Introduction
Yes-associated protein (YAP or YAP1) is an
oncoprotein encoded by the YAP gene in the human chromosome
11q22 (1). YAP is one of the
downstream proteins in the Hippo signaling pathway (1). It functions in cooperation with
transcriptional coactivator with PDZ-binding motif (TAZ) (2). These two proteins are responsible for
cell proliferation control and have important regulatory functions
in regeneration, organ development and stem cell self-renewal. YAP
and TAZ both are regulated by several mechanisms, including the
microenvironment and extracellular signals. YAP is also able to
inhibit apoptosis in cells through the activation of survival
signaling such as survivin/baculoviral IAP repeat containing 5
(2). Recent investigations suggest
that YAP crosstalks with numerous signaling pathways and has more
functions in cancer than previously assumed.
Structural domains of YAP protein
YAP consists of 488 amino acids and several
structural domains (3) (Fig. 1). The most important domains are a TEA
DNA-binding domain and two WW domains (2). The first one binds to the TEA domain
(TEAD) family of transcription factors, while the second one binds
to a transcriptional coactivator, which in turn, binds to the PPxY
motif present on transcription factors (4). In addition, the PDZ-binding motif
regulates the function of YAP by localizing YAP to the specific
nuclear foci and is essential for YAP-mediated cellular
transformation (5). The SRC homolog
3-binding motif regulates the activity of YAP through binding to
p53 binding protein 2, in cooperation with the WW1 domain (6).
Regulation of YAP
The regulation of YAP occurs mostly through the
regulation of the Hippo signaling pathway (2). Large tumor suppressor 1 (LATS1) and
LATS2 are the inhibitors of the YAP/TAZ complex. When activated,
these two proteins phosphorylate YAP and localize it in the
cytoplasm (2). YAP is tightly
regulated by multiple factors, including extracellular signals and
the microenvironment (5).
Regulation by extracellular
signals
One of the most important regulators of YAP is
G-protein coupled receptor (GPCR), which consists of three
subunits, α, β, γ, among which, Gα11, Gα12,
Gα13, Gαi, Gαo and Gαq
activate YAP and TAZ, while Gαs inhibits YAP/TAZ
activity (7). GPCR activates YAP by
inducing F-actin polymerization through Rho GTPase, and thus
inhibit LATS kinase activity (7).
There are two types of ligands-agonists that activate GPCR: i)
Lipoprotein(a), sphingosine-1-phosphate and thrombin, which inhibit
cell proliferation; and ii) glucagon and epinephrine, which induce
cell proliferation (8) (Fig. 2). In addition, Wnt signaling can
regulate YAP activity by separating YAP from the β-catenin
destruction complex, and thus causing accumulation of nuclear YAP
(Fig. 2) (9). Leukemia inhibitory factor receptor and
epidermal growth factor can activate YAP by dissociating it from
Hippo kinase (7).
 | Figure 2.Regulation of the Yes-associated
protein/transcriptional coactivator with PDZ-binding motif complex.
YAP, Yes-associated protein; TAZ, transcriptional coactivator with
PDZ-binding motif; GPCR, G-protein-coupled receptor; LPA,
lipoprotein(a); S1P, sphingosine-1-phosphate; APC, adenomatous
polyposis coli; cAMP, cyclic adenosine monophosphate; PKA, protein
kinase A; LATS, large tumor suppressor; AMOT, angiomotin; VGLL4,
vestigial-like family member 4; TEAD, TEA domain; P,
phosphorylation. |
Regulation by microRNA (miRNA)
Increasing evidence suggest that miRNAs regulate
YAP, including miRNA 31, which is considered as an oncogene that
acts through YAP during cancer progression (10). The overexpression of miRNA 31 leads to
anchorage-independent uncontrolled growth in vitro and
increases the potential of tumor formation in vivo (10). The expression level of miRNA 31 was
much more increased in patients with high risk of recurrence,
compared with those with low risk of recurrence (10). According to Mitamura et al
(10), miRNA 31 suppresses the
luciferase activity of messenger (m)RNA combined with LATS2 3′
untranslated region (UTR). This promoted an increase in the nuclear
level of YAP and moreover, enhanced the transcription of oncogenes
such as cyclin D1.
