Introduction
Epithelial ovarian cancer (EOC) is the most lethal
and the seventh most common gynecologic malignancy in women
worldwide (1,2). EOC represents 90% of ovarian malignant
tumors (3) and is an extremely
heterogeneous group of neoplasms that exhibit a wide range of tumor
morphology, clinical manifestations and underlying genetic
alterations (4). The most common
histological type, high-grade ovarian serous carcinoma (OSC), is
characterized by tumor protein p53 mutations and breast cancer 1
and 2 dysfunction (5). This type of
tumor is aggressive and usually diagnosed at an advanced stage, and
although OSC frequently responds to surgery and platinum-based
chemotherapy, it usually recurs (6).
Ovarian clear cell carcinoma (OCCC) is a more unusual histotype of
EOC, which is known to be intrinsically chemoresistant and is
associated with poor prognosis in advanced stages (6). Molecular alterations of OCCC are not
well known, presenting a challenge to treat this type of tumor.
However, OCCC is characterized by a unique histology, de
novo expression of hepatocyte nuclear factor-1β transcription
factor and somatic mutations of AT-rich interaction domain 1A gene,
and loss of expression (7,8). In previous years, other genetic
alterations and epigenetic modulation of signaling pathways have
been reported in OSC and OCCC (9),
including the overexpression of Notch pathway elements and histone
deacetylases (HDAC) (10). The Notch
pathway has multiple roles in cell fate determination, since it
regulates cell proliferation, differentiation, survival and
apoptosis (11,12). This signaling pathway is deregulated
in human hematological malignancies and solid tumors (13,14), and
it is also implicated in angiogenesis (15,16). Notch
signaling is a juxtacrine pathway composed by Notch receptors
(Notch1-4) and two classes of ligands, Delta-like (Dll) 1, 3 and 4
and serrate-like Jagged 1 and 2 (17–20).
Notch signaling is initiated by the binding of
Delta/Jagged ligands to Notch receptors. Through several
proteolytic cleavages, the Notch intracellular domain (NICD) is
released and activates the transcription of target genes, hairy
enhancer of split (Hes) family proteins, Hes-related
proteins (Hey) (21) as well
as cell cycle regulators, including p21cip1/waf1
(11), cyclin D1 and 3
(22), c-myc (23) and human epidermal growth factor
2 (24).
Epigenetic alterations are also involved in the
repression of tumor suppressor genes and promotion of tumorigenesis
in ovarian cancers, and HDAC inhibitor (HDACi) drugs are an
attractive therapeutic approach (25). HDACis inhibit cancer cell growth in
vitro and in vivo, revert oncogene-transformed cell
morphology, induce apoptosis and enhance cell differentiation
(26). Vorinostat (suberoylanilide
hydroxamic acid) is a HDACi (27)
that was FDA approved in 2006 for the treatment of cutaneous T-cell
lymphoma, and it has demonstrated interesting results in in
vitro models of ovarian cancer (28). However, to the best of our knowledge,
there has been no previous study addressing the effect of HDACi,
and in particular of vorinostat, on Notch signaling in ovarian
cancer. Therefore, the aim of the present study was to investigate
the modulation of the Notch pathway by vorinostat in ovarian cancer
cell lines.
Materials and methods
Cell lines and cell culture
conditions
OCCC ES2 (CRL-1978) and OSC OVCAR3 (HTB-161) cell
lines were obtained from American Type Culture Collection
(Manassas, VA, USA). The cells were incubated at 37°C in a
humidified atmosphere containing 5% CO2 in McCoy's 5A
Modified Medium (Sigma-Aldrich, St. Louis, MO, USA) supplemented
with 10% fetal bovine serum (FBS) and 1% antibiotic-antimycotic
(AA) (Invitrogen™; Thermo Fisher Scientific, Inc., Waltham, MA,
USA). The cells were cultured to 80–100% confluence prior to
detachment by incubation with 1X 0.05% trypsin-EDTA (Invitrogen™;
Thermo Fisher Scientific, Inc.) at room temperature. For the
various assays, cell number was determined using a Bürker counting
chamber. Vorinostat (catalog no., CAS 149647-78-9; Cayman Chemical,
Ann Arbor, MI, USA) was used at 5 µM to treat cells.
