Introduction
Prostate cancer (PCa) is the most common malignant
cancer in men and the second leading cause of cancer in men in the
western world (1). Patients usually
respond initially to androgen deprivation therapy but eventually
relapse and metastasize because of the development of castration
resistant prostate cancer (CRPC).
Statins are commonly prescribed cholesterol-lowering
drug that inhibit 3-hydroxy-3-methylglutaryl-coenzyme A (HMG-CoA)
reductase. In recent years, statins use has been reported to be
associated with a reduced risk of PCa, particularly reducing the
reduced risk of advanced or metastatic PCa (2). A previous study reviewing the clinical
data of 1351 cases after radical prostatectomy showed less
aggressive histological features in men taking statins (3). The mechanisms for these epidemiological
observations are partially understood. In vitro experiments
have demonstrated that simvastatin induces apoptosis in CRPC cells
by inhibiting nuclear factor-κB pathway (4). It was reported that mevastatin and
simvastatin can downregulate androgen receptor (AR) expression and
prostate specific antigen (PSA) secretion, thus exhibiting an
anti-proliferation effect on PCa cells (5). Brown et al (6) found that statins, including
atorvastatin, mevastatin, simvastatin and rosuvastatin, reduced the
migration and colony formation of PC-3 cells co-cultured with bone
marrow stroma cells. Because of the high prevalence of PCa and lack
of potent chemoprevention strategies, the application of statins on
the prevention and treatment of PCa is promising, and would be
beneficial to public health.
Epithelial-mesenchymal transition (EMT) is a highly
conserved process that allows polarized, immobile epithelial cells
to trans-differentiate to those with motile mesenchymal phenotypes.
Accumulating evidence indicates that EMT is a critical initial step
and a hallmark of cancer cell migration, invasion and metastasis
(7). Transforming growth factor-β1
(TGF-β1) is widely recognized as an inducer and a chief regulator
of EMT in a variety of cell types, including cancer cells (8). It was reported that overproduction of
TGF-β1 was associated with high grade, metastasis and poor clinical
outcome in PCa, and TGF-β1 may promote tumor progression by
stimulating metastasis (9). At
present, to the best of our knowledge, no data has been reported to
demonstrate the relationship between simvastatin and EMT in PCa.
The present study is the first to investigate the effects of
simvastatin on the TGF-β1-induced EMT in PCa cells. DU145 PCa cells
were treated with simvastatin, at non-apoptotic concentrations, and
the expression levels of the epithelial markers E-cadherin and
mesenchymal markers such as N-cadherin and vimentin were assessed.
Wound-healing and transwell assays were used to investigate the
effects of simvastatin on the motility and invasion of PCa cancer
cells. Further studies investigated the effects of simvastatin on
TGF-β1-induced EMT by assessing the p38 Mitogen-Activated Protein
Kinases (p38 MAPK) pathway, non-canonical Smad signaling. The
present study aimed to determine whether simvastatin has potential
as an anti-metastatic agent for PCa.
Materials and methods
Cell culture and reagents
Human DU145 cells were purchased from the American
Type Culture Collection (Manassas, VA, USA) and cultured in
RPMI-1640 medium supplemented with 10% fetal bovine serum (FBS)
(ThermoFisher Scientific, Inc., Schuylerville, NY, USA), and 100
units/ml penicillin and 0.1 mg/ml streptomycin (Thermofisher
Scientific, Inc.) at 37°C in a humidified 5% CO2
incubator. Simvastatin was obtained from Sigma-Aldrich (Santa
Clara, CA, USA) and dissolved at a concentration of 50 µM in
dimethyl sulfoxide (DMSO) (stored in aliquots at −20°C). The
maximum final concentration of DMSO was <0.1% for each
treatment, and was also used as a control. Recombinant human TGF-β1
was purchased from PeproTech (Rocky Hill, CT, USA) and used at a
final concentration of 5 ng/ml. The cells were incubated in
serum-free medium overnight, and then treated with TGF-β1 for 48 h
to induce EMT.
Cell viability assay
DU145 cells (3×103 cells/well) were
seeded in 96-well plates and incubated overnight. After serum
starvation for 24 h, the cells were treated with 5 ng/ml TGF-β1 and
various concentrations of simvastatin for 48 h. Cell viability was
measured using a Cell Counting Kit-8 (CCK-8, Dojindo Molecular
Technologies, Japan) according to the manufacturer's instructions.