Regulation by phosphorylation and
methylation
Phosphorylation is the main pathway to inhibit YAP
activity (7). As shown in Fig. 1, multiple sites of YAP can be
phosphorylated. Nuclear Dbf2-related/LATS kinases regulate YAP1
through its phosphorylation at Serine 127, and promotes its
exclusion from the nucleus into the cytoplasm and its degradation
(11). Consequently, the
transcription of pro-growth genes is inhibited. The artificial
depletion of these kinases in the colon results in the formation of
cancer (11). The phosphorylation of
YAP at Serine 127 promotes binding of 14-3-3 protein to YAP and,
thus, localizes it in the cytoplasm (4). Another kinase that promotes binding of
14-3-3 protein to YAP is Akt kinase (12). The inhibition of YAP by Akt causes
inhibition of transcription factors, including p53, which regulates
the pro-apoptotic gene B-cell lymphoma (Bcl)-2 associated X
protein. Thus, the phosphorylation and consequent inhibition of YAP
by Akt suppresses the induction of pro-apoptotic gene expression
upon cell damage (12). Another
molecule that regulates YAP function through phosphorylation is
protein kinase C ζ (PKCζ), which plays an important role in the
retention of YAP in the cytoplasm (13). PKCζ phosphorylates YAP at Serine 109
and Threonine 110, which are highly conserved residues among
different species (13). In
leucine-rich repeat-containing G-protein coupled receptor
5-positive intestinal stem cells, the inactivating mutation of YAP
causes increased tumorigenic and regenerative activities (13).
YAP methylation is another regulatory mechanism that
localizes YAP in the cytoplasm and inhibits its function (14). The methylation of YAP is induced by
Su(var)3–9 and enhancer of zeste (SET)7, a SET-domain-containing
lysine methyltransferase. Monomethylation of YAP at Lysine 494
occurs in parallel with YAP phosphorylation at Serine 127, which
promotes the cytoplasmic localization of YAP (15) (Fig.
1).
Regulation by metabolism
The sterol regulatory element-binding
protein/mevalonate signaling pathway, an important cellular
metabolic pathway, is able to regulate the activity of YAP.
According to Sorrentino et al (16), the inhibition of its rate-limiting
enzyme (3-hydroxy-3-methylglutaryl-coenzyme A reductase) opposes
YAP/TAZ nuclear localization. The production of geranylgeranyl
pyrophosphate generated by the mevalonate pathway is required for
the activation of Rho GTPase (16).
This GTPase is responsible for the polymerization of F-actin, which
removes angiomotin (AMOT) from its complex with YAP, and thus
allows YAP to translocate to the nucleus and to induce the
transcription of pro-growth genes (7).
Regulation by the
microenvironment
The microenvironment plays an important role in the
regulation of YAP activity. Recent studies revealed that the
stiffness of the extracellular matrix can cause changes in the
levels of YAP in the nucleus (17).
When the cells are placed into soft matrix, the cytoplasmic level
of YAP increased and the cell proliferation rate decreased, and
vice versa, when the cells were subjected to a stiff environment,
the cytoplasmic YAP moved to the nucleus and induced cell
proliferation (18). When cells
undergo pressure, to balance the tension, they create an opposite
force with the help of the cytoskeleton (17). Thus, when F-actin polymerization
happens, the nuclear level of YAP increases and the cells start to
proliferate (17). According to
Mana-Capelli et al, AMOT links these two processes (19). AMOT is an inhibitor of YAP, and can
act on it either directly or through activating LATS1 and LATS2
(19). When F-actin polymerization
occurs, it competes with YAP for the binding to AMOT, and thus
weakens the inhibitory effect of AMOT on YAP (18). Additionally, F-actin is also able to
suppress the activity of LATS1/2, and thus further increases the
nuclear level of YAP (18).
