Introduction
Epithelial ovarian cancer (EOC) is the seven most
frequent type of cancer in women and the eighth cause of mortality
from cancer in women worldwide (1).
In contrast to the continuous development in molecular
characterization of a number of neoplasms, the progress made in
understanding the molecular background of ovarian cancer is
limited. This could be due to the complexity of the disease, but
also due to certain limitations of study designs and experimental
data collection (2).
The sodium-hydrogen antiporter 3 regulator 1
(SLC9A3R1) gene is located on chromosome 17q25.1, consists of six
exons and encodes Na+/H+ exchanger regulatory
factor 1 (NHERF1). The isolated protein has a molecular weight of
50–53 kD, contains 357 amino acids and is structured in three
protein domains (3,4). NHERF1 has two PDZ domains (PDZ1 and
PDZ2) located in tandem (PSD-95/Dlg/ZO1), mediating protein-protein
interaction (5), and a C-terminal
ezrin-binding (EB) domain that binds to the ezrin-radixin-moesin
(ERM) family of proteins (6).
NHERF1 is expressed primarily at the plasma membrane
of polarized epithelia, including that of the kidney, intestine,
colon, lungs and uterus. The main function of this adaptor protein
is stabilization of protein complexes at the plasma membrane
connecting signaling pathways and structural proteins to the cell
cytoskeleton (7). NHERF1 binds to
β-catenin through PDZ2, and stabilizes the interaction between
β-catenin and E-cadherin in the adherent junction of epithelial
cells (8,9). In the absence of NHERF1, β-Catenin
accumulates in the cytoplasm and E-Cadherin localization at the
cell membrane is reduced, resembling the process of epithelial to
mesenchymal-like transition (EMT). EMT is observed in normal
embryonic development and is recreated during tumor progression
(10–12).
NHERF1 has been extensively studied at the protein
level, principally in its interactions at the cell membrane, but
its gene regulation remains largely unexplored. Thus far, only a
few gene mutations associated with human cancer have been
characterized. For instance, one previous study (13) in breast cancer showed that the
combination of the intragenic mutation rate of 48 breast cancer
cell lines and 37 primary breast tumors was 4%. Two missense
mutations were described. One of them, a somatic sequence variant
of AAG→AAC in the NHERF1 PDZ2 domain that produces a switch in
codon 172 (Lys to Asn), was found in primary breast cancer. The
other, a missense mutation in codon 180 of exon 2 (CGG→TGG) with a
replacement of Arg to Trp, which corresponds to a conserved basic
residue in the PDZ2 domain, was found in the MDA-MB-231 breast
cancer cell line. Two of the mutations that occur in the PDZ2
domain (codons 172 and 182) decreased the interaction of NHERF1
with SYK (spleen tyrosine kinase), a tumor suppressor gene in the
mammary gland. Additionally, the mutation in codon 180 disrupted
the interaction with another tumor suppressor gene (Merlin), which
shows the importance of the integrity of the PDZ2 motif in NHERF1
tumor suppressor activity in breast cancer (13). A recent study performed by The Cancer
Genome Atlas research network analyzed the DNA sequence from coding
genes in 316 high-grade serous ovarian adenocarcinomas. No
mutations were detected in the coding sequence of the SLCA9AR1 gene
despite changes in expression levels and copy number amplification
in 7.6% of the cases; splicing sites were not selected for the
analysis (14).
The present study reports the results of mutation
analysis in the SLC9A3R1 gene that revealed the presence of splice
mutations in 8 out of 31 screened EOC samples (25.8%). To the best
of our knowledge, this is the first study on SLC9A3R1 point
mutations in EOC. Further studies in a larger cohort of ovarian
cancer patients will determine the predictive and prognostic value
of this mutation.
Materials and methods
Patients and tumor samples
The analysis was performed in 31 EOC tumor
adenocarcinoma samples (25 high-grade serous, 4 undifferentiated, 1
clear cell and 1 endometrioid sample) from patients who had
undergone primary surgery in the Department of Gynecological
Oncology, Medical University of Gdansk (Gdansk, Poland) between
1995 and 1996, and between 2002 and 2004. Informed consent was
obtained from all patients, and the study was approved by the
Medical Review Board of Gdansk Medical University. The patients
treated between 1995 and 1996 received 6 cycles of postoperative
chemotherapy combination of cisplatin (75 mg/m2) and
cyclophosphamide (750 mg/m2) every 3 weeks. The patients
treated between 2002 and 2004 received 6 cycles of postoperative
chemotherapy combination of cisplatin (75 mg/m2) and
paclitaxal 175 mg/m2 (over 3 h) every 3 weeks. Only 3 cases did not
receive any adjuvant treatment due to a poor performance status (PS
3/4). The disease was classified according to the histological
grade (G1-G3) and the International Federation of Gynecology and
Obstetrics stage (I–IV) (15).
Residual disease was defined by the diameter of the largest tumor
left in the abdominal cavity after cytoreductive surgery for
advanced stages. Patients with and without a family history of the
disease were included in the study. The samples of fresh tumor were
immediately frozen at −80°C for molecular analysis; a portion of
each tumor was fixed in formalin and embedded in paraffin. Tissue
sections (5 µm) were obtained from the blocks and stained with
hematoxylin and eosin for histopathological analysis.
Molecular screening
The DNA from the ovarian tumors was extracted from
fresh tumor tissues by standard phenol-chloroform procedures. The
sequences of exons 2 and 3, and the flanking sequences of the
SLC9A3R1 gene were amplified with specific primers (13). The sequences were as follows: Exon 2
forward, 5′-AAT TGC TGT GTA GGG ATC TAG-3′ and reverse, 5′-GGA AGA
GAG CGA GAA GCA TC-3′ (322-bp product); and exon 3 forward, 5′ACT
GCA AAC TGG CTG AGA AC-3′ and reverse, 5′-TGG CTC ACA TCC CTG ACT
TG-3′ (331-bp product). The PCR reaction was carried with the
following conditions: 30 ng of DNA/sample in presence of 1.5 mM
MgCl2, (95°C for 5 sec, followed by 95°C for 30 sec,
gradient 56–63°C for 30 sec and 72°C for 30 sec repeated 34 cycles,
and a final amplification step at 72°C for 7 min) using the Taq DNA
recombinant polymerase (Fermentas, Thermo Scientific, Waltham, MA,
USA) according to the manufacturer's instructions. Samples
harboring the mutation were re-amplified using high fidelity
polymerase (Thermo Scientific) to ensure the result was accurate.
Following PCR amplification, the PCR products were cleaned using
the Axyprep-96 PCR Cleanup kit following the manufacturer's
instruction (Axigen, Corning, Tewksbury, MA, USA). The PCR products
were prepare for sequencing using the Big Day reaction. Briefly,
the PCR products were amplified with the forward or reverse primers
separately according to the manufacturer's instruction. Following
the PCR amplification, the product was cleaned using the
ExTerminator Nucleotide Terminators Removal kit (A&A
Biotechnology, Gdynia, Poland), according to the manufacturer's
instructions, and sequenced directly by bi-directional sequencing
(ABI Prism 3130; Applied Biosystems, Life Technologies, Foster
City, CA, USA). Electropherograms were analyzed by the free BioEdit
Sequence Alignment Editor program (16).
Bioinformatics analyses
To predict the splicing signals in wild-type and
mutated DNA sequences, the mutations were analyzed using the
bioinformatics Human Splicing Finder (HSF) free program (17). The software allows the comparison of a
wild-type and mutant sequence in order to predict the impact of in
the splicing process.
Results
Mutational analysis
Cases characteristics of the 31 patients with EOC
included in the study are presented in Table I. The sequences of exons 2 and 3,
together with the flanking intronic sequences of the SLC9A3R1 gene
that codes for the PDZ2 domain of the NHERF1 protein, were
analyzed. In total, 8 out of the 31 analyzed samples (25.8%) were
found to carry a potentially harmful alteration located in the
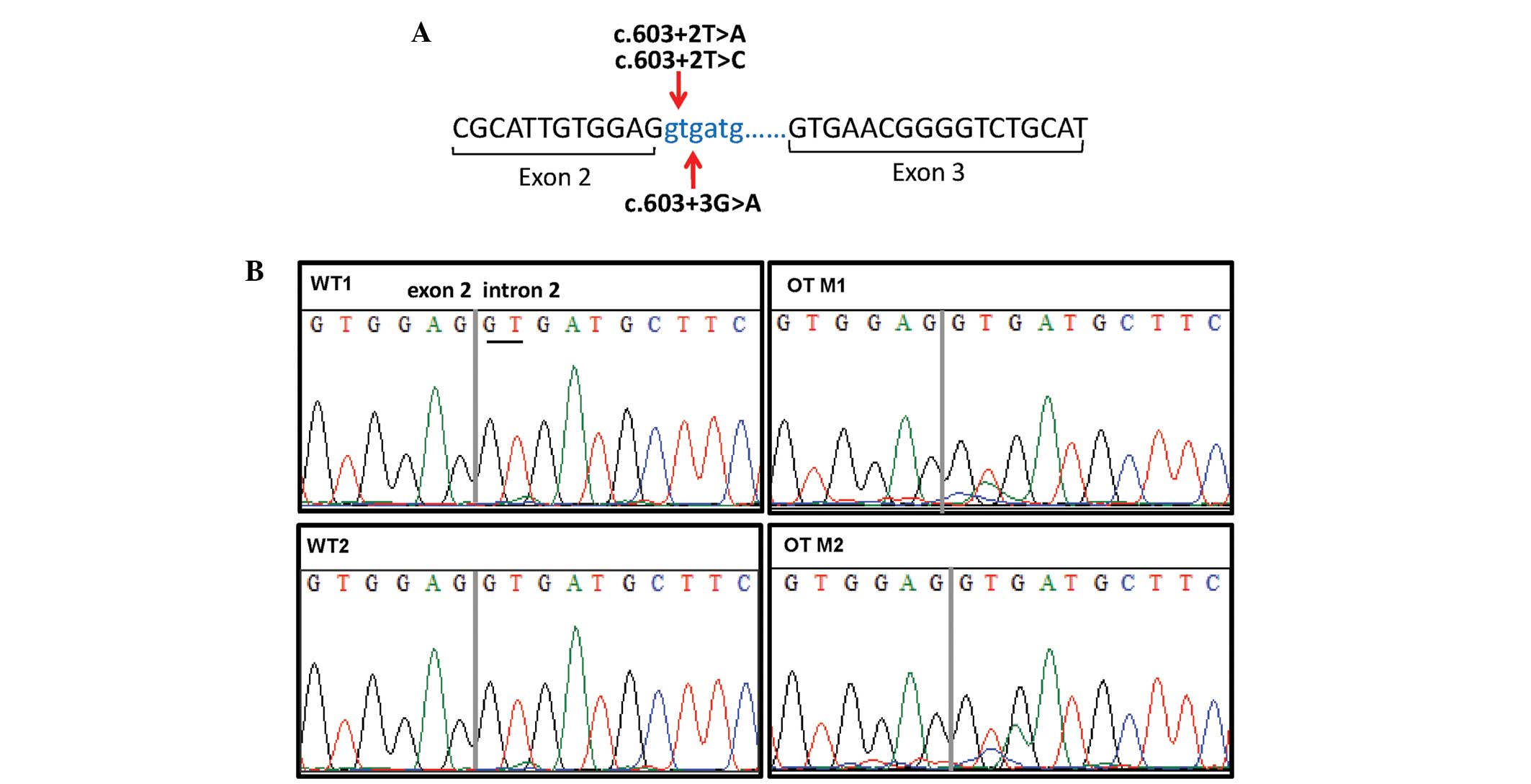
splicing donor site of intron 2 (Fig.
1; Table II). While 3 samples
displayed two different substitutions in the +2 position
(c.603+2T>A; c.603+2T>C), 5 other samples exhibited
co-occurrence of two substitutions (c.603+2T>C; c.603+3G>A)
located in the same splicing donor site (in the +2 and +3
positions) (Table I). Moreover,
reported alterations were only identified in the tumor tissue of
the tested cohort of EOC, no alterations were found in the blood of
the patients indicating that they were somatic mutations. All
identified alterations were located in the consensus sequence of
the splice donor site of intron 2, suggesting a detrimental effect
on the splicing process.
 | Table I.Characteristic of the 31 patients with
epithelial ovarian carcinoma. |
Table I.
Characteristic of the 31 patients with
epithelial ovarian carcinoma.
| Case no. | Tumor | FHOC | Age, years | CA-125,
U/mla | EOC
Histologyb | Grade | FIGO stage | Residual disease,
cmc | OS time, months |
|---|
| 1 | 2 | Negative | 63 | >600 | Serous | G3 | IIIC | >5 | 12.3 |
| 2 | 21 | Negative | 58 | >600 | Serous | G1 | IIIC | >5 | 10.1 |
| 3 | 23 | Negative | 42 | 1161 | Undifferentiated | G3 | IIIC | <1 | 106.7 |
| 4 | 32 | Negative | 74 | 4207 | Serous | G3 | IIIC | 1–5 | 6.9 |
| 5 | 37 | Positive | 55 | 399 | Serous | G3 | IIIC | 1–5 | 33.2 |
| 6 | 40 | Negative | 48 | No data | Serous | G1 | IIIC | <1 | 7.2 |
| 7 | 49 | Negative | 41 | No data | Serous | G2 | IIIC | <1 | 63.0 |
| 8 | 56 | Positive | 46 | 654 | Serous | G3 | IIIC | <1 | 104.3 |
| 9 | 58 | Negative | 77 | No data | Serous | G2 | IIIC | <1 | 0.3 |
| 10 | 63 | Negative | 60 |
14 | Serous | G1 | IC | 0 | 32.5 |
| 11 | 74 | Positive | 45 | 599 | Serous | G2 | IV | >5 | 19.6 |
| 12 | 93 | Negative | 75 | 319 | Serous | G3 | IIIC | 1–5 | 19.9 |
| 13 | 102 | Negative | 32 |
326 | Clear cell | G2 | IIC | 0 | 130.9 |
| 14 | 118 | Negative | 79 | 7368 | Serous | G2 | IIIC | >5 |
35.0 |
| 15 | 127 | Negative | 47 |
198 |
Undifferentiated | G3 | IIIB | <1 | 100.4 |
| 16 | 135 | Negative | 55 | >600 | Serous | G1 | IIIC | 1–5 |
16.9 |
| 17 | 137 | Negative | 63 | No data | Serous | G1 | IIIC | <1 |
17.8 |
| 18 | 150 | Negative | 56 |
403 | Serous | G3 | IIIC | <1 |
0.2 |
| 19 | 153 | Positive | 48 | 1089 | Serous | G3 | IIIC | <1 |
87.3 |
| 20 | 157 | Negative | 37 | >600 | Serous | G2 | IIIC | <1 |
21.5 |
| 21 | 165 | Negative | 59 |
10 | Serous | G2 | IIIC | >5 |
14.6 |
| 22 | 170 | Positive | 78 | No data | Serous | G1 | IIIC | >5 |
18.0 |
| 23 | 172 | Negative | 54 | 1001 | Serous | G2 | IIIC | 1–5 |
94.3 |
| 24 | 182 | Negative | 73 |
563 |
Undifferentiated | G3 | IV | <1 |
26.6 |
| 25 | 200 | Negative | 73 | 4851 | Serous | G3 | IV | <1 |
7.0 |
| 26 | 211 | Positive | 82 |
802 | Serous | G2 | IIIA | <1 |
41.7 |
| 27 | 218 | Negative | 88 | No data | Serous | G1 | IV | <1 | No FU |
| 28 | 219 | Negative | 54 |
311 |
Undifferentiated | G3 | IIIC | 1–5 |
84.5 |
| 29 | 257 | Negative | 62 |
300 | Serous | G2 | IIIC | >5 |
25.8 |
| 30 | 766 | Positive | 46 |
156 | Endometrioid | G3 | IIB | 0 |
43.3 |
| 31 | 792 | Negative | 36 |
136 | Serous | G1 | IIIB | 0 | 106.9 |
| Table II.Mutations in the splicing donor site
of the SCLC9A3R1 gene in ovarian cancer. |
Table II.
Mutations in the splicing donor site
of the SCLC9A3R1 gene in ovarian cancer.
| Case no. | Tumor | FHOC | Age at diagnosis,
years | CA-125,
U/mla | EOC
histologyb | Grade | FIGO stage | Residual disease,
cmc | OS time,
months | Splice donor site
mutation in SLC9A3R1 gene |
|---|
| 1 | 2 | Negative | 63 | >600 | Serous | G3 | IIIC | >5 |
12,3 | c.603+2T>A |
| 2 | 21 | Negative | 58 | >600 | Serous | G1 | IIIC | >5 |
10,1 | c.603+2T>C |
| 7 | 49 | Negative | 41 | no data | Serous | G2 | IIIC | <1 |
63,0 | c.603+2T>C,
c.603+3G>A |
| 10 | 63 | Negative | 60 | 14 | Serous | G1 | IC | NA |
32,5 | c.603+2T>C,
c.603+3G>A |
| 14 | 118 | Negative | 79 | 7368 | Serous | G2 | IIIC | >5 |
35,0 | c.603+2T>C,
c.603+3G>A |
| 15 | 127 | Negative | 47 |
198 |
Undifferentiated | G3 | IIIB | <1 | 100,4 | c.603+2T>C,
c.603+3G>A |
| 20 | 157 | Negative | 37 | >600 | Serous | G2 | IIIC | <1 |
21,5 | c.603+2T>C |
| 26 | 211 | Positive | 82 |
802 | Serous | G2 | IIIA | <1 |
41,7 | c.603+2T>C,
c.603+3G>A |
Bioinformatics analysis
The mutations were analyzed using the bioinformatics
HSF (version 2.4.1), which compares the wild-type and mutated
sequences to the consensus splice site sequences from the HSF
database. The ‘consensus splicing site’ in the database was
determined previously by the analysis of data extracted from
Ensembl containing ~22,000 genes and 46,000 transcripts of Homo
sapiens, which includes introns and exons of all human genes
(17). The impact of the mutation is
analyzed by using matrices from the study by Shapiro and Senepathy
(18) where a consensus value is
attributed to each sequence. If the difference between the
sequences is >10%, the program predicts a significant effect in
the splicing process. The analysis showed that two of the mutations
in the +2 splicing site (c.603+2T>A and c.603+2T>C) could
exhibit a significant effect in the splicing process. By contrast,
the mutation in the +3 position (c.603+3G>A) did not appreciably
modify the site (Table III).
| Table III.Bioinformatics analyses. |
Table III.
Bioinformatics analyses.
|
|
| Human splice
findera |
|
|---|
|
|
|
|
|
|---|
| No. of cases | Gene SLC9A3R1
mutations | WT CVb | Mut CV | ΔCV, % | Software
prediction |
|---|
| 7c | c.603+2T>C | 82.83 | 56 | −32.4 | ΔCV reduction
>10%, significant effect in ‘splicing’ |
| 1d | c.603+2T>A | 82.83 | 56 | −32.4 | ΔCV reduction
>10%, significant effect in ‘splicing’ |
| 5e | c.603+3G>A | 82.83 | 83.99 |
1.4 | ΔCV reduction
<10%, splicing site not affected |
Discussion
The present study analyzed the sequence of exons 2
and 3 of the SLC9A3R1 gene, which encodes the PDZ2 domain of the
NHERF1 protein. Through this domain, NHERF1 binds to β-Catenin and
stabilizes the interaction with E-cadherin at cell-cell junctions
(9). The PDZ2 domain also has a
significant role in the regulation of the conformational state of
NHERF1 by an intramolecular interaction with the C-terminal EB
region, which is able to mask other protein domains in order to
bind to other partner proteins (19).
The present study found two intronic mutations in
the donor splicing site of exon 2 of the SLC9A3R1 gene that, to the
best of our knowledge, had not been previously described and could
affect the expression of the NHERF1 isoforms. Point mutations in
splicing recognition sites are a major cause of splicing defects,
such as exon skipping of one or more adjacent exons, or inclusion
of the intronic sequence, and are frequently found in different
diseases (21–25).
It has been reported that the alteration of splicing
could have a huge impact during tumorigenesis, as several genes
express cancer-specific splicing isoforms (26–28). It
was previously shown that one of the tumor suppressor mechanisms
displayed by the PDZ2 domain of NHERF1 was the selective
stabilization of the interaction between β-catenin and E-cadherin,
which contributes to the maintenance of the structure of polarized
epithelial cells. In the absence of NHERF1 expression, the
β-catenin/E-cadherin association is disrupted and leads to
decreased β-catenin at the plasma-membrane localization, reduced
expression of E-cadherin at the cell-cell junction and cell
transformation (9,29). The potential disruption of the PDZ2
domain as a result of the mutation could modify the interaction of
NHERF1 with proteins that interact directly with the PDZ2 domain,
such as β-catenin, as well it possibly affecting the regulation of
the conformational state of the protein, and its binding to
phosphatase and tensin homolog and ERM proteins (19).
In summary, mutations of splicing recognition sites
of the SLC9A3R1 gene in malignant ovarian tumors may potentially
affect the behavior of cancer cells. The present study found
mutations in early low-grade and advanced (G1-G3) EOC tumors,
however, future studies are required in order to understand the
clinical implications of these mutations in the prognosis of
ovarian cancer patients.
Acknowledgements
The authors are especially grateful to Dr Alberto R.
Kornblihtt for his contribution to the analysis of the results and
to Dr Guillermo Juvenal for useful discussions. This study was
supported by a grant from the Medical University of Gdansk: ST-2,
Polish National Science Centre projects: 2011/02/A/NZ2/00017, the
National Research Council of Argentina and the National Agency for
the Promotion of Science and Technology of Argentina (PICT 0087,
2008).
References
|
1
|
Ferlay J, Soerjomataram I, Ervik M,
Dikshit R, Eser S, Mathers C, Rebelo M, Parkin DM, Forman D and
Bray F: GLOBOCAN 2012 v1.0, Cancer Incidence and Mortality
Worldwide: IARC CancerBase No. 11 (Internet). International Agency
for Research on Cancer (Lyon, France). 2013.http://globocan.iarc.frAccessed on. Dec
13–2013
|
|
2
|
Tinker AV, Boussioutas A and Bowtell DD:
The challenges of gene expression microarrays for the study of
human cancer. Cancer Cell. 9:333–339. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Weinman EJ, Steplock D, Wang Y and
Shenolikar S: Characterization of a protein cofactor that mediates
protein kinase A regulation of the renal brush border membrane
Na(+)/H+ exchanger. J Clin Invest. 95:2143–2149. 1995. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Reczek D, Berryman M and Bretscher A:
Identification of EBP50: A PDZ-containing phosphoprotein that
associates with members of the ezrin-radixin-moesin family. J Cell
Biol. 139:169–179. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Cho KO, Hunt CA and Kennedy MB: The rat
brain postsynaptic density fraction contains a homolog of the
Drosophila discs-large tumor suppressor protein. Neuron. 9:929–942.
1992. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Saotome I, Curto M and McClatchey AI:
Ezrin is essential for epithelial organization and villus
morphogenesis in the developing intestine. Dev Cell. 6:855–864.
2004. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Bretscher A, Chambers D, Nguyen R and
Reczek D: ERM-Merlin and EBP50 protein families in plasma membrane
organization and function. Annu Rev Cell Dev Biol. 16:113–143.
2000. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Shibata T, Chuma M, Kokubu A, Sakamoto M
and Hirohashi S: EBP50, a beta-catenin-associating protein,
enhances Wnt signaling and is over-expressed in hepatocellular
carcinoma. Hepatology. 38:178–186. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Kreimann EL, Morales FC, de Orbeta-Cruz J,
Takahashi Y, Adams H, Liu TJ, McCrea PD and Georgescu MM: Cortical
stabilization of beta-catenin contributes to NHERF1/EBP50 tumor
suppressor function. Oncogene. 26:5290–5299. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Hayashi Y, Molina JR, Hamilton SR and
Georgescu MM: NHERF1/EBP50 Is a new marker in colorectal cancer.
Neoplasia. 12:1013–1022. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Thiery JP: Epithelial-mesenchymal
transitions in tumour progression. Nat Rev Cancer. 2:442–454. 2002.
View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Hazan RB, Qiao R, Keren R, Badano I and
Suyama K: Cadherin switch in tumor progression. Ann NY Acad Sci.
1014:155–163. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Dai JL, Wang L, Sahin AA, Broemeling LD,
Schutte M and Pan Y: NHERF1 (Na+/H+ exchanger regulatory factor)
gene mutations in human breast cancer. Oncogene. 23:8681–8687.
2004. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Cancer Genome Atlas Research Network:
Integrated genomic analyses of ovarian carcinoma. Nature.
474:609–615. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Scully RE: World Health Organization
International Histological Classification of Tumours (2nd). New
York: Springer. 1999.
|
|
16
|
Hall TA: BioEdit: A user-friendly
biological sequence alignment editor and analysis program for
Windows 95/98/NT. Nucleic Acids Symp Ser. 41:95–98. 1999.
|
|
17
|
Desmet FO, Hamroun D, Lalande M,
Collod-Béroud G, Claustres M and Béroud C: Human splicing finder:
An online bioinformatics tool to predict splicing signals. Nucleic
Acids Res. 37:e672009. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Shapiro MB and Senapathy P: RNA splice
junctions of different classes of eukaryotes: Sequence statistics
and functional implications in gene expression. Nucleic Acids Res.
15:7155–7174. 1987. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Morales FC, Takahashi Y, Momin S, Adams H,
Chen X and Georgescu MM: NHERF1/EBP50 head-to-tail intramolecular
interaction masks the association with PDZ-domain ligands. Mol Cell
Biol. 27:2527–2537. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Pauler DK, Menon U, McIntosh M, Symecko
HL, Skates SJ and Jacobs IJ: Factors influencing serum CA125II
levels in healthy postmenopausal women. Cancer Epidemiol Biomarkers
Prev. 10:489–493. 2001.PubMed/NCBI
|
|
21
|
Venables JP: Aberrant and alternative
splicing in cancer. Cancer Res. 64:7647–7654. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Cooper DN and Krawczak M: Human Gene
Mutation. (Oxford, UK). Bios Scientific Publishers. 111–127.
1993.
|
|
23
|
Cáceres JF and Kornblithtt AR: Alterative
splicing: Multiple control mechanisms and involvement in human
disease. Trends Genet. 18:186–193. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Baralle D and Baralle M: Splicing in
action: Assessing disease causing sequence changes. J Med Genet.
42:737–748. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Fu XD: Towards a splicing code. Cell.
119:736–738. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Blencowe BJ: Alternative splicing: New
insights from global analyses. Cell. 126:37–47. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Brinkman BM: Splice variants as cancer
biomarkers. Clin Biochem. 37:584–594. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Kalnina Z, Zayakin P, Silina K and Linē A:
Alterations of pre-mRNA splicing in cancer. Genes Chromosomes
Cancer. 42:342–357. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Nelson WJ and Nusse R: Convergence of Wnt,
beta-catenin, and cadherin pathways. Science. 303:1483–1487. 2004.
View Article : Google Scholar : PubMed/NCBI
|