The absorbance was measured using a VARIOSCAN FLASH (ThermoFisher
Scientific, Inc.).
Western blot analysis
DU145 cells were pre-treated with simvastatin (0, 5
or 10 µM) for 2 h followed by incubation with or without 5 ng/ml
TGF-β1 for another 48 h. Total protein extracts were prepared using
a Protein Extraction kit (KeyGEN BioTECH, Beijing, China). Protein
samples were separated by 10% SDS-PAGE and transferred to PVDF
membrane (Millipore, Bedford, UK). Blots were blocked for 1 h with
5% nonfat milk in Tris-buffered saline/Tween (TBST, 0.05% Tween-20
in TBS), and then probed overnight at 4°C with primary antibodies
(Smad2/3 Antibody Sampler Kit; cat. no. 12747; dilution, 1:1,000).
All primary antibodies in this study were from Cell Signaling
Technologies, Inc., (Beverly, MA, USA). After incubation with
HRP-conjugated goat anti-rabbit IgG secondary antibodies (cat. no.
7074; dilution, 1:2,000; Cell Signaling Technologies Inc.) for 1 h
at room temperature, the membranes were then developed using a
chemiluminescence kit (Millipore) with G: BOX Chemi XL1. GENESys
(Syngene, Cambridge, UK) and analyzed by Image J software 1.48
(imagej.nih.gov/).
Wound healing assay
DU145 cells were seeded at a density of
2×105 cells/ml in 6-well plates and cultured to
confluence. After serum starvation for 24 h, cell monolayers were
scratched with a sterilized 200 ml pipette tip and washed 3 times
with phosphate-buffered saline (PBS). Cells were further treated
with 5 µM or 10 µM simvastatin and 5 ng/ml TGF-β1. The ability of
cells to migrate into the cleared section was observed and images
were captured using the IX73 inverted microscope system for
advanced live cell imaging (Olympus Corporation, Tokyo, Japan) at
48 h.
Transwell invasion assay
After serum starvation for 24 h, DU145 cells were
pre-treated with 5 ng/ml TGF-β1 and 5 µM or 10 µM simvastatin for
48 h. The filter was coated with 15 µl of matrigel (BD Biosciences,
Franklin Lakes, NJ, USA). The pretreated cells were trypsinized and
resuspended in serum-free medium and seeded at a density of
1×104 cells/well onto the top chamber. Next, 500 µl of
culture medium containing 10% FBS was added to the bottom chamber.
After incubation for 36 h, non-invading cells were carefully
removed with a cotton swab. The filter membrane was fixed with 4%
paraformaldehyde (Beijing Solarbio Science & Technology Co.,
Ltd., Beijing, China) for 15 min and stained with 0.1% crystal
violet (Beijing Solarbio Science & Technology Co., Ltd.) for 15
min. The migrated cells were counted in 5 randomly selected fields
at 200× magnification using the IX73 inverted microscope (Olympus
Corporation).
Statistical analysis
The mean values from the experiments were pooled for
statistical analysis. One-way analysis of variance followed by a
least significant difference test or Student's t-test was performed
to determine the differences between groups. All statistical
analysis was performed by the SPSS statistics 17.0 software (SPSS
Inc., Chicago, IL, USA). P<0.05 was considered to indicate a
statistically significant difference.
Results
Simvastatin blocks TGF-β1-induced EMT
in DU145 cells
To investigate simvastatin on TGF-β1-induced EMT in
DU145 cells, cell viability assay was performed to determine the
cytotoxic effects of simvastatin. Figure
1 shows that simvastatin had a dose dependent cytotoxic effect
on DU145 cells. The calculated IC50 value was 50.90 mM
at 48 h treatment. After 48 h treatment, no significant differences
were observed on cell viability at concentrations up to 10 µM in
the presence of TGF-β1, so 5 and 10 µM simvastatin were used in the
following experiments.
TGF-β1 has been widely recognized as an inducer of
EMT in a variety of types of epithelial cell (8). In the present study, TGF-β1 stimulation
was used to induce the occurrence of EMT in DU145 cells, to
identify whether TGF-β1-induced EMT is suppressed by simvastatin.