Function of YAP in cancer
As an oncoprotein, YAP controls transcription
factors that regulate cell division and cell cycle, such as TEAD
family members (7). TEAD proteins are
associated with the transcription cofactor vestigial-like protein 4
(VGLL4) in steady state, which suppresses target gene expression
(20). When the Hippo signaling
pathway is activated, YAP is phosphorylated, which prevents YAP
from passing through the nuclear pores and reaching TEAD proteins
(20). When the pathway is
inactivated, YAP moves to the nucleus, displaces VGLL4 from TEAD
proteins and initiates cell division. In numerous cancers,
including hepatocellular carcinoma (HCC), ovarian cancer and
non-small cell lung cancer, elevated nuclear level of YAP was
observed, suggesting an association between YAP function and
uncontrolled cell division (21).
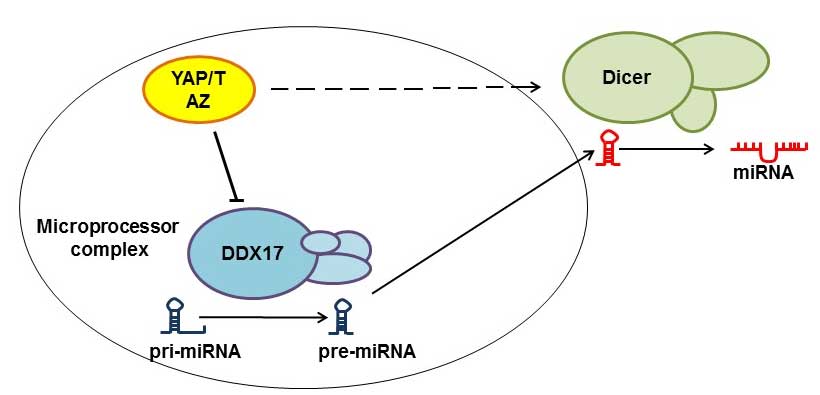
Regulation of miRNA biogenesis
The suppression of miRNAs has been proposed to
promote tumorigenesis (22). In order
for miRNAs to become functional, they should first be subjected to
the Microprocessor and Dicer complexes in the form of primary miRNA
transcripts. Recent studies have demonstrated that YAP regulates
miRNA production through diverting DEAD box helicase 17 (DDX17)
from the Microprocessor complex in a cell-density dependent manner.
When the cell density is low, YAP localizes in the nucleus and
removes DDX17 from the Microprocessor complex, and when the cell
density is high, YAP moves to the cytoplasm and DDX17 becomes free
to combine with the Microprocessor complex (22). However, nuclear YAP/TAZ also regulates
Dicer complexes, and thus mediates the formation of specific miRNAs
(23). In summary, the YAP/TAZ
complex may have opposite regulatory effects on different miRNAs
(Fig. 3).
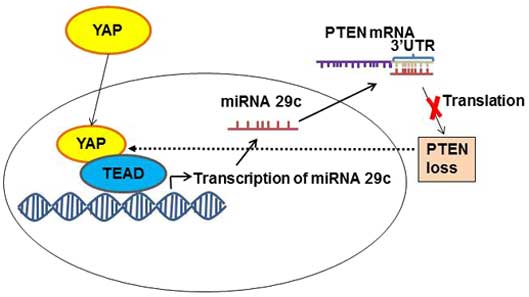
Regulation of phosphatase and tensin homolog
(PTEN)
PTEN, as a tumor suppressor protein, inhibits the
phosphatidylinositol 3-kinase-mechanistic target of rapamycin
signaling pathway, and is responsible for controlling organ size
and cell proliferation (24). YAP
regulates this pathway through PTEN. YAP inhibits PTEN activity by
activating transcription factors of the TEAD family to express
miRNA 29c (24) (Fig. 4). This miRNA 29c regulates PTEN
function by targeting PTEN 3′UTR (24). When the level of activated YAP is
increased, the level of PTEN decreases, and vice versa, when YAP is
phosphorylated and inhibited, PTEN function is restored and
oncogene transcription is inhibited (24). Therefore, mutations in the Hippo
signaling pathway, which lead to an increase in the nuclear level
of YAP, also cause the inhibition of its natural tumor suppressor,
PTEN, and thus facilitate tumor formation.