Immunofluorescence
Cells were cultured on glass slides with a 0.2%
gelatin coating in McCoy's 5A Modified Medium supplemented with 10%
FBS, 1% AA until 80% confluence, and were then fixed in 2%
paraformaldehyde for 15 min at 4°C. Blocking was performed with
0.2% (w/v) bovine serum albumin (BSA; catalog no., A9647;
Sigma-Aldrich) in 1X phosphate-buffered saline (PBS) for 1 h at
room temperature, and incubated with primary antibodies at 4°C
overnight [dilution, 1:100 in 0.2% (w/v) BSA in 1X PBS]. The
primary antibodies were as follows: Rabbit polyclonal anti-human
Notch1 extracellular (catalog no., ABS90; EMD Millipore, Billerica,
MA, USA), rabbit monoclonal anti-Notch1 cleaved (catalog no., 4147;
Cell Signaling Technology, Inc., Danvers, MA, USA), rabbit
polyclonal anti-Notch2 cleaved (catalog no., 07-1234; EMD
Millipore) and rabbit polyclonal anti-Notch4 (catalog no., N5163;
Sigma-Aldrich). The cells were incubated with Alexa
Fluor® 488 goat anti-rabbit secondary antibody (catalog
no., A-11008; Invitrogen™, Thermo Fisher Scientific, Inc.) for 2 h
at room temperature. The slides were mounted in VECTASHIELD
Antifade Mounting Medium with DAPI (catalog no., H-1200; Vector
Laboratories, Inc., Burlingame, CA, USA) and examined by standard
fluorescence microscopy using an Axio Imager microscope (Zeiss
GmbH, Jena, Germany). Images were acquired with AxioVision software
(version 4.5; Zeiss GmbH) and processed with ImageJ software
(version 1.44p; imagej.nih.gov/ij/).
Reverse transcription-quantitative
polymerase chain reaction (qPCR)
Total RNA was isolated from cells cultured in
complete McCoy's with or without (control conditions) vorinostat,
using RNeasy Mini Extraction kit (catalog no., 74104, Qiagen, Inc.,
Valencia, CA, USA), according to the manufacturer's protocol. cDNA
synthesis was performed with 1 µg total RNA, using random hexamers
(catalog no., 11034731001; Roche Diagnostics, Indianapolis, IN,
USA) and SuperScript II™ (200 U; Invitrogen™, Thermo Fisher
Scientific, Inc.), according to the manufacturer's protocol. The
PCR amplification conditions were as follows: 95°C for 2 min, 95°C
for 10 min, followed by 45 cycles of 95°C for 15 sec and 60°C for 1
min. Dissociation curve conditions were as follows: 95°C for 15 sec
and 60°C for 15 sec. qPCR was performed using an ABI PRISM 7900HT
Sequence Detection System (Applied Biosystems®; Thermo
Fisher Scientific, Inc.) with Power SYBR Green PCR Master Mix
(catalog no., 4367659; Applied Biosystems®, Thermo
Fisher Scientific, Inc.). The primers sequences used were as
follows: Notch1, TGGCGGGAAGTGTGAAGCGG (forward) and
GTGCGAGGCACGGGTTGGG (reverse); Notch2, CCATATGCTTCAGCCGGGATAC
(forward) and GTCTCACATTTCTGCCCTGTG (reverse); Notch3,
CTGCAAGGACCGAGTCAATGG (forward) and CGTCCACGTTGCGATCACAC (reverse);
Nocth4, CCACCTTTCACCTCTGCCTC (forward) and ACCTCACAGTCTGGGCCTAT
(reverse); Dll1, ATGCCTTCGGCCACTTCAC (forward) and
CACATCCAGGCAGGCAGAT (reverse); Dll3, GAACCCGTGTGCCAATGGAG (forward)
and GTAGGCAGAGTAGGGTCTG (reverse); Dll4, GTGGGTCAGAACTGGTTATTGGA
(forward) and TGACAGATGACCCGGTAAGAGT (reverse); Jagged1,
CGGCTTTGCCATGTGCTT (forward) and TCTTCCTCCATCCCTCTGTCA (reverse);
Jagged2, GTCGTCATCCCCTTCCAGTTC (forward) and CTCATTCGGGGTGGTATCGTT
(reverse); Hey1, GAAGTTGCGCGTTATCTGAG (forward) and
GTTGAGATGCGAAACCAGTC (reverse); Hey2, TCGCCTCTCCACAATTCAG (forward)
and TGAATCCGCATGGGCAAACG (reverse); Hes1, CGGAGGTGCTTCACTGTCAT
(forward) and ACGACACCGGATAAACCAAA (reverse); Hes5,
GAGAAAAACCGACTGCGGAAG (forward) and GACAGCCATCTCCAGGATGTC
(reverse); Hes6, GAAGTGCTGGAGCTGACGG (forward) and
CGAGCAGATGGTTCAGGAGC (reverse). All samples were run in triplicate.
