
the data were normalized to the β-actin loading controls. All
western blotting analyses were performed three times.
Statistical analysis
Results obtained from a minimum of three independent
experiments are expressed as the mean ± standard deviation.
Student's t-test and a one-way analysis of variance test were used
to determine the differences between control and treatment groups.
Data analyses were performed using SPSS statistical software
(version 15.0; SPSS, Inc., Chicago, IL, USA). P<0.05 was used to
indicate a statistically significant difference.
Results
Sensitivity of NSCLC cell lines to
EGFR and MEK1/2 inhibitors
MTT assays were used to evaluate the
anti-proliferative effects of gefitinib (0, 0.25, 0.5, 2.5, 5, 25
and 50 µM) as a single agent on EGFR-TKI-sensitive PC-9 (mutant
EGFR/wild-type K-Ras) and EGFR-TKI-resistant A549 (wild-type
EGFR/mutant K-Ras) NSCLC cell lines. Dose-dependent growth
inhibitory effects of gefitinib were observed in the two NSCLC cell
lines (Fig. 1). It was shown that the
EGFR-TKI-sensitive PC-9 cells were more sensitive to gefitinib than
the EGFR-TKI-resistant A549 cells. The IC50 in the PC-9
and A549 cells after 72 h was 4.1 and 10.4 µM, respectively.
The MEK1/2-dependent proliferation of the NSCLC cell
lines was examined by treating cells with the newly developed
MEK1/2 inhibitor, AZD6244 (AstraZeneca) (Fig. 2). EGFR-TKI-sensitive PC-9 (mutant
EGFR/wild-type K-Ras) and EGFR-TKI-resistant A549 (wild-type
EGFR/mutant K-Ras) NSCLC cell lines were selected to test the
inhibitory effect of AZD6244. Cell proliferation was analyzed by
MTT assay in cells treated with 0.001, 0.01, 0.1, 1 and 10 µM of
the AZD6244. The results showed that the lung cancer cell lines
tested were all resistant to AZD6244 monotherapy (Fig. 3), with an IC50 of >10
µM. Thus, the EGFR-TKI-resistant A549 (wild-type EGFR/mutant K-Ras)
NSCLC cell line was selected for the combination treatment
experiments.
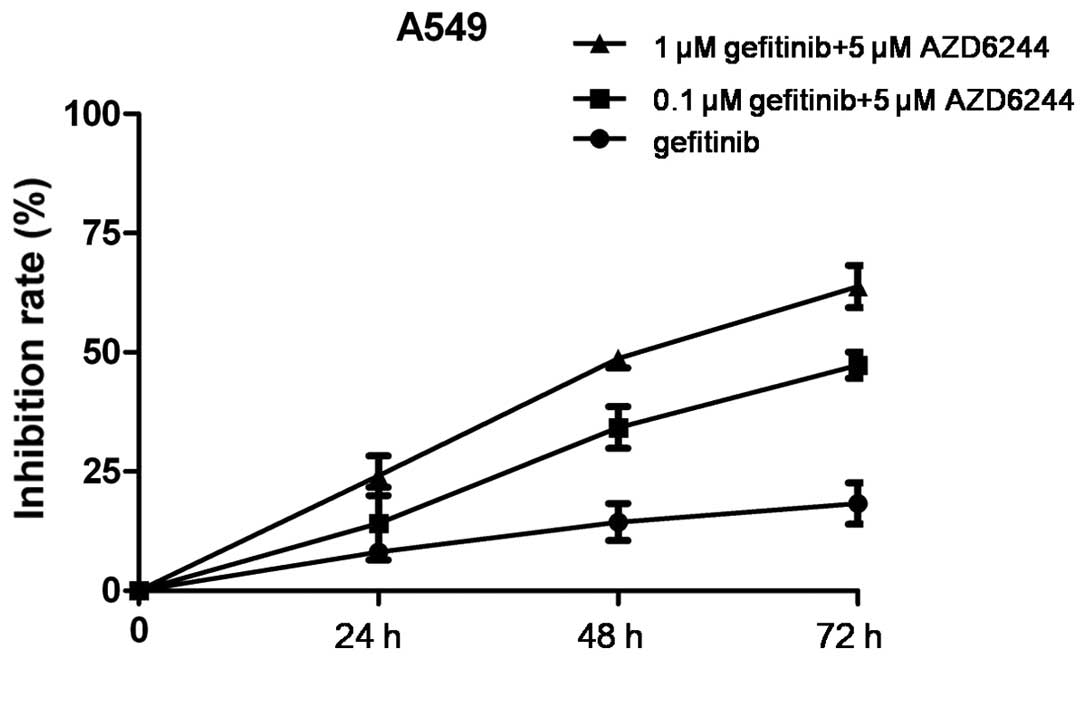
Synergistic growth inhibitory effect
of gefitinib and AZD6244 in NSCLC cells
Since the KRAS-RAF-MEK-ERK cascade is the main
downstream path way of EGFR signaling, we hypothesized that the
dual inhibition of EGFR and MEK1/2 may enhance the therapeutic
efficacy of each agent. Therefore, the effect of gefitinib (0.1 or
1 µM), alone or in combination with 5 µM AZD6244, was examined. The
growth inhibitory effect of the combination treatment was greater
than that of gefitinib alone in the EGFR-TKI-resistant A549 cells
(Fig. 4).
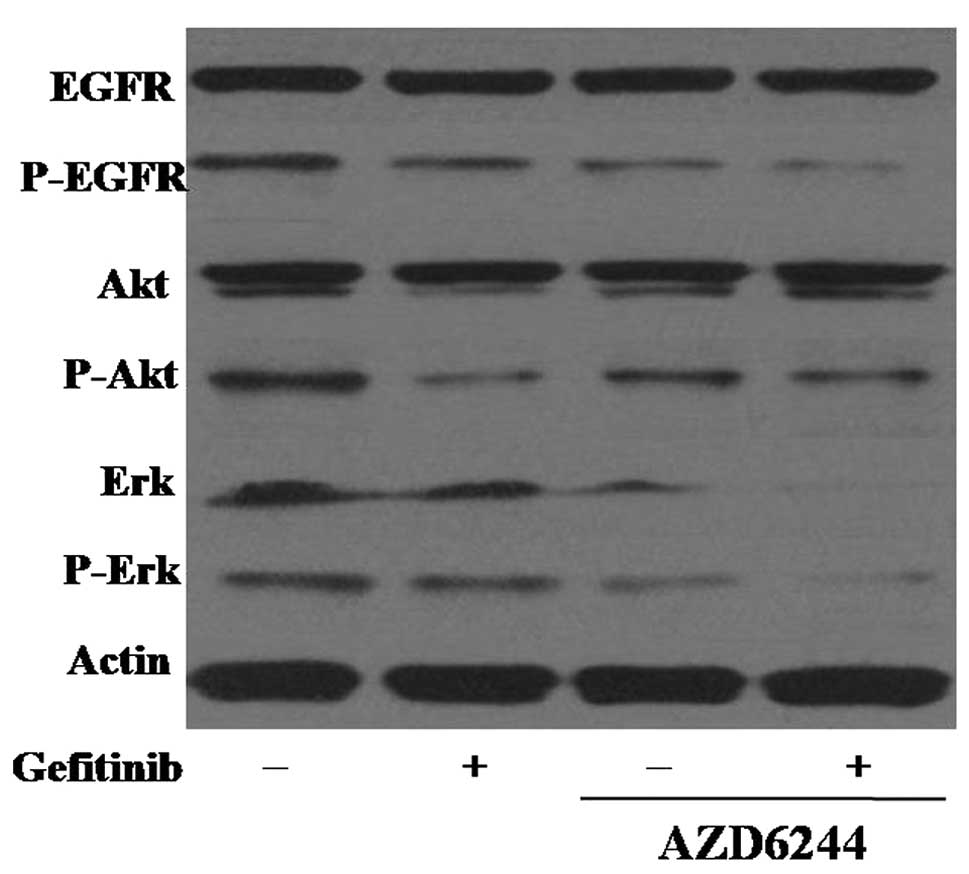
Effects of AZD6244 in combination with
gefitinib on the EGFR signaling pathway
The effects of AZD6244 and gefitinib on the EGFR
pathway in gefitinib-sensitive and -resistant cell lines were
determined using western blot analysis. In gefitinib-sensitive PC-9
cells, 0.25 µM gefitinib was sufficient to completely
dephosphorylate EGFR, but the inhibitory effect of gefitinib on the
phosphorylation of EGFR in the EGFR-TKI-resistant A549 cells was
lower than that in the PC-9 cells. The combination treatment of
gefitinib and AZD6244 in the A549 cells increased the inhibition of
EGFR phosphorylation, suggesting that gefitinib in combination with
AZD6244 demonstrated superior efficacy in gefitinib-resistant NSCLC
cells.
To further assess the potential synergistic
mechanisms of gefitinib and AZD6244, the effects of a combination
of the two agents on the downstream Akt and ERK signaling pathways
were detected by western blot analysis in the A549 cells. As the
MEK/ERK and PI3K/Akt pathways are essential for cell proliferation
and survival, the effect of combined treatment with gefitinib and
AZD6244 on Akt phosphorylation was investigated. It was found that
the treatment of A549 cells with gefitinib alone reduced the
expression level of the activated form of Akt, and that the
combination of the two drugs showed stronger inhibition of Erk and
p-Erk (Fig. 5).
Discussion
Despite significant advances in the development of
therapeutic agents against lung cancer, the mortality rate of lung
cancer patients remains high. Recently, targeted anticancer drugs,
including EGFR-TKIs, have been approved for the treatment of NSCLC
(10). However, not all patients
benefit from this therapy due to primary or acquired resistance,
which are usually caused by the activation of alternative signaling
pathways.
The mechanisms of resistance to EGFR inhibitors may
be varied, with one of the most important mechanisms being the
activation of signaling pathways downstream of EGFR. Overexpression
of downstream effectors, including PI3K/Akt, results in the
persistent activation of the PI3K/Akt and MAPK pathways, and the
consequent development and maintenance of an EGFR-resistant
phenotype (11). Therefore,
inhibiting EGFR alone may not be sufficient for antitumor effects,
and the development of a combination of different signaling pathway
inhibitors is a promising strategy in NSCLC patients.
When considering recent promising studies on highly
selective small molecules, MEK1/2 inhibitors have been developed
and have shown clinical activity in a number of malignancies,
including NSCLC (12,13). In the present study, the MEK1/2
inhibitor, AZD6244, in combination with gefitinib was selected to
investigate the efficacy of this treatment in NSCLC cell lines,
particularly in gefitinib-resistant cells. The present study was
performed in EGFR-TKI-sensitive PC-9 (EGFR mutant/wild-type K-Ras)
and EGFR-TKI-resistant A549 (wild-type EGFR/mutant K-Ras) human
NSCLC cells. Synergy was observed for the combination of gefitinib
and varying doses of AZD6244 in gefitinib-resistant cell lines. The
results reported in the present study demonstrate that the combined
inhibition of EGFR and MEK results in a more effective growth
blockade in NSCLC cell lines compared with either agent alone. The
strongest synergism in the NSCLC cells was observed upon
simultaneous administration of gefitinib and AZD6244 in the two
cell lines.
ERK, the only known target of MEK1/2, has multiple
targets, which are involved in cell proliferation, survival,
mitosis and migration (14,15). AZD6244 is a potent, selective, and
orally available MEK1/2 inhibitor. Previous studies have shown that
the inhibition of MEK1/2 can induce apoptosis by inhibiting
ERK-mediated B-cell lymphoma 2 phosphorylation and stabilization
(16,17). Currently available MEK1/2 inhibitors
have shown moderate single-agent activity in various tumors
(18). The inability of MEK
inhibitors to suppress the phosphorylation of MEK1/2 and ERK1/2
completely indicates that ERK activity may be regulated by multiple
signaling pathways and that MEK1/2 inhibitors could be combined
with standard chemotherapy drugs. A recent study showed that
AZD6244 and docetaxel have a synergistic effect in advanced
KRAS-mutated NSCLC. In the present study, it was found that the
treatment of A549 cells with gefitinib alone reduced the expression
level of the activated form of Akt, and that the combination of the
two drugs showed stronger inhibition of Erk and p-Erk.
The present study provided evidence that a
combination treatment with gefitinib and AZD6244 is effective for
overcoming acquired EGFR-TKI resistance in lung cancer cells
through the inhibition of ERK activity. The study suggested that
EGFR-TKI combined with MEK1/2 inhibitors, such as AZD6244, may be
beneficial to those patients with NSCLC who develop a primary or
acquired resistance to EGFR-TKIs.
Acknowledgements
This study was supported by a grant from the Wenzhou
Science and Technology Bureau (no. Y20130041).
References
|
1
|
Mok TS, Wu YL, Thongprasert S, Yang CH,
Chu DT, Saijo N, Sunpaweravong P, Han B, Margono B, Ichinose Y, et
al: Gefitinib or carboplatin-paclitaxel in pulmonary
adenocarcinoma. N Engl J Med. 361:947–957. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Scagliotti GV, Parikh P, von Pawel J,
Biesma B, Vansteenkiste J, Manegold C, Serwatowski P, Gatzemeier U,
Digumarti R, Zukin M, et al: Phase III study comparing cisplatin
plus gemcitabine with cisplatin plus pemetrexed in
chemotherapy-naive patients with advanced-stage non-small-cell lung
cancer. J Clin Oncol. 26:3543–3551. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Sandler A, Gray R, Perry MC, Brahmer J,
Schiller JH, Dowlati A, Lilenbaum R and Johnson DH:
Paclitaxel-carboplatin alone or with bevacizumab for non-small-cell
lung cancer. N Engl J Med. 355:2542–2550. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Zhou C, Wu YL, Chen G, Feng J, Liu XQ,
Wang C, Zhang S, Wang J, Zhou S, Ren S, et al: Erlotinib versus
chemotherapy as first-line treatment for patients with advanced
EGFR mutation-positive non-small-cell lung cancer (OPTIMAL,
CTONG-0802): A multicentre, open-label, randomised, phase 3 study.
Lancet Oncol. 12:735–742. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Ono M and Kuwano M: Molecular mechanisms
of epidermal growth factor receptor (EGFR) activation and response
to gefitinib and other EGFR-targeting drugs. Clin Cancer Res.
12:7242–7251. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Costa DB, Kobayashi S, Tenen DG and
Huberman MS: Pooled analysis of the prospective trials of gefitinib
monotherapy for EGFR-mutant non-small cell lung cancers. Lung
Cancer. 58:95–103. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Pao W, Miller VA, Politi KA, Riely GJ,
Somwar R, Zakowski MF, Kris MG and Varmus H: Acquired resistance of
lung adenocarcinomas to gefitinib or erlotinib is associated with a
second mutation in the EGFR kinase domain. PLoS Med. 2:e732005.
View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Sos ML, Rode HB, Heynck S, Peifer M,
Fischer F, Klüter S, Pawar VG, Reuter C, Heuckmann JM, Weiss J, et
al: Chemogenomic profiling provides insights into the limited
activity of irreversible EGFR Inhibitors in tumor cells expressing
the T790M EGFR resistance mutation. Cancer Res. 70:868–874. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Engelman JA, Zejnullahu K, Mitsudomi T,
Song Y, Hyland C, Park JO, Lindeman N, Gale CM, Zhao X, Christensen
J, et al: MET amplification leads to gefitinib resistance in lung
cancer by activating ERBB3 signaling. Science. 316:1039–1043. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Zhang H, Xu Y, Filipovic A, Lit LC, Koo
CY, Stebbing J and Giamas G: SILAC-based phosphoproteomics reveals
an inhibitory role of KSR1 in p53 transcriptional activity via
modulation of DBC1. Br J Cancer. 109:2675–2684. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Li J, Pan YY and Zhang Y: Synergistic
interaction between sorafenib and gemcitabine in EGFR-TKI-sensitive
and EGFR-TKI-resistant human lung cancer cell lines. Oncol Lett.
5:440–446. 2013.PubMed/NCBI
|
|
12
|
Kim KB, Kefford R, Pavlick AC, Infante JR,
Ribas A, Sosman JA, Fecher LA, Millward M, McArthur GA, Hwu P, et
al: Phase II study of the MEK1/MEK2 inhibitor Trametinib in
patients with metastatic BRAF-mutant cutaneous melanoma previously
treated with or without a BRAF inhibitor. J Clin Oncol. 31:482–489.
2013. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Metro G, Chiari R, Baldi A, De Angelis V,
Minotti V and Crinò L: Selumetinib: A promising pharmacologic
approach for KRAS-mutant advanced non-small-cell lung cancer.
Future Oncol. 9:167–177. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Meloche S and Pouysségur J: The ERK1/2
mitogen-activated protein kinase pathway as a master regulator of
the G1- to S-phase transition. Oncogene. 26:3227–3239. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Balmanno K and Cook SJ: Tumour cell
survival signalling by the ERK1/2 pathway. Cell Death Differ.
16:368–377. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Bodoky G, Timcheva C, Spigel DR, La Stella
PJ, Ciuleanu TE, Pover G and Tebbutt NC: A phase II open-label
randomized study to assess the efficacy and safety of selumetinib
(AZD6244 [ARRY-142886]) versus capecitabine in patients with
advanced or metastatic pancreatic cancer who have failed first-line
gemcitabine therapy. Invest New Drugs. 30:1216–1223. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Hainsworth JD, Cebotaru CL, Kanarev V,
Ciuleanu TE, Damyanov D, Stella P, Ganchev H, Pover G, Morris C and
Tzekova V: A phase II, open-label, randomized study to assess the
efficacy and safety of AZD6244 (ARRY-142886) versus pemetrexed in
patients with non-small cell lung cancer who have failed one or two
prior chemotherapeutic regimens. J Thorac Oncol. 5:1630–1636. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Kim EY, Kim A, Kim SK and Chang YS:
AZD6244 inhibits cisplatin-induced ERK1/2 activation and
potentiates cisplatin-associated cytotoxicity in K-ras G12D
preclinical models. Cancer Lett. 358:85–91. 2015. View Article : Google Scholar : PubMed/NCBI
|