Regulation of senescence
YAP is able to control senescence tightly through
its activity. According to Fausti et al (25), the accumulation of YAP correlates with
slower cell division rate and accelerated senescence. The authors
stated that the loss of Werner syndrome ATP-dependent helicase
protein activity in Werner syndrome activates the ataxia
telangiectasia mutated (ATM)-YAP-promyelocytic leukemia protein
(PML)-p53 axis, and thus induces the senescence process. ATM kinase
is required for YAP-PML complex formation, and this complex
promotes p53 activation (25).
However, according to Xie et al (26), the senescence process of the cell is
caused by downregulation of YAP levels, and the silencing of the
YAP gene causes inhibition of cell proliferation and induces
senescence. According to that report, YAP regulates the expression
of cell division protein kinase 6 (Cdk6), an important protein in
the regulation of cell cycle progression. The results demonstrated
that introduction of either YAP or Cdk6 into YAP-knocked down cells
inhibited senescence and restored the proliferative abilities of
the cells (26). In summary, there
are different points of view about the regulation of cell
senescence, and further studies are required to clarify this
mechanism.
Regulation of Ras-dependent oncogenesis
Overexpression of proteins from the Ras family leads
to uncontrolled cell growth and cancer, such as pancreatic ductal
adenocarcinoma (27). Elevated level
of YAP was observed in K-Ras suppression-resistant cancer cells
(28). Recently, YAP was identified
as the main compensator of cell proliferation for loss of K-Ras
signaling in K-Ras-dependent cancers (29). K-Ras and YAP proteins converge on the
transcription factor FOS, and activate the transcription of the
genes responsible for the regulation of the epithelial-mesenchymal
transition, which initiates metastasis in cancer cells (28).
YAP function in prostate cancer
progression
Androgen receptor (AR), as a transcription factor,
plays a central role in prostate cancer progression through
regulating the transcription of cell proliferation-related genes.
Recent studies have shown that YAP is able to bind and activate
nuclear AR, thus promoting cell proliferation (30). Binding of AR to YAP occurs via the PDZ
binding domain and the coiled-coiled protein-protein interaction
domain (30). The YAP complex can
activate androgen-dependent transcription of AR target genes, which
has the same DNA binding site. However, enhanced level of YAP mRNA
was observed in androgen-insensitive prostate cancer cells,
compared with androgen-sensitive cells (31). Furthermore, immunohistochemistry
revealed increased levels of activated YAP in castration-resistant
prostate tumors, compared with hormonal-responsive prostate tumors
(31). Further studies demonstrated
that YAP overexpression was sufficient to cause the transition of
cells from an androgen-sensitive to an androgen-insensitive
phenotype in vitro, and YAP conferred castration resistance
in vivo (31). Extracellular
signal-regulated kinase ribosomal S6 kinase signaling appeared to
be regulated by YAP in cell survival, migration and invasion in
androgen-insensitive cells, and thus, the downregulation of YAP in
prostate cancer cells greatly reduced their migratory and invasive
rates, and in androgen-deprivation conditions, it inhibited cell
division (31). In summary, in
prostate cells with deregulated Hippo signaling pathway, YAP can
promote cancer cells to lose their sensitivity to androgen and
transform into uncontrolled dividing cells that are insensitive to
androgen but sensitive to other proliferative signals such as those
of the MET signaling pathway (32)
(Fig. 5).
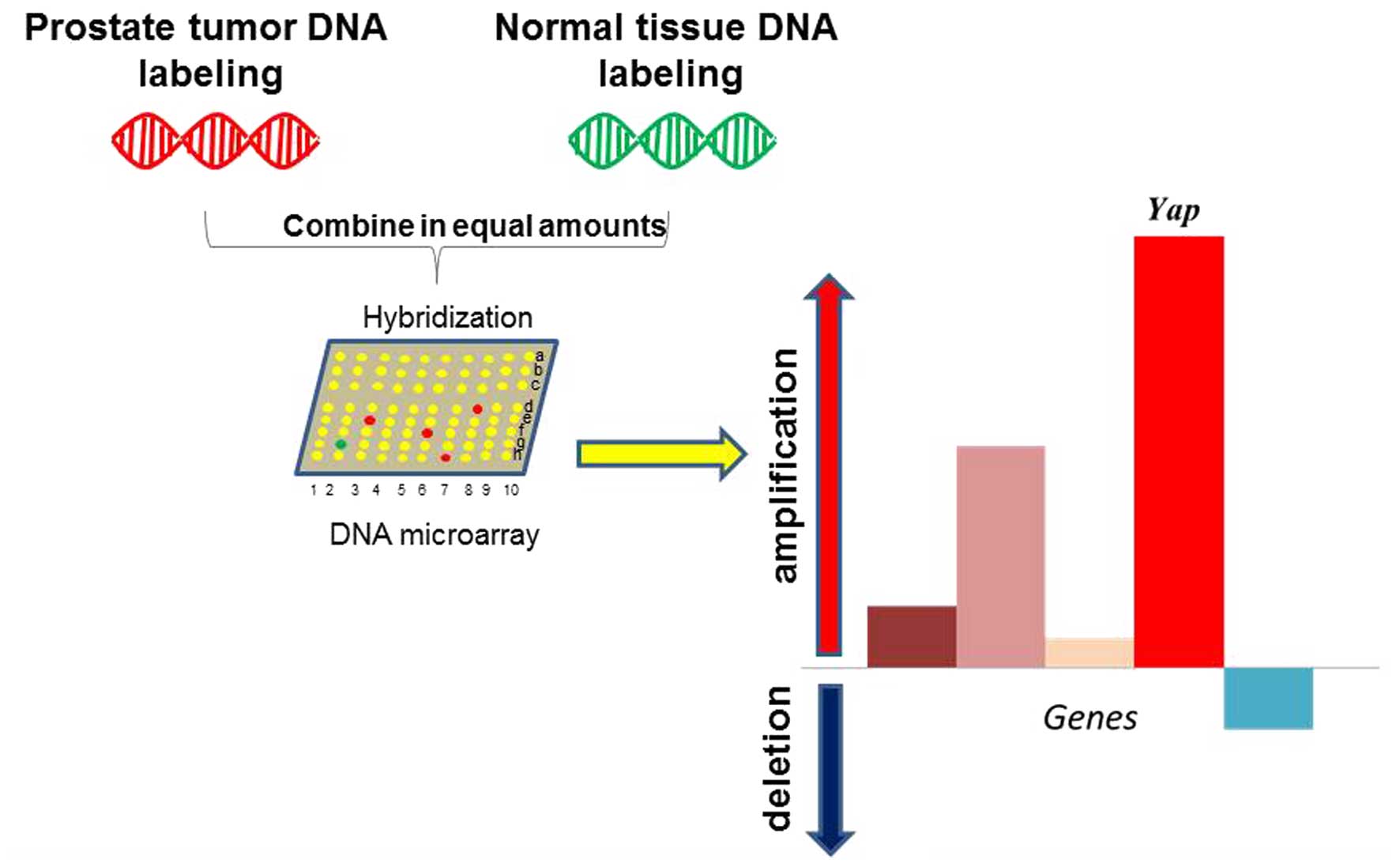
Notably, the mouse Yap gene copy number was
significantly increased in a mouse model of prostate cancer driven
by PTEN/p53 loss (33). Yap
was the top ranked gene amplified by whole genome screening in this
model (33) (Fig. 6). As PTEN and p53 are frequently
mutated or inactivated in several cancers, particularly in prostate
cancer, this finding suggests that YAP may act as a master
regulator in prostate cancer upon oncogenic insults stimulation.
Furthermore, YAP may provide feedback regulation through the
YAP/PTEN axis (Fig. 4). However, the
detailed mechanism of this feedback loop and its clinical relevance
must be further investigated.
Implications for clinical targeted
therapy
As aforementioned, YAP is involved in the control of
proliferation, and is regulated by different mechanisms. Due to its
very important role in tumorigenesis, this protein could be used as
a target for novel avenues of cancer therapy. A recent study has
suggested several approaches to suppress the activity of YAP in
cancer cells with increased cell proliferation rate (34). One of the molecules that competes for
YAP activity in the nucleus is VGLL4 (34). This molecule functions as an
antagonist and competes with YAP for binding to TEAD family
members, and thus can inhibit the activity of YAP. More precisely,
VGLL4′s tandem Tondu domains inhibit the activity of YAP (33). YAP plays a major role in the formation
of HCC (35). In addition, small
interfering RNA-lipid nanoparticles, which target and inactivate
YAP protein, can decrease the levels of activated YAP protein
(35). This causes HCC tumor
regression and restores hepatocyte differentiation (35).
Currently, numerous single treatments using
anticancer drugs have little effect on cancer cells growth. By
contrast, the combination of several drugs is able to cause cancer
cells to stop proliferating and induce apoptosis (36). As an example, the combination of the
drugs trametinib and vemurafenib, which inhibit regulatory proteins
in the mitogen-activated protein kinase signaling pathway, promotes
cancer cells to stop proliferating, but still is not sufficient to
cause tumor cell death (37). The
introduction of RNA interference specific to YAP, which indirectly
induces the expression of the pro-survival gene Bcl-extra large,
reduces cell proliferation and induces cell apoptosis (36). Thus, YAP protein plays a major role in
drug resistance through the regulation of the expression of genes
responsible for cancer cell survival. Therefore, targeting YAP may
sensitize cancer cells to chemotherapy drugs.
In summary, YAP, as a downstream target of the Hippo
signaling pathway, can crosstalk with various biological signaling
pathways. Directly targeting of YAP signaling would be a novel
avenue for the treatment of aggressive cancer and reversing drug
resistance.
References
|
1
|
Lorenzetto E, Brenca M, Boeri M, Verri C,
Piccinin E, Gasparini P, Facchinetti F, Rossi S, Salvatore G,
Massimino M, et al: YAP1 acts as oncogenic target of 11q22
amplification in multiple cancer subtypes. Oncotarget. 5:2608–2621.
2014. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Shen Z and Stanger BZ: YAP regulates
S-Phase entry in endothelial cells. PLoS One. 10:e01175222015.
View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Zhao B, Li L, Lei Q and Guan KL: The
Hippo-YAP pathway in organ size control and tumorigenesis: An
updated version. Genes Dev. 24:862–874. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Kanai F, Marignani PA, Sarbassova D, Yagi
R, Hall RA, Donowitz M, Hisaminato A, Fujiwara T, Ito Y, Cantley LC
and Yaffe MB: TAZ: A novel transcriptional co-activator regulated
by interactions with 14-3-3 and PDZ domain proteins. EMBO J.
19:6778–6791. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Shimomura T, Miyamura N, Hata S, Miura R,
Hirayama J and Nishina H: The PDZ-binding motif of Yes-associated
protein is required for its co-activation of TEAD-mediated CTGF
transcription and oncogenic cell transforming activity. Biochem
Biophys Res Commun. 17:917–931. 2014. View Article : Google Scholar
|
|
6
|
Espanel X and Sudol M: Yes-associated
protein and p53-binding protein-2 interact through their WW and SH3
domains. J Biol Chem. 276:14514–14523. 2001.PubMed/NCBI
|
|
7
|
Moroishi T, Hansen CG and Guan KL: The
emerging roles of YAP and TAZ in cancer. Nat Rev Cancer. 276:73–79.
2015. View
Article : Google Scholar
|
|
8
|
Zhou X, Wang Z, Huang W and Lei QY: G
protein-coupled receptors: Binding the gap from the extracellular
signals to the Hippo pathway. Acta Biochim Biophys Sin (Shanghai).
47:10–15. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Azzolin L, Panciera T, Soligo S, Enzo E,
Bicciato S, Dupont S, Bresolin S, Frasson C, Basso G, Frasson C, et
al: YAP/TAZ incorporation in the β-catenin destruction complex
orchestrates the Wnt response. Cell. 158:157–170. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Mitamura T, Watari H, Wang L, Kanno H,
Kitagawa M, Hassan MK, Kimura T, Tanino M, Nishihara H, Tanaka S
and Sakuragi N: MicroRNA 31 functions as an endometrial cancer
oncogene by suppressing Hippo tumor suppressor pathway. Mol Cancer.
13:972014. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Zhang L, Tang F, Terracciano L, Hynx D,
Kohler R, Bichet S, Hess D, Cron P, Hemmings BA, Hergovich A and
Schmitz-Rohmer D: NDR functions as a physiological YAP1 kinase in
the intestinal epithelium. Curr Biol. 25:296–305. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Basu S, Totty NF, Irwin MS, Sudol M and
Downward J: Akt phosphorylates the Yes-associated protein, YAP, to
induce interaction with 14-3-3 and attenuation of p73-mediated
apoptosis. Mol Cell. 11:11–23. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Llado V, Nakanishi Y, Duran A, ReinaCampos
M, Shelton PM, Linares JF, Yajima T, Campos A, AzaBlanc P, Leitges
M, et al: Repression of intestinal stem cell function and
tumorigenesis through direct phosphorylation of β-catenin andYap by
PKCζ. Cell Rep. pii:S2211–1247. 2015.
|
|
14
|
Oudhoff MJ, Freeman SA, Couzens AL,
Antignano F, Kuznetsova E, Min PH, Northrop JP, Lehnertz B,
BarsyteLovejoy D, Vedadi M, et al: Control of the hippo pathway by
Set7-dependent methylation of Yap. Dev Cell. 26:188–194. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Hergovich A and Hemmings BA: Mammalian
NDR/LATS protein kinases in hippo tumor suppressor signaling.
Biofactors. 35:338–345. 2009. View
Article : Google Scholar : PubMed/NCBI
|
|
16
|
Sorrentino G, Ruggeri N, Specchia V,
Cordenonsi M, Mano M, Dupont S, Manfrin A, Ingallina E, Sommaggio
R, Vedadi M, et al: Metabolic control of YAP and TAZ by the
mevalonate pathway. Nat Cell Biol. 16:357–366. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Low BC, Pan CQ, Shivashankar GV,
Bershadsky A, Sudol M and Sheetz M: YAP/TAZ as mechanosensors and
mechanotransducers in regulating organ size and tumor growth. FEBS
Lett. 588:2663–2670. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Halder G, Dupont S and Piccolo S:
Transduction of mechanical and cytoskeletal cues by YAP and TAZ.
Nat Rev Mol Cell Biol. 13:591–600. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
ManaCapelli S, Paramasivam M, Dutta S and
McCollum D: Angiomotins link F-actin architecture to Hippo pathway
signaling. Mol Biol Cell. 25:1676–1685. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Johnson R and Halder G: The two faces of
Hippo: Targeting the Hippo pathway for regenerative medicine and
cancer treatment. Nat Rev Drug Discov. 13:63–79. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Harvey KF, Zhang X and Thomas DM: The
Hippo pathway and human cancer. Nat Rev Cancer. 13:246–257. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Mori M, Triboulet R, Mohseni M,
Schlegelmilch K, Shrestha K, Camargo FD and Gregory RI: Hippo
signaling regulates Microprocessor and links cell density-dependent
miRNA biogenesis to cancer. Cell. 156:893–906. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Chaulk SG, Lattanzi VJ, Hiemer SE, Fahlman
RP and Varelas X: The Hippo pathway effectors TAZ/YAP regulate
dicer expression and microRNA biogenesis through Let-7. J Biol
Chem. 289:1886–1891. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Tumaneng K, Schlegelmilch K, Russell RC,
Yimlamai D, Basnet H, Mahadevan N, Fitamant J, Bardeesy N, Camargo
FD and Guan KL: YAP mediates crosstalk between the Hippo and PI
(3)K-TOR pathways by suppressing PTEN via miR-29. Nat Cell Biol.
14:1322–1329. 2012. View
Article : Google Scholar : PubMed/NCBI
|
|
25
|
Fausti FI, Di Agostino S, Cioce M, Bielli
P, Sette C, Pandolfi PP, Oren M, Sudol M, Strano S and Blandino G:
ATM kinase enables the functional axis of YAP, PML and p53 to
ameliorate loss of Werner protein-mediated oncogenic senescence.
Cell Death Differ. 20:1498–1509. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Xie Q, Chen J, Feng H, Peng S, Adams U,
Bai Y, Huang L, Li J, Huang J, Meng S, et al: YAP/TEAD-mediated
transcription controls cellular senescence. Cancer Res.
73:3615–3624. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Kapoor A, Yao W, Ying H, Hua S, Liewen A,
Wang Q, Zhong Y, Wu CJ, Sadanandam A, Hu B, et al: Yap1 activation
enables bypass of oncogenic Kras addiction in pancreatic cancer.
Cell. 158:185–197. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Shao DD, Xue W, Krall EB, Bhutkar A,
Piccioni F, Wang X, Schinzel AC, Sood S, Rosenbluh J, Kim JW, et
al: KRAS and YAP1 converge to regulate EMT and tumor survival.
Cell. 158:171–184. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Greten FR: YAP1 takes over when oncogenic
K-Ras slumbers. Cell. 158:11–12. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Alptekin A, Abali GK, Wang Q and Bekir C:
Regulation of androgenic signaling by yes-associated protein, YAP,
in prostate cancer cells. Cancer Res. 73(8): Supplement. 7592013.
View Article : Google Scholar
|
|
31
|
Zhang L, Yang S, Chen X, Stauffer S, Yu F,
Lele SM, Fu K, Datta K, Palermo N, Chen Y and Dong J: The hippo
pathway effector, YAP, regulates motility, invasion and
castration-resistant growth of prostate cancer cells. Mol Cell
Biol. 35:1350–1362. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Xie Y, Lu W, Liu S, Yang Q, Carver BS, Li
E, Wang Y, Fazli L, Gleave M and Chen Z: Crosstalk between nuclear
MET and SOX9/β-catenin correlates with castration-resistant
prostate cancer. Mol Endocrinol. 28:1629–1639. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Wanjala J, Taylor BS, Chapinski C,
Hieronymus H, Wongvipat J, Chen Y, Nanjangud GJ, Schultz N, Xie Y,
Liu Sr, et al: Identifying actionable targets through integrative
analyses of GEM model and human prostate cancer genomic profiling.
Mol Cancer Ther. 14:278–288. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Jiao S, Wang H, Shi Z, Dong A, Zhang W,
Song X, He F, Wang Y, Zhang Z, Wang W, et al: A peptide mimicking
VGLL4 function acts as a YAP antagonist therapy against gastric
cancer. Cancer Cell. 25:166–180. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Fitamant J, Kottakis F, Benhamouche S,
Tian HS, Chuvin N, Parachoniak CA, Nagle JM, Perera RM, Lapouge M,
Deshpande V, et al: YAP inhibition restores hepatocyte
differentiation in advanced HCC, leading to tumor regression. Cell
Rep. pii:S2211–S1247. 2015.
|
|
36
|
Lin L, Sabnis AJ, Chan E, Olivas V, Cade
L, Pazarentzos E, Asthana S, Neel D, Yan JJ, Lu X, et al: The Hippo
effector YAP promotes resistance to RAF- and MEK-targeted cancer
therapies. Nat Genet. 47:250–256. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Eroglu Z and Ribas A: Combination therapy
with BRAF and MEK inhibitors for melanoma: Latest evidence and
place in therapy. Ther Adv Med Oncol. 8:48–56. 2016.PubMed/NCBI
|