Data were analyzed in SDS 2.4.1 software (Applied Biosystems;
Thermo Fisher Scientific, Inc.) and the relative expression of each
gene was quantified by comparative quantification cycle (Cq) method
(∆∆Ct) (29) using hypoxanthine
phosphoribosyltransferase gene (HPRT) as an endogenous
reference gene.
Statistical analysis
Statistical analysis was performed using Student's
t-test with GraphPad Prism software (version 5.03; GraphPad
Software, Inc., La Jolla, CA, USA). P<0.05 was considered to
indicate a statistically significant difference.
Results
Effects of vorinostat treatment on
Notch signaling in ovarian cancer cells
In order to investigate the effect of HDACi
vorinostat on the Notch signaling pathway, OCCC ES2 and OSC OVCAR3
cell lines were exposed to vorinostat for various periods of time.
The results demonstrated that vorinostat exposed OCCC ES2 cells
express increased levels of Notch2 (P<0.010), 3 and 4
(P<0.001) mRNAs, whereas Notch1 expression remained unchanged
(Fig. 1A). Increased levels of Notch1
NICD in control and vorinostat exposed cells was observed using
immunofluorescence. In Fig. 1B,
immunofluorescence revealed that vorinostat exposure increased
Notch2 and 4 protein expression but not extracellular Notch1.
Concerning Notch ligands in ES2 cells, vorinostat induced an
increased expression of Dll1, Dll3 and Dll4 (P<0.001), whereas
Jagged1 and 2 levels remained the same compared with control cells
(Fig. 1C). The significant increase
in mRNA levels of Notch downstream genes Hes1 (P<0.010),
Hes5 (P<0.001), Hey1 (P<0.010) and Hey2
(P<0.050), in cells exposed to vorinostat, confirmed the
activation of Notch pathway in this cell line (Fig. 1D). In OVCAR3 cells, vorinostat also
significantly increased Notch2, Notch3 and Notch4 (P<0.010), and
slightly decreased Notch1 mRNA levels (Fig. 2A). Protein expression of Notch2 and 4
was also higher in cells exposed to vorinostat; however,
extracellular and NICD Notch1 levels were not increased (Fig. 2B). For Notch ligands, it was observed
that there was a significant increase in Dll1 (P<0.010), Jagged1
and Jagged2 (P<0.001) mRNA levels in OVCAR3 cells exposed to
vorinostat (Fig. 2C), whereas Dll3
and 4 mRNA expression was not increased. Regarding Notch downstream
targets, it was observed that vorinostat statistically increased
Hey1, Hey2 (P<0.001), Hes1 (P<0.050) and
Hes5 (P<0.001), mRNA levels compared with control cells
(Fig. 2D); however, Hes6
levels were not increased.