The effect of simvastatin on TGF-β1-induced EMT was evaluated by
checking the well-established markers using western blotting. As
shown in Fig. 2, DU145 cells treated
with 5 ng/ml TGF-β1 for 48 h led to significant reduction in the
epithelial phenotype marker E-cadherin and an increase in the
mesenchymal phenotype markers N-cadherin and Vimentin. However,
when combined with simvastatin (5 or 10 µM), expression of
E-cadherin was significantly increased, and the expression levels
of N-cadherin and vimentin were significantly reduced compared with
the TGF-β1 alone group (P<0.01). These findings indicate that
simvastatin inhibited the effects of TGF-β1-induced EMT in DU145
cells.
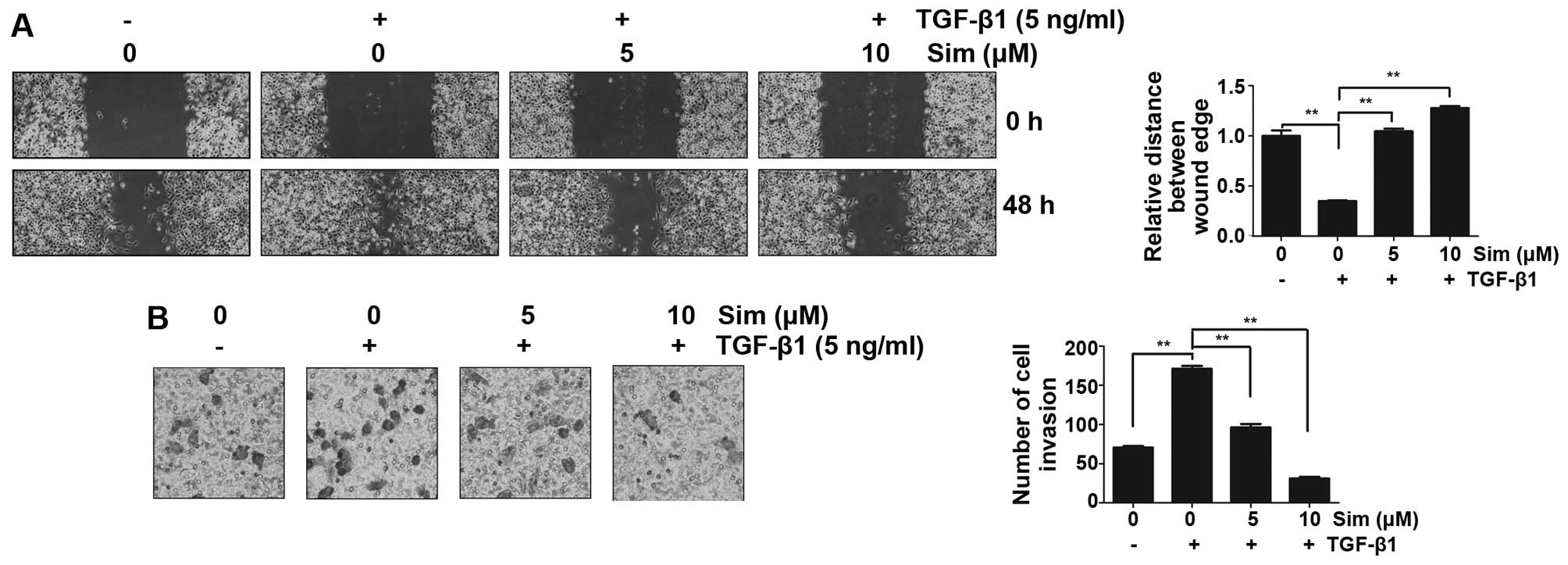
Simvastatin inhibits TGF-β1-induced
migration and invasion
To explore the potential role of simvastatin in
prostate cancer invasion and metastasis by inhibiting
TGF-β1-induced EMT, the effects of simvastatin on cellular
migration and invasion were evaluated using a wound-healing assay
and a matrigel invasion assay in vitro, respectively. The
wound healing assay revealed that TGF-β1 treatment stimulated DU145
cells to migrate and close the wound (P<0.01 versus no TGF-β1
control), while co-incubation with simvastatin (5 or 10 µM)
distinctly reversed TGF-β1-mediated migration (Fig. 3A; P<0.01 versus TGF-β1 alone). The
invasion assay demonstrated that TGF-β1 treatment markedly enhanced
the ability of DU145 cells to cross the basement membrane matrix
(P<0.01 versus no TGF-β1 control). In the matrigel invasion
assay, simvastatin (5 or 10 µM) significantly inhibited the
TGF-β1-induced invasion of DU145 cells across the gelatin-coated
membrane (Fig. 3B; P<0.01 versus
TGF-β1 alone). These results indicate that simvastatin is an
effective inhibitor of cell migration and invasion in
TGF-β1-induced DU145 cells.
Simvastatin inhibits TGF-β1-induced
EMT-transcriptional factors
To determine whether simvastatin inhibits Snail and
Slug induction via TGF-β1, the expression levels of Snail and Slug
were measured using Western blotting. TGF-β1 treatment for 48 h led
to significant increase in the expression of Snail and Slug
(P<0.01). However, Snail and Slug were significantly inhibited
by 10 µM simvastatin compared with TGF-β1 alone group cells
(P<0.01) (Fig. 4). Simvastatin
reduced TGF-β1-induced Snail and Slug levels at the concentration
of 10 µM. These results show that TGF-β1-induced expression of EMT
inducting transcription factors, Snail and Slug, were inhibited by
10 µM simvastatin.
Simvastatin has no effect on
TGF-β1/Smads signaling
Because simvastatin could inhibit the TGF-β1-induced
EMT in DU145 cells, the effects of simvastatin on Smad2 and Smad3,
two important mediators of the TGF-β/Smads signaling, were studied
to investigate whether simvastatin could modulate the canonical
TGF-β/Smads pathway. The levels of phosphorylated and total Smad2,
as well as phosphorylated and total Smad3, were detected using
western blotting. As is shown in Fig.
5, TGF-β1 triggers phosphorylation of Smad2 and Smad3, while
simvastatin has no effects on TGF-β1-induced phosphorylation of
Smad2 and Smad3, even at 10 µM. These data indicated that the
inhibition of TGF-β1-induced EMT by simvastatin is not through the
TGF-β/Smad signaling pathway.
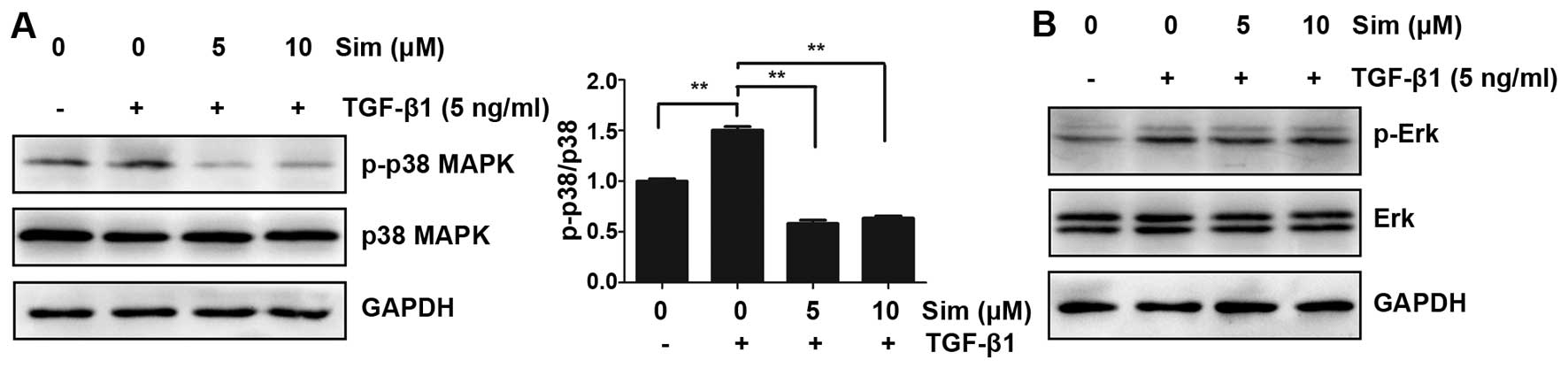
Simvastatin attenuates TGF-β1-induced
p38 phosphorylation in DU145 cells
Besides canonical Smads signaling, TGF-β1 can
mediate the EMT process independently of Smads. The
mitogen-activated protein kinase (MAPK) signaling pathway is
induced by TGF-β1, and can modulate the outcome of TGF-β1-induced
responses (10). The above results
indicated that simvastatin had no effect on TGF-β1/Smad signaling,
so the influence of simvastatin on MAPK signaling, including p38
MAPK and Erk1/2, was next investigtaed. As shown in Fig. 6, TGF-β1 induced the phosphorylation of
p38 MAPK and Erk1/2 protein in DU145 cells, and this TGF-β1-induced
p38 MAPK phosphorylation was strongly inhibited by the addition of
5 or 10 µM simvastatin, whereas no major difference in Erk1/2
expression was demonstrated (Fig. 6).
These data indicated that simvastatin may inhibit TGFb1-induced EMT
through the p38 MAPK pathway, not through TGF-β1/Smad
signaling.
Discussion
EMT comprises a conversion in cell phenotype and
serves crucial roles in the development of multiple organs and in
tissue repair. It also adversely contributes to organ fibrosis and
carcinoma invasion and metastasis (7). In epithelial cancers, EMT is
characterized by a switch in cell membrane cadherins (from
E-cadherin to N-cadherin) and a shift from apical-basal to
front-back end polarity. During EMT, tumor cells lose cell-cell
adhesion and acquire motility and invasion. These tumor cells that
have gone through EMT are thought to be responsible for seeding
distant dissemination from primary tumors (11). EMT is orchestrated by a set of
pleiotropically acting transcription factors, such as Snail
(12), Slug (13), Zeb1 (14) and Twist (15), which can directly repress mediators of
epithelial adhesion, including the hallmark of EMT: E-cadherin.
TGF-β signaling has been implicated as major inducer
of EMT (16). High production and
activity of TGF-β1 are associated with highly aggressive cancer and
poorly prognostic patients (17). In
tumor cell lines, TGF-β1 signaling has been reported to induce EMT
via up-regulating EMT-TFs, such as Snail and Slug, thereby allowing
cancer cells to become more motile and invasive (18,19). In
mouse tumor models, activation of the TGF-β1 pathway has been shown
to induce EMT and promote cells to spread to distant organs in mice
(20,21).
Statins, which can inhibit HMG-CoA reductase, are
frequently prescribed medications for treating high cholesterol. In
addition to their cholesterol-lowing effect, epidemiological
evidence suggests that statins may be associated with a lower risk
of aggressive prostate cancer (2).
Therefore, it was hypothesized that statins may have
anti-metastasis effect in PCa. Recently, statins has been shown to
attenuate TGF-β1-induced EMT in EMT-related diseases, such as lung
fibrosis (22), renal fibrosis
(23), atherosclerotic renal artery
stenosis (24), and postoperative
complications associated with EMT of lens epithelial cells
(25). These findings suggested that
statins may be a promising therapeutic strategy to treat
EMT-related disorders, including cancer metastasis. The present
study focused on exploring the effect of simvastatin on
TGF-β1-induced EMT in prostate cancer cells. It was demonstrated
that simvastatin inhibited TGF-β1-induced EMT in DU145 cells at a
concentration of 5 and 10 µM, and suppressed cell migration and
invasion, although at these concentrations it exerted no influence
on cell proliferation. These data indicate that simvastatin could
be an effective antagonizer on metastasis.
A previous study reported that stimulation of TGF-β1
could induce EMT in prostate cancer (26). In the present study, DU145 cells
treated with 5 ng/ml TGF-β1 for 48 h exhibited reduced levels of
E-cadherin and increased expression of N-cadherin and vimentin.
Thus, an EMT model in DU145 cells to study the therapeutic effect
of simvastatin on this pathological process was successfully
established. Simvastatin pretreatment prior to TGF-β1 stimulation
in DU145 cells inhibited TGF-β1-induced EMT characteristics, the
loss of E-cadherin and the increase of N-cadherin and vimentin.
Also, simvastatin inhibited the TGF-β1-induced migration and
invasion of DU145 cells (Fig. 3).
EMT programs are orchestrated by a set of
pleiotropically transcription factors, including Snail and Slug,
which suppress expression of epithelial markers, induce expression
of mesenchymal markers, and promote the dissociation of cell
adhesion, thereby allowing the migration of cancer cells. After
treatment for 48 h, TGF-β1 induced Snail and slug expression in
DU145 cells (Fig. 4). The Fig. 4 also showed that expression of
TGF-β1-induced upregulation of Snail and Slug, were inhibited by 10
µM simvastatin. Both Snail and Slug belong to the super-family of
zinc finger transcription factors, which can bind proximal promoter
sequences of E-cadherin, namely E-box elements, thus repressing its
transcription (27). So the present
study next sought to elucidate the upstream regulatory mechanisms
underlying the alteration in Snail and Slug expression.
TGF-β1 induces EMT via two specific pathways, the
canonical Smad signaling pathway and a non-Smad signaling pathway
(28). Binding of TGF-β1 to
heterotetrameric complexes of type I (TβRI) and type II TGF-β
receptors (TβRII) leads to the phosphorylation/activation of the
TβRI. With the assistance of adapter proteins, TβRI binds and
phosphorylates the Smad2 and/or Smad3. The phosphorylated Smad2 or
Smad3 will then associate with Smad4, translocate into the nucleus,
and activate target gene expression through interaction with other
transcription factors, such as snail and slug (29). Yang et al (22) reported that simvastatin attenuated
TGF-β1 induced EMT in human alveolar epithelial cells associated
with modulation of TGF-β1-Smad2/3 pathway. Other studies have also
reported simvastatin inhibited TGF-β1 induced Smad2/3
phosphorylation during intestinal fibroblast (30) and myofibroblast differentiation in
nasal polyp-derived fibroblasts (31), in which EMT also plays an important
role. The present study focused on the influence of simvastatin on
TGF-β1 mediated Smad2/3 signaling and found that simvastatin did
not affect TGF-β1 induced phosphorylation of Smad2 and Smad3.
Notably, the results suggested that simvastatin abrogated
TGF-β1-induced EMT and cell migration and invasion in DU145 cells
not through canonical TGF-β1-Smads signaling.
TGF-β1 can also transduct independently of Smads.
TGF-β1 induces the MAPK signaling pathway and can modulate the
outcome of TGF-β1-induced responses (10). In particular, p38 MAPK has been shown
to mediate Smad-independent TGF-β1 responses (32). To further investigate the underlying
mechanism of simvastatin inhibition on TGF-β1 induced EMT, p38 and
Erk1/2 MAPK signaling were investigated. It was demonstrated that
TGF-β1 induced phosphorylation of both p38 and Erk1/2 (Fig. 6) after 48 h of incubation. The
activation of p38 was significantly reduced in the presence of 5
and 10 µM simvastatin, whereas Erk1/2 phosphorylation was
unaffected by simvastatin, even at 10 µM. These data suggested that
p38 MAPK signaling involved in action of simvastatin inhibiting the
TGF-β1 induced EMT in DU 145 cells. A similar mechanism of statins
attenuating EMT via inhibition of p38 MAPK activation was also
reported in human tendon fibroblast cells (33). These data clearly demonstrate that the
specific mechanism involved in the effect of statins on TGF-β1
induced EMT depends on the cancer cell type.
Overproduction of TGF-β1 is associated with poor
clinical outcome in prostate cancer, and TGF-β1 may promote tumor
progression by stimulating metastasis (9). Previous studies have demonstrated that
statins reduced advanced PCa risk but not overall PCa risk
(2,34,35). The
present study is the first to show the inhibitory effect of
simvastatin on TGF-β1 induced EMT and in TGF-β1-induced DU145
cells. This indicates that simvastatin attenuated TGF-β1 induced
EMT associated with modulation of P38 MAPK signaling, not through
the canonical Smad-dependent pathway. These data could to some
degree explain why statins exhibit a protective effect in PCa and
reduce advanced PCa risk.
In summary, the present study demonstrated that
simvastatin was able to suppress PCa cell migration and invasion
in vitro by inhibiting TGF-β1 induced EMT. These effects may
have been mediated by the inhibition of p38 MAPK phosphorylation.
These findings provide a novel insight into the actions of
simvastatin as an inhibitor of EMT and cancer metastasis in
PCa.
Acknowledgements
The present study was supported by a grant from the
Beijing Natural Science Foundation (grant no. 7122183).
References
|
1
|
Jemal A, Siegel R, Xu J and Ward E: Cancer
statistics, 2010. CA Cancer J Clin. 60:277–300. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Platz EA, Leitzmann MF, Visvanathan K,
Rimm EB, Stampfer MJ, Willett WC and Giovannucci E: Statin drugs
and risk of advanced prostate cancer. J Natl Cancer Inst.
98:1819–1825. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Loeb S, Kan D, Helfand BT, Nadler RB and
Catalona WJ: Is statin use associated with prostate cancer
aggressiveness? BJU Int. 105:1222–1225. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Park YH, Seo SY, Lee E, Ku JH, Kim HH and
Kwak C: Simvastatin induces apoptosis in castrate resistant
prostate cancer cells by deregulating nuclear factor-κB pathway. J
Urol. 189:1547–1552. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Yokomizo A, Shiota M, Kashiwagi E, Kuroiwa
K, Tatsugami K, Inokuchi J, Takeuchi A and Naito S: Statins reduce
the androgen sensitivity and cell proliferation by decreasing the
androgen receptor protein in prostate cancer cells. Prostate.
71:298–304. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Brown M, Hart C, Tawadros T, et al: The
differential effects of statins on the metastatic behaviour of
prostate cancer. Br J Cancer. 106:1689–1696. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Thiery JP, Acloque H, Huang RY and Nieto
MA: Epithelial-mesenchymal transitions in development and disease.
Cell. 139:871–890. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Zavadil J and Böttinger EP: TGF-beta and
epithelial-to-mesenchymal transitions. Oncogene. 24:5764–5774.
2005. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Wikström P, Stattin P, Franck-Lissbrant I,
Damber JE and Bergh A: Transforming growth factor beta1 is
associated with angiogenesis, metastasis and poor clinical outcome
in prostate cancer. Prostate. 37:19–29. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Javelaud D and Mauviel A: Crosstalk
mechanisms between the mitogen-activated protein kinase pathways
and Smad signaling downstream of TGF-beta: Implications for
carcinogenesis. Oncogene. 24:5742–5750. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Thiery JP: Epithelial-mesenchymal
transitions in tumour progression. Nat Rev Cancer. 2:442–454. 2002.
View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Cano A, Pérez-Moreno MA, Rodrigo I,
Locascio A, Blanco MJ, del Barrio MG, Portillo F and Nieto MA: The
transcription factor snail controls epithelial-mesenchymal
transitions by repressing E-cadherin expression. Nat Cell Biol.
2:76–83. 2000. View
Article : Google Scholar : PubMed/NCBI
|
|
13
|
Bolós V, Peinado H, Pérez-Moreno MA, Fraga
MF, Esteller M and Cano A: The transcription factor Slug represses
E-cadherin expression and induces epithelial to mesenchymal
transitions: A comparison with Snail and E47 repressors. J Cell
Sci. 116:499–511. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Eger A, Aigner K, Sonderegger S, Dampier
B, Oehler S, Schreiber M, Berx G, Cano A, Beug H and Foisner R:
DeltaEF1 is a transcriptional repressor of E-cadherin and regulates
epithelial plasticity in breast cancer cells. Oncogene.
24:2375–2385. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Yang MH, Hsu DS, Wang HW, et al: Bmi1 is
essential in Twist1-induced epithelial-mesenchymal transition. Nat
Cell Biol. 12:982–992. 2010. View
Article : Google Scholar : PubMed/NCBI
|
|
16
|
Yang J and Weinberg RA:
Epithelial-mesenchymal transition: At the crossroads of development
and tumor metastasis. Dev Cell. 14:818–829. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
González-Santiago AE, Mendoza-Topete LA,
Sánchez-Llamas F, Troyo-Sanromán R and Gurrola-Díaz CM: TGF-β1
serum concentration as a complementary diagnostic biomarker of lung
cancer: Establishment of a cut-point value. J Clin Lab Anal.
25:238–243. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Comijn J, Berx G, Vermassen P, Verschueren
K, van Grunsven L, Bruyneel E, Mareel M, Huylebroeck D and van Roy
F: The two-handed E box binding zinc finger protein SIP1
downregulates E-cadherin and induces invasion. Mol Cell.
7:1267–1278. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Peinado H, Quintanilla M and Cano A:
Transforming growth factor beta-1 induces snail transcription
factor in epithelial cell lines: Mechanisms for epithelial
mesenchymal transitions. J Biol Chem. 278:21113–21123. 2003.
View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Janda E, Lehmann K, Killisch I, et al: Ras
and TGF [beta] cooperatively regulate epithelial cell plasticity
and metastasis: Dissection of Ras signaling pathways. J Cell Biol.
156:299–313. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Oft M, Heider KH and Beug H: TGFbeta
signaling is necessary for carcinoma cell invasiveness and
metastasis. Curr Biol. 8:1243–1252. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Yang T, Chen M and Sun T: Simvastatin
attenuates TGF-β1-induced epithelial-mesenchymal transition in
human alveolar epithelial cells. Cell Physiol Biochem. 31:863–874.
2013. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Patel S, Mason RM, Suzuki J, et al:
Inhibitory effect of statins on renal epithelial-to-mesenchymal
transition. Am J Nephrol. 26:381–387. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Agrotis A: Simvastatin, an inhibitor of
epithelial-to-mesenchymal transition in experimental
atherosclerotic renovascular disease? J Hypertens. 26:1553–1555.
2008. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Urakami C, Kurosaka D, Tamada K, et al:
Lovastatin alters TGF-β-induced epithelial-mesenchymal transition
in porcine lens epithelial cells. Curr Eye Res. 37:479–485. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Ao M, Williams K, Bhowmick NA and Hayward
SW: Transforming growth factor-beta promotes invasion in
tumorigenic but not in nontumorigenic human prostatic epithelial
cells. Cancer Res. 66:8007–8016. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Hotz B, Arndt M, Dullat S, Bhargava S,
Buhr HJ and Hotz HG: Epithelial to mesenchymal transition:
Expression of the regulators snail, slug and twist in pancreatic
cancer. Clin Cancer Res. 13:4769–4776. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Moustakas A and Heldin CH: Non-Smad
TGF-beta signals. J Cell Sci. 118:3573–3584. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Shi Y and Massagué J: Mechanisms of
TGF-beta signaling from cell membrane to the nucleus. Cell.
113:685–700. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Burke JP, Watson RW, Murphy M, Docherty
NG, Coffey JC and O'Connell PR: Simvastatin impairs smad-3
phosphorylation and modulates transforming growth factor
beta1-mediated activation of intestinal fibroblasts. Br J Surg.
96:541–551. 2009. View
Article : Google Scholar : PubMed/NCBI
|
|
31
|
Park IH, Park SJ, Cho JS, Moon YM, Moon
JH, Kim TH, Lee SH and Lee HM: Effect of simvastatin on
transforming growth factor beta-1-induced myofibroblast
differentiation and collagen production in nasal polyp-derived
fibroblasts. Am J Rhinol Allergy. 26:7–11. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Yu L, Hébert MC and Zhang YE: TGF-beta
receptor-activated p38 MAP kinase mediates Smad-independent
TGF-beta responses. EMBO J. 21:3749–3759. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Meyer-Ter-Vehn T, Katzenberger B, Han H,
Grehn F and Schlunck G: Lovastatin inhibits TGF-beta-induced
myofibroblast transdifferentiation in human tenon fibroblasts.
Invest Ophthalmol Vis Sci. 49:3955–3960. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Jacobs EJ, Rodriguez C, Bain EB, Wang Y,
Thun MJ and Calle EE: Cholesterol-lowering drugs and advanced
prostate cancer incidence in a large US cohort. Cancer Epidemiol
Biomarkers Prev. 16:2213–2217. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Murtola TJ, Tammela TL, Lahtela J and
Auvinen A: Cholesterol-lowering drugs and prostate cancer risk: A
population-based case-control study. Cancer Epidemiol Biomarkers
Prev. 16:2226–2232. 2007. View Article : Google Scholar : PubMed/NCBI
|