Introduction
Malignant melanoma is the most aggressive type of
skin cancer, and its incidence and mortality have increased
steadily over the last 50 years to 3% of all tumors (1). Prognosis is poor once melanoma has
metastasized, with a median survival time of 4–6 months (1,2). The B-Raf
inhibitor dabrafenib, in combination with MEK inhibitor trametinib
were approved in 2013 by the Food and Drug Administration as a
treatment strategy for unresectable or metastatic BRAF-mutated
melanoma. Compared with BRAF-inhibitor monotherapy, combined
therapy offers an improved response rate for the treatment of
advanced melanoma. However, similar to monotherapy, associated
toxicity and tumor resistance and progression are still observed in
the majority of patients (3).
Previous studies have revealed that activating BRAF
kinase mutations drive oncogenesis in a wide variety of
malignancies, most notably so in melanoma (~70% of cases). These
mutations reduce the activation state of the Raf-MEK-ERK
mitogen-activated protein kinase signalling pathway, which is
involved in the growth of Raf-mutated melanoma (4), and B-Raf has been exploited as a novel
therapeutic target for melanoma in recent years. The present
authors have previously demonstrated that the MEK/ERK inhibitor
U0126 is capable of inhibiting the proliferation of the A375 human
malignant melanoma cell line in vitro. However, the cells
develop resistance to U0126, preventing a durable response
(5), a phenomenon shared with other
MEK/ERK inhibitors (6). Various
studies have demonstrated that inhibiting the nuclear factor
(NF)-κB signaling pathway increases the cytotoxicity of anticancer
agents, thus, reducing multiple drug resistance in the tumor
(7,8).
However, there are no reports on the synergistic effect of MEK/ERK
and NF-κB inhibitors on melanoma cell proliferation in the current
literature.
The aim of the current study is to evaluate the
effects of the NF-κB inhibitor BMS-35541 and the MEK/ERK inhibitor
U0126, alone or in combination, on the proliferation and apoptosis
of human melanoma cells in vitro. This may increase the
field of applications for MEK-targeted therapy.
Materials and methods
Cell culture
The A375 human malignant melanoma cell line was
purchased from the Cell Bank of the Chinese Academy of Sciences
(Shanghai, China). The cells were cultured in Dulbecco's modified
Eagle's medium (GE Healthcare Life Sciences, Logan, UT, USA) and
supplemented with 10% fetal bovine serum (FBS; GE Healthcare Life
Sciences, Little Chalfont, UK), 100 U penicillin and 100 µg
streptomycin at 37°C in a humid atmosphere of 5% CO2.
The study protocol was approved by Ethics Committee of the
Affiliated Hospital of Qingdao University (Qingdao, China).
Proliferation inhibition assay
A375 cells were seeded into 96-well plates at a
density of 1,000 cells/well. Following incubation in 10% FBS medium
for 24 h, the cells were then incubated with either 150 µl/well
dimethyl sulphoxide (DMSO; vehicle control), the ERK inhibitor
U0126 (1,5 or 10 µmol/l), the NF-κB inhibitor BMS-345541 (1,5 or 10
µmol/l), or U0126 and BMS-345541 in combination (5 µmol/l). At
timepoints of 12, 24, 48 and 72 h following treatment, cell
viability was assessed using the
3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyl-tetrazolium bromide (MTT)
assay (9). Optical density values
were determined and the rate of inhibition of cellular
proliferation was calculated using the following equation: (1 -
mean ODexperimental group) / (mean ODcontrol
group) × 100% (9). The combined
effect of U0126 and BMS-345541 was determined as previously
described (10). DMSO, U0126,
BMS-345541 and MTT were all purchased from Sigma-Aldrich (St.
Louis, MO, USA). The proliferation inhibition assay was performed
three times.
Flow cytometry
A375 cells were seeded into six-well plates and
treated with inhibitors, as described in the proliferation assay.
At 12, 24, 48 and 72 h after treatment, cells were harvested and
stained with fluorescein isothiocyanate-labeled annexin V and
propidium iodide (PI; Sigma-Aldrich). Cell cycle progression and
apoptosis was analyzed using a FACSCalibur™ flow cytometer and
CellQuest™ software (BD Biosciences, San Jose, CA, USA).
Western blotting
A375 cells were plated onto dishes and treated with
various inhibitors, as described in the proliferation assay. At the
end of the designated culture period, cells were washed twice in
ice-cold phosphate-buffered saline (PBS) and then lyzed in ice-cold
radioimmunoprecipitation assay lysis buffer. Cell homogenates were
obtained following the removal of non-soluble debris by
centrifugation at a high speed for 20 min at 4°C. Homogenized
proteins (20 µg) were loaded into a 12% polyacrylamide gel,
separated by SDS-PAGE and transferred onto a nitrocellulose
membrane using a Transblot® apparatus at 100 V for 90 min. The
membrane was sequentially incubated with polyclonal mouse
anti-human Beclin-2 (cat no. ZM-0010; 1:400 dilution) and
monoclonal mouse anti-human GAPDH (cat no. ab8245; 1:1,000
dilution) primary antibodies, followed by horseradish
peroxidase-conjugated goat anti-mouse IgG (cat no. ZDR-5307;
1:10,000 dilution) secondary antibody. All antibodies used were
diluted in Tris-buffered saline with Tween 20 and purchased from
OriGene Technologies, Inc. (Beijing, China). The membranes were
washed with PBS three times for 10 min during and after
immunolabelling. Protein expression was quantified using ImageJ
software (National Institutes of Health, Bethesda, MD, USA).
Statistical analysis
Data were analyzed by one-way analysis of variance
and Student's t-test, using SPSS version 10.0 statistical software
(SPSS Inc., Chicago, IL, USA). P<0.05 was considered to
represent a statistically significant difference.
Results
Cell viability
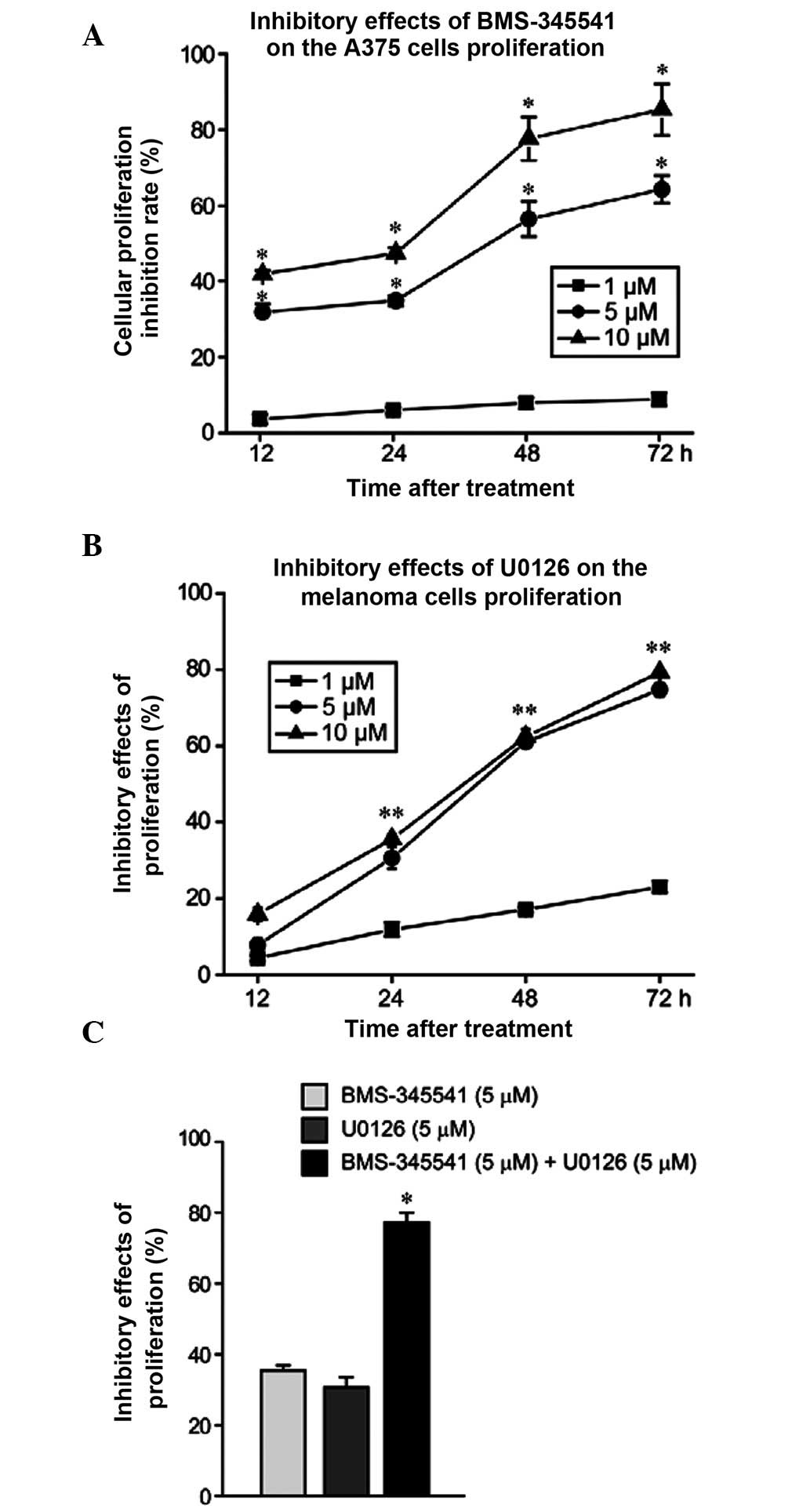
At all timepoints, BMS-345541 demonstrated a
significantly higher ability to suppress A375 cell proliferation
when used at 5 and 10 µmol/l compared with 1 µmol/l (P<0.05,
Fig. 1A). By contrast, the inhibitory
effect of U0126 was not significantly different between 5 and 10
µmol/l and 1 µmol/l at 12 h (P>0.05), but became significantly
different at 24, 48 and 72 h (P<0.05; Fig. 1B). The half maximal inhibitory
concentration at 24 h was ~5 µmol/l for both drugs. When BMS-345541
and U0126 were used in combination, the inhibition rate of cell
proliferation was significantly higher than when they were used
alone (P<0.05; Fig. 1C).
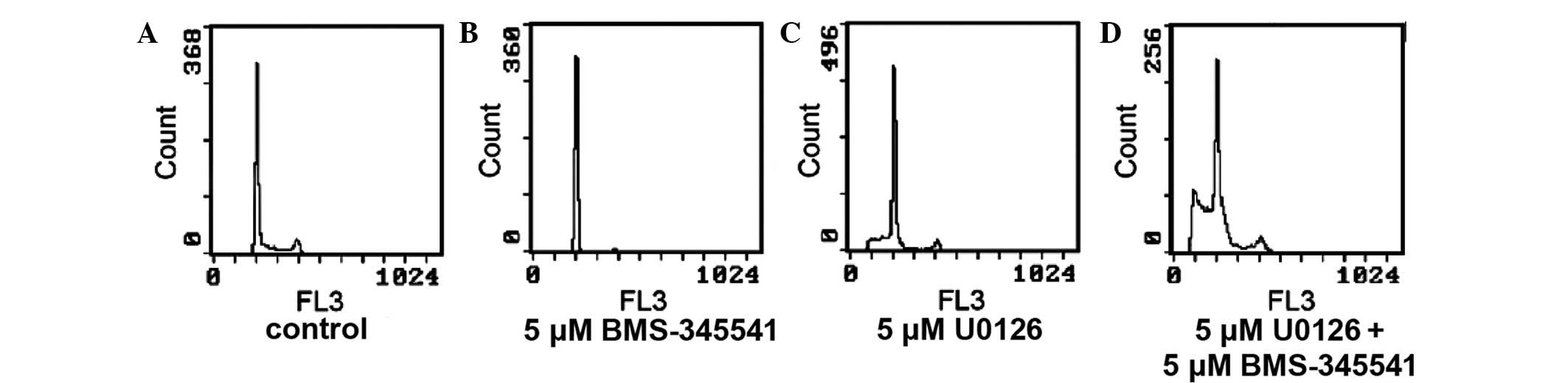
Cell cycle progression
Compared with the untreated controls (Fig. 2A), treatment with BMS-345541 for 24 h
(Fig. 2B) significantly increased the
proportion of A375 cells in G1 (62.97% vs. 69.13; P<0.01) and G2
(15.6 vs. 7.57%; P<0.01), but there was a significant reduction
in the proportion of cells in S phase (29.47% vs. 15.27;
P<0.01). U0126 significantly increased the percentage of cells
in G1 phase (84.80 vs. 62.97%; P<0.01) and decreased the
percentage of cells in S phase (11.07 vs. 29.47%; P<0.01) and G2
phase (4.13 vs. 7.57%; P>0.01). In the combination group, the
proportion of cells in G1 phase fell between those of the
BMS-345541 group and the U0126 group (73.43%; P<0.01); the
percentage of G2 phase cells was marginally higher than in the
BMS-345541 group (16.3 vs. 15.6%); and the percentage of S phase
cells reduced to 6.83% (P<0.01).
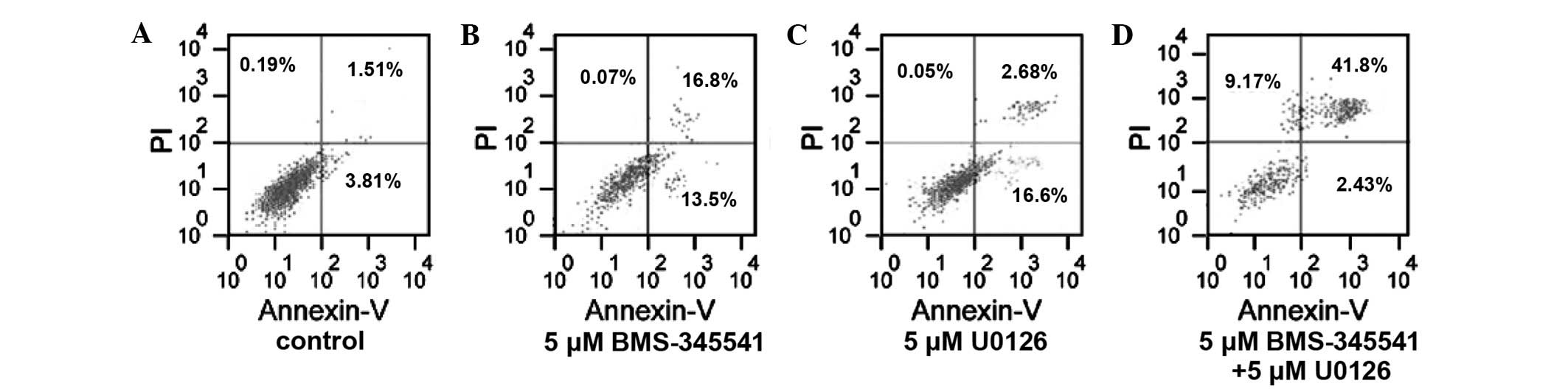
Cell apoptosis
Apoptosis was induced more frequently in the A375
cells treated by BMS-345541 in combination with U0126 than when
used with either inhibitor alone. The apoptosis rate in the
BMS-345541 (5 µmol/l) and U0126 (5 µmol/l) increased by 24.98%
(t=9.74, P<0.01) and 13.96% (t=15.82, P<0.01), respectively,
compared with the control group. Furthermore, the apoptosis rate of
the combination group (5 µmol/l BMS-345541 plus 5 µmol/l U0126)
increased by 38.91%, which was significantly more than that of the
control group (t=8.15, P<0.01; Fig.
3).
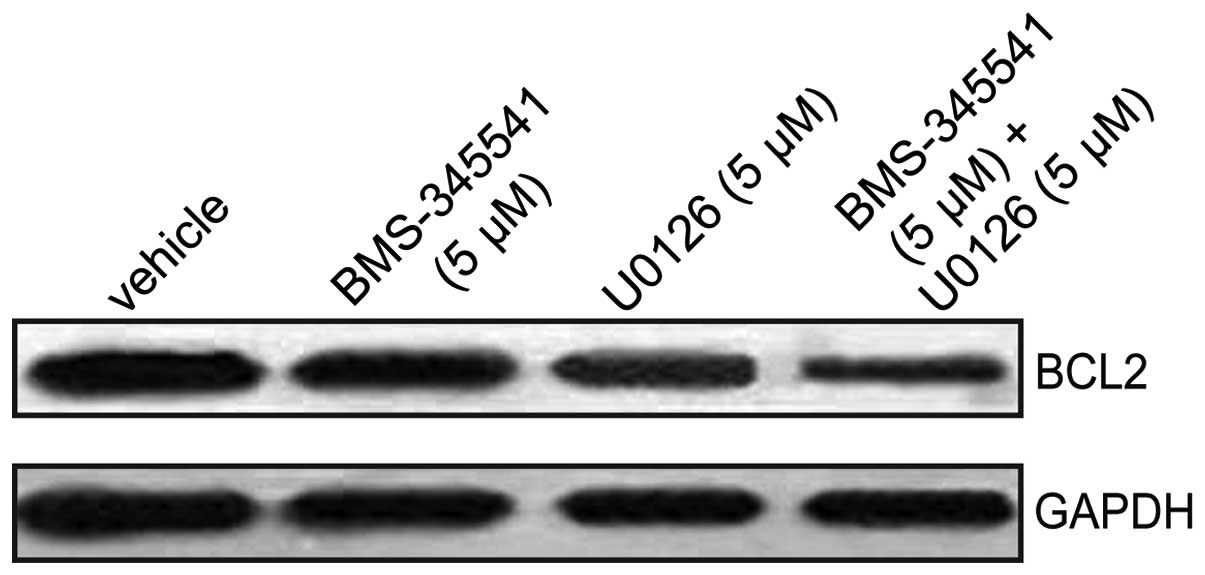
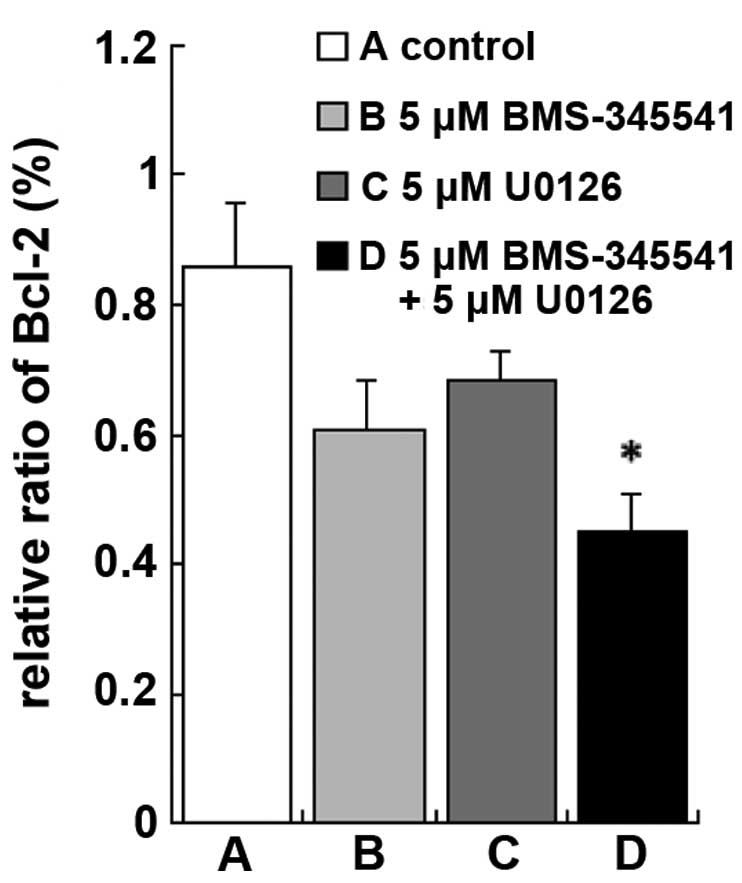
Changes in Bcl-2 protein expression
levels
To further explore the mechanism by which BMS-345541
and U0126 induce A375 cell apoptosis, western blotting was used to
measure the protein expression levels of Bcl-2, a known regulator
of apoptosis. Treatment with BMS-345541, in combination with U0126
resulted in a significant reduction in Bcl-2 protein expression
compared with the control, BMS-345541 (5 µmol/l) and U0126 (5
µmol/l) (Figs. 4 and 5; P<0.01). These results indicate that
BMS-345541 in combination with U0126 strongly inhibited
proliferation of A375 cells by activating the intrinsic apoptosis
signaling pathway.
Discussion
The Raf/MEK/ERK protein kinase cascade is an
important intracellular signaling pathway that influences a number
of fundamental cellular processes. Aberrant activation of the
pathway is a major cause of cancer cell growth (11). Ras, a member of this protein network
is mutated and active in ~30% of all cases of cancer and B-Raf is
the most commonly mutated kinase in human cancer (~70% of
melanomas) (11,12). MEK inhibition is consequently an
important and logical target, however, proof of concept has yet to
be identified in clinical trials. In accordance with previous
studies, the results of the current study demonstrated that
treatment with the MEK inhibitor U0126 resulted in greater
induction of A375 melanoma cell apoptosis (13,14).
However, the invariable development of resistance to these agents
(including U0126 and other MEK inhibitors) represents a significant
clinical obstacle to their long-term efficacy (6,13,14).
Activation of NF-κB is considered to confer
resistance to cytotoxic therapies and allow an escape from
apoptosis. The inhibitor of κB kinase complex (IKK) is the
essential upstream protein kinase in the classical NF-κB activating
pathway (15–17). In the present study, BMS-345541, a
highly selective inhibitor of IKK, was used to explore the role of
NF-κB in the network of apoptosis (18,19). In
the present study, BMS-345541 exhibited a concentration-dependent
inhibition of melanoma cell survival in vitro. However,
melanoma cells exhibited no greater sensitivity to BMS-345541 than
to U0126. When BMS-345541 was used as a co-treatment with U0126,
strong synergistic activity was generated, which indicates that
combining NF-κB and MEK inhibition may be a promising approach for
treating melanoma with acquired drug resistance.
The molecular mechanism of the U0126-induced
antitumor effect and its synergistic effects with BMS-345541 was
subsequently explored. The present study demonstrated that U0126
plus BMS-345541 combination treatment enhanced apoptosis, induced
cell cycle arrest, and inhibited the expression of Bcl-2. A
previous study demonstrated that BMS-345541 results in accumulation
of BE-13 and DND-41 cells in the G2/M phase, and that U0126 results
in G1/S phase cell cycle arrest in K562 leukemia cells (18). Consistent with these findings, the
present study demonstrated that in A375 melanoma cells, BMS-345541
predominantly blocked cells in G2 phase, U0126 mainly blocked cells
in G1 phase, and U0126 plus BMS-345541 blocked cells in G1 and G2
phase, and significantly inhibited tumor cell proliferation and
consequently induced apoptosis.
The balance of pro-apoptotic (Bax) and
anti-apoptotic (Bcl-2) proteins modulates intrinsic cell death
following apoptotic insult (20,21).
Therefore, Bcl-2 expression is the key step to protect cells from
apoptosis in melanoma. It has a crucial role in chemoresistance in
various human cancers (22,23). The present study detected expression
of Bcl-2 at 24 h after treatment with the inhibitors. In accordance
with previous studies, BMS-345541 and U0126 downregulated
expression of Bcl-2, leading to reversal of chemoresistance and
enhancement of apoptosis (24,25).
The major question addressed by the current study is
whether combination of IKK and MEK inhibitors improves the efficacy
of chemotherapy and enhances inhibition of cell proliferation. The
present study demonstrates for the first time that U0126 in
combination with BMS-345541 inhibits the proliferation of human
melanoma cells, and that the combined effect involves G1 and G2
phase arrest, as well as downregulation of Bcl-2 expression. The
curative effect of the majority of single agents that target
melanoma is insufficient. Thus, based on the results of the current
study, we propose that therapy with NF-κB and ERK pathway
inhibitors may become a novel, improved treatment strategy for
patients with melanoma.
Previous studies have revealed that activation of
the ERK pathway may promote cell cycle progression from G1 to S
phase, eventually modulating the expression of downstream nuclear
transcription factors including NF-κB, activator protein-1 and
signal transducer and activator of transcription 3 (26). Furthermore, blocking the
phosphorylation of ERK proteins may lead to NF-κB inactivation
(27).
In conclusion, the results of the present study
demonstrated that although melanoma cells were no more sensitive to
BMS-345541 alone than to U0126 alone, strong synergistic activity
was generated by their combination. This may indicate that MEK
inhibitor U0126 induces A375 melanoma cell apoptosis through an
NF-κB-independent mechanism. However, the exact underlying
molecular mechanisms remain to be elucidated.
Acknowledgements
The present study was supported by the Natural
Science Foundation of Shandong Province (grant no.
ZR2010HM022).
References
|
1
|
Bai J, Xie X, Lei Y, et al: Ocular
albinism type 1 induced melanoma cell migration is mediated through
the RAS/RAF/MEK/ERK signaling pathway. Molecular Medicine Reports.
10:491–495. 2014.PubMed/NCBI
|
|
2
|
Aris M and Barrio MM: Combining
Immunotherapy with Oncogene-Targeted Therapy: A New Road for
Melanoma Treatment. Front Immunol. 6:462015. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Queirolo P, Picasso V and Spagnolo F:
Combined BRAF and MEK inhibition for the treatment of BRAF-mutated
metastatic melanoma. Cancer Treatment Reviews. 4:1–8. 2015.
|
|
4
|
Tuveson DA, Weber BL and Herlyn M: BRAF as
a potential therapeutic target in melanoma and other malignancies.
Cancer Cell. 4:95–98. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Ning L, Xiao J and Min GP: The effects of
the MEK inhibitor on the proliferation of human melanoma cells.
Chin J Dermatovenereology. 26:961–965. 2012.(In Chinese).
|
|
6
|
Acquaviva J, Smith DL, Jimenez JP, et al:
Overcoming acquired BRAF inhibitor resistance in melanoma via
targeted inhibition of Hsp90 with ganetespib. Mol Cancer Ther.
13:353–363. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Deng LL, Shao YX, Lv HF, Deng HB and Lv
FZ: Over-expressing CYLD augments antitumor activity of TRAIL by
inhibiting the NF-κB survival signaling in lung cancer cells.
Neoplasma. 59:18–29. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Song B, Bian Q, Shao CH, Li G, Liu AA,
Jing W, Liu R, Zhang YJ, Zhou YQ, Hu XG and Jin G: Ulinastatin
reduces the resistance of liver cancer cells to epirubicin by
inhibiting autophagy. PLoS One. 10:1–15. 2015. View Article : Google Scholar
|
|
9
|
Zhao B, Fang GJ, Zhu J, et al: The
computing method of IC50 in determining cell
proliferation inhibition rate by MTT method. Anhui Med Pharm J.
11:834–836. 2007.(In Chinese).
|
|
10
|
Yeh YA, Herenyiova M and Weber G:
Quercetin: Synergisticaction with carboxamidotriazole in human
breast carcinoma cells. Life Sci. 57:1285–1292. 1995. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Duffy A and Kummar S: Targeting
mitogen-activated protein kinase kinase (MEK) in solid tumors.
Target Oncol. 4:267–273. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Pratilas CA, Hanrahan AJ, Halilovic E, et
al: Genetic predictors of MEK dependence in non-small cell lung
cancer. Cancer Res. 68:9375–9383. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Sadaria MR, Yu JA, Meng X, et al:
Secretory phospholipase A2 mediates human esophageal adenocarcinoma
cell growth and proliferation via ERK 1/2 pathway. Anticancer Res.
33:1337–1342. 2013.PubMed/NCBI
|
|
14
|
Walters DM, Lindberg JM, Adair SJ, et al:
Inhibition of the growth of patient-derived pancreatic cancer
xenografts with the MEK inhibitor trametinib is augmented by
combined treatment with the epidermal growth factor receptor/HER2
inhibitor lapatinib. Neoplasia. 15:143–155. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Madonna G, Ullman CD, Gentilcore G,
Palmieri G and Ascierto PA: NF-κB as potential target in the
treatment of melanoma. J Transl Med. 10:532012. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Wang Y, Zhou Y, Jia G, Han B, Liu J, Teng
Y, et al: Shikonin suppresses tumor growth and synergizes with
gemcitabine in a pancreatic cancer xenograft model: Involvement of
NF-κB signaling pathway. Biochem Pharmacol. 88:322–333. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Schmid JA and Birbach A: IkappaB kinase
beta(IKKbeta/IKK2/IKBKB) - a key molecule in signaling to the
transcription factor NF-kappaB. Cytokine Growth Factor Rev.
19:157–165. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Buontempo F, Chiarini F, Bressanin D, et
al: Activity of the selective IκB kinase inhibitor BMS-345541
against T-cell acute lymphoblastic leukemia: Involvement of FOXO3a.
Cell Cycle. 11:2467–2475. 2012. View
Article : Google Scholar : PubMed/NCBI
|
|
19
|
Yang J, Amiri KI, Burke JR, Schmid JA and
Richmond A: BMS-345541 targets inhibitor of kappaB kinase and
induces apoptosis in melanoma: Involvement of nuclear factor kappaB
and mitochondria pathways. Clin Cancer Res. 12:950–960. 2006.
View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Nesic-Taylor O, Cittelly D, Ye Z, et al:
Exogenous Bcl-xL fusion protein spares neurons after spinal cord
injury. J Neurosci Res. 79:628–637. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Han X, Lu M, Wang S, Lv D and Liu H:
Targeting IKK/NF-κB pathway reduces infiltration of inflammatory
cells and apoptosis after spinal cord injury in rats. Neurosci
Lett. 511:28–32. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Zhang H, Cai X, Wang Y, et al:
microRNA-143, down-regulated in osteosarcoma, promotes apoptosis
and suppresses tumorigenicity by targeting Bcl-2. Oncol Rep.
24:1363–1369. 2010.PubMed/NCBI
|
|
23
|
Wu DW, Wu TC, Wu JY, Cheng YW, Chen YC,
Lee MC, Chen CY and Lee H: Phosphorylation of paxillin confers
cisplatin resistance in non-small cell lung cancer via activating
ERK-mediated Bcl-2 expression. Oncogene. 33:4385–4395. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Berger A, Quast SA, Plötz M, Kammermeier A
and Eberle J: Sensitization of melanoma cells for TRAIL-induced
apoptosis by BMS-345541 correlates with altered phosphorylation and
activation of Bax. Cell Death Dis. 4:e4772013. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Xie J, Jin B, Li DW, Shen B, Cong N, Zhang
TZ and Dong P: ABCG2 regulated by MAPK pathways is associated with
cancer progression in laryngeal squamous cell carcinoma. Am J
Cancer Res. 4:698–709. 2014.PubMed/NCBI
|
|
26
|
Sullivan RJ and Atkins MB:
Molecular-targeted therapy in malignant melanoma. Expert Rev
Anticancer Ther. 9:567–581. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Zhang H, Zhang S, He H, Zhao W, Ren K,
Chen J and Shao RG: RasGAP-derived peptide 38GAP potentiates the
cytotoxicity of cisplatin through inhibitions of Akt, ERK and NF-κB
in colon carcinoma HCT116 cells. Cancer Lett. 308:62–70. 2011.
View Article : Google Scholar : PubMed/NCBI
|