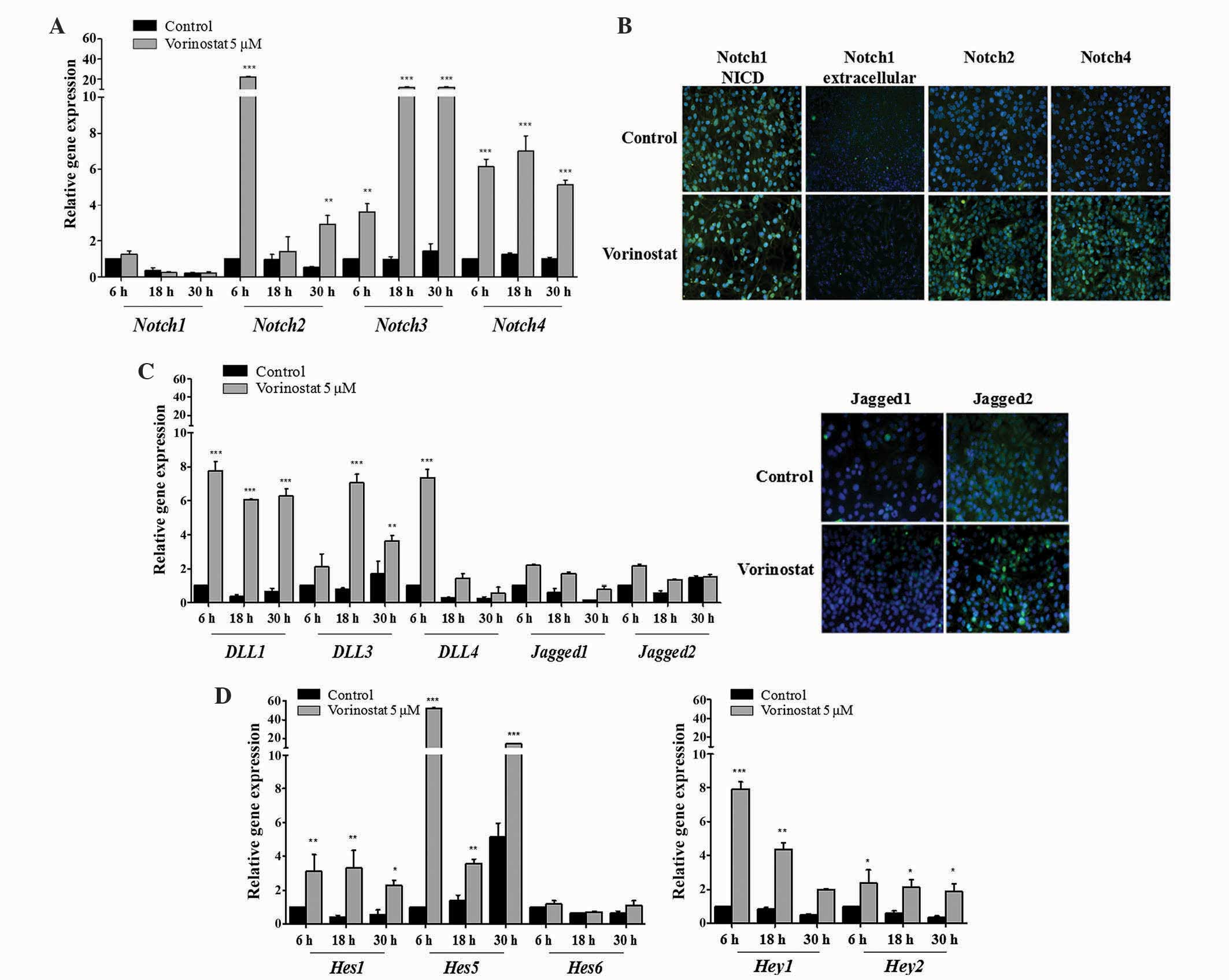 | Figure 1.Vorinostat increases the mRNA
expression of Notch receptors, Dll/Jagged ligands and
Hey/Hes downstream target genes. Ovarian clear cell
carcinoma ES2 cells were grown in the absence and presence of
vorinostat (5 µM) for 6, 18 and 30 h, following starvation, with
medium supplemented with 1% fetal bovine serum. (A) Notch receptor
expression levels in ES2 cells. RT-qPCR revealed that Notch2,
Notch3 and Notch4 mRNA expression was increased following
vorinostat exposure, whereas Notch1 was not differentially
expressed. (B) Representative staining of Notch receptors by
immunofluorescence. It was observed that there was an increase of
Notch2 and Notch4 protein following exposure to vorinostat
(magnification, ×200). Nuclei are stained with
4′,6-diamidino-2-phenylindole (blue) and Notch receptors with
fluorescein isothiocyanate (green). (C) Notch ligand expression
levels in ES2 cells. RT-qPCR indicated that Dll1, Dll3, Dll4 mRNA
expression levels in cells treated with vorinostat were increased
compared with cells without vorinostat treatment (magnification,
×200). Jagged1 and Jagged2 mRNA expression levels were not
significantly different. (D) Notch downstream target gene
expression levels. RT-qPCR revealed that Hes1, Hes5,
Hey1 and Hey2 mRNA expression levels in cells treated
with vorinostat were increased compared with cells without
vorinostat treatment. Hes6 had a similar expression under
all conditions. RT-qPCR was normalized to the hypoxanthine
phosphoribosyltransferase gene. Data are presented as the mean ±
standard deviation of triplicate experiments. *P<0.05;
**P<0.01; ***P≤0.001 vs. control cells. Dll, Delta-like;
RT-qPCR, reverse transcription-quantitative polymerase chain
reaction; Hes, hairy enhancer of split; Hey,
Hes-related proteins. |
 | Figure 2.Vorinostat increases the mRNA
expression of Notch receptors, ligands and downstream targets in
ovarian serous carcinoma OVCAR3 cell line. The cells were grown in
the absence and presence of vorinostat (5 µM) for 6, 18 and 30 h,
following starvation, with medium supplemented with 1% fetal bovine
serum. (A) Notch receptor expression levels in OVCAR3 cells.
RT-qPCR indicated that Notch2, Notch3 and Notch4 mRNA levels were
increased following vorinostat exposure. (B) Representative
staining of Notch receptors by immunofluorescence. Vorinostat
slightly increased the protein expression of Notch receptors 2 and
4, and decreased Notch1 expression following exposure to vorinostat
(magnification, ×200). Nuclei are stained with
4′,6-diamidino-2-phenylindole (blue) and Notch receptors with
fluorescein isothiocyanate (green). (C) Notch ligand expression
levels in OVCAR3 cell line. RT-qPCR revealed that Dll1, Jagged1 and
Jagged2 mRNA expression levels in cells treated with vorinostat
were increased compared with cells not treated with vorinostat
(magnification, ×200). Dll3 and Dll4 mRNA expression levels were
not differentially expressed. (D) Notch downstream target gene
expression levels. RT-qPCR revealed that Hes1, Hes5,
Hey1 and Hey2 mRNA expression levels in cells treated
with vorinostat were increased compared with cells without
vorinostat treatment. Hes6 mRNA expression levels were not
affected by vorinostat treatment. RT-qPCR was normalized to
hypoxanthine phosphoribosyltransferase. Data are presented as the
mean ± standard deviation of triplicate experiments. *P<0.05;
**P<0.01; ***P≤0.001 vs. control cells. Dll, Delta-like;
RT-qPCR, reverse transpiration-quantitative polymerase chain
reaction; Hes, hairy enhancer of split; Hey,
Hes-related proteins. |
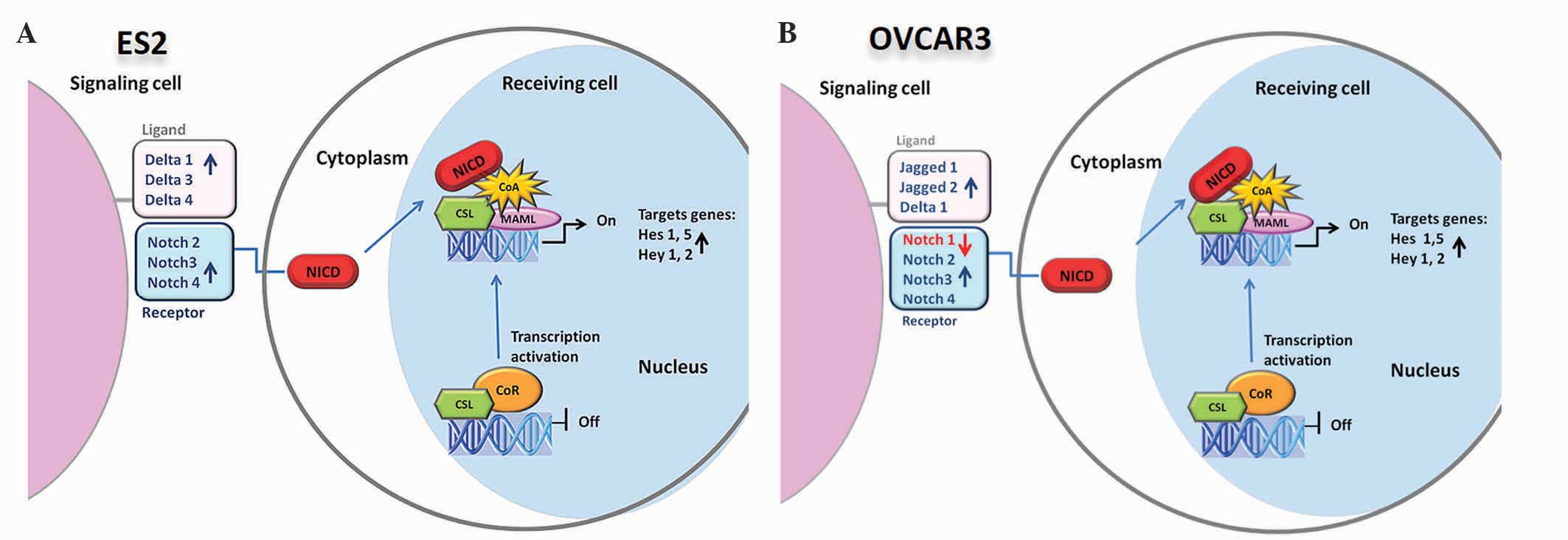
Overall, the present results reveal that vorinostat
activated the Notch pathway in OCCC and OSC cell lines; however,
this activation was through a different expression panel of Notch
ligands in the two cell lines. In OCCC, the activation of Notch
pathway appears to occur through Dll1, Dll2 and Dll3, whereas in
OSC Dll1 and Jagged 1 and 2 ligand families appear to be involved.
Nevertheless, the present results revealed that the activation of
Notch pathway by vorinostat, in OCCC and OSC cell lines, culminated
in the increased expression of the same downstream targets,
Hey1, Hey2, Hes1 and Hes5.
Discussion
The Notch signaling pathway is an important cell
signaling system that is activated in various types of cancer,
including ovarian carcinoma (5,30,31). Notch1 overexpression has been
demonstrated in several studies to promote ovarian cancer cell
proliferation (32–34), and this is primarily associated with
increased levels of Notch1 NICD (33). In the literature, Notch3 has also been
demonstrated to be active in certain ovarian cancer cell lines and
its overexpression was described in 20% of OSC (35,36). In
addition, Notch3 was revealed to increase following tumor
chemotherapy, and its overexpression is associated with tumor
aggressiveness and poor prognosis of patients (36).
The present study aimed to investigate the effect of
vorinostat, an HDAC inhibitor, on the Notch signaling pathway in
ovarian cancer cells. For this purpose, the present study used cell
lines from two different histological types of EOC: OSC, the most
prevalent and aggressive EOC type; and OCCC, which is a rare type
of EOC that is highly resistant to chemotherapy (37).
No significant alteration in Notch1 mRNA levels
following vorinostat exposure was identified in the two different
ovarian cancer cell lines; however, high levels of cleaved Notch1
were detected in OCCC cell line (ES2). In addition, Notch3 was
demonstrated to be upregulated following vorinostat exposure in the
two cell lines. Furthermore, the present study revealed that
vorinostat activates the Notch pathway through specific Notch
ligands in OCCC and OSC cell lines; Dll1, 3 and 4 are activated in
OCCC, while Dll1 and Jagged1 and 2 are activated in OSC. The
present results are supported by a previous studies that
demonstrated altered Notch signaling in OSC, due to increased
expression of Jagged1 and 2 ligands (5,38).
The Hes and Hey gene families are the
best characterized canonical Notch target genes, and the activation
of Notch signaling upregulates their transcription (39); therefore, Hes and Hey
mRNA expression may be considered as markers for Notch activation.
The present study demonstrated that Hes and Hey Notch
target genes are overexpressed in EOC cells following exposure to
vorinostat. The present data reveals that vorinostat induces the
overexpression of Hes and Hey Notch target genes in
EOC cells, due to Notch signaling activation. In addition, the
redundancy of Notch, Delta and Jagged elements expressed in EOC
cell lines was shown (Fig. 3). Hence,
no matter which panel of receptors or ligands is expressed on the
cell membrane, the downstream target genes are always expressed in
the presence of vorinostat. This upregulation can aid our
understanding of the mechanism underlying the failure of vorinostat
therapy in ovarian carcinoma.
In conclusion, the present findings illustrate the
redundancy of Notch pathway in ovarian cancer, and suggest that
disruption of histone acetylation may not be a useful therapeutic
strategy in these carcinomas.
References
|
1
|
Reinhardt MJ: Gynecologic tumorsPET in
Oncology. Dresel S: 170. Springer; Berlin: pp. 141–150. 2008,
View Article : Google Scholar
|
|
2
|
Bast RC, Hennessy B and Mills GB: The
biology of ovarian cancer: New opportunities for translation. Nat
Rev Cancer. 9:415–428. 2009. View
Article : Google Scholar : PubMed/NCBI
|
|
3
|
Siegel R, Naishadham D and Jemal A: Cancer
statistics, 2013. CA Cancer J Clin. 63:11–30. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Smolle E, Taucher V, Pichler M, Petru E,
Lax S and Haybaeck J: Targeting signaling pathways in epithelial
ovarian cancer. Int J Mol Sci. 14:9536–9555. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Cancer Genome Atlas Research Network.
Integrated genomic analyses of ovarian carcinoma. Nature.
474:609–615. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
DelCarmen MG, Birrer M and Schorge JO:
Clear cell carcinoma of the ovary: A review of the literature.
Gynecol Oncol. 126:481–490. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Ayhan A, Mao TL, Seckin T, Wu CH, Guan B,
Ogawa H, Futagami M, Mizukami H, Yokoyama Y, Kurman RJ and Shih
IeM: Loss of ARID1A expression is an early molecular event in tumor
progression from ovarian endometriotic cyst to clear cell and
endometrioid carcinoma. Int J Gynecol Cancer. 22:1310–1305. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Guan B, Wang TL and Shih IM: ARID1A, a
factor that promotes formation of SWI/SNF-mediated chromatin
remodeling, is a tumor suppressor in gynecologic cancers. Cancer
Res. 71:6718–6727. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Kotsopoulos IC, Papanikolaou A,
Lambropoulos AF, Papazisis KT, Tsolakidis D, Touplikioti P and
Tarlatzis BC: Serous ovarian cancer signaling pathways. Int J
Gynecol Cancer. 24:410–417. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Jin KL, Pak JH, Park JY, Choi WH, Lee JY,
Kim JH and Nam JH: Expression profile of histone deacetylases 1, 2
and 3 in ovarian cancer tissues. J Gynecol Oncol. 19:185–190. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Ranganathan P, Weaver KL and Capobianco
AJ: Notch signalling in solid tumours: A little bit of everything
but not all the time. Nat Rev Cancer. 11:338–351. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Takebe N, Nguyen D and Yang SX: Targeting
notch signaling pathway in cancer: Clinical development advances
and challenges. Pharmacol Ther. 141:140–149. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Nickoloff BJ, Osborne BA and Miele L:
Notch signaling as a therapeutic target in cancer: A new approach
to the development of cell fate modifying agents. Oncogene.
22:6598–6608. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Aster JC and Blacklow SC: Targeting the
Notch pathway: Twists and turns on the road to rational
therapeutics. J Clin Oncol. 30:2418–2420. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Li JL and Harris AL: Notch signaling from
tumor cells: A new mechanism of angiogenesis. Cancer Cell. 8:1–3.
2005. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Dufraine J, Funahashi Y and Kitajewski J:
Notch signaling regulates tumor angiogenesis by diverse mechanisms.
Oncogene. 27:5132–5137. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Fleming RJ, Purcell K and
Artavanis-Tsakonas S: The NOTCH receptor and its ligands. Trends
Cell Biol. 7:437–441. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Lendahl U: A growing family of Notch
ligands. Bioessays. 20:103–107. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Gray GE, Mann RS, Mitsiadis E, Henrique D,
Carcangiu ML, Banks A, Leiman J, Ward D, IshHorowitz D and
Artavanis-Tsakonas S: Human ligands of the Notch receptor. Am J
Pathol. 154:785–794. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Yamaguchi E, Chiba S, Kumano K, Kunisato
A, Takahashi T, Takahashi T and Hirai H: Expression of Notch
ligands, Jagged1, 2 and Delta1 in antigen presenting cells in mice.
Immunol Lett. 81:59–64. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Kao HY, Ordentlich P, KoyanoNakagawa N,
Tang Z, Downes M, Kintner CR, Evans RM and Kadesch T: A histone
deacetylase corepressor complex regulates the Notch signal
transduction pathway. Genes Dev. 12:2269–2277. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Ronchini C and Capobianco AJ: Induction of
cyclin D1 transcription and CDK2 activity by Notch (ic):
Implication for cell cycle disruption in transformation by Notch
(ic). Mol Cell Biol. 21:5925–5934. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Weng AP, Millholland JM, YashiroOhtani Y,
Arcangeli ML, Lau A, Wai C, Del Bianco C, Rodriguez CG, Sai H,
Tobias J, et al: c-Myc is an important direct target of Notch1 in
T-cell acute lymphoblastic leukemia/lymphoma. Genes Dev.
20:2096–2109. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Hirose H, Ishii H, Mimori K, Ohta D,
Ohkuma M, Tsujii H, Saito T, Sekimoto M, Doki Y and Mori M: Notch
pathway as candidate therapeutic target in Her2/Neu/ErbB2
receptor-negative breast tumors. Oncol Rep. 23:35–43.
2010.PubMed/NCBI
|
|
25
|
Takai N and Narahara H: Histone
deacetylase inhibitor therapy in epithelial ovarian cancer. J
Oncol. 2010:4584312010. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Bolden JE, Peart MJ and Johnstone RW:
Anticancer activities of histone deacetylase inhibitors. Nat Rev
Drug Discov. 5:769–784. 2006. View
Article : Google Scholar : PubMed/NCBI
|
|
27
|
Richon VM: Cancer biology: mechanism of
antitumour action of vorinostat (suberoylanilide hydroxamic acid),
a novel histone deacetylase inhibitor. Br J Cancer. 95(Suppl 1):
S2–S6. 2006. View Article : Google Scholar
|
|
28
|
Cooper AL, Greenberg VL, Lancaster PS, Van
Nagell JR Jr, Zimmer SG and Modesitt SC: In vitro and in vivo
histone deacetylase inhibitor therapy with suberoylanilide
hydroxamic acid (SAHA) and paclitaxel in ovarian cancer. Gynecol
Oncol. 104:596–601. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Livak KJ and Schmittgen TD: Analysis of
relative gene expression data using real-time quantitative PCR and
the 2(−Delta Delta C(T)) Method. Methods. 25:402–408. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Bolós V, Grego-Bessa J and de la Pompa JL:
Notch signaling in development and cancer. Endocr Rev. 28:339–363.
2007. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
McAuliffe SM, Morgan SL, Wyant GA, Tran
LT, Muto KW, Chen YS, Chin KT, Partridge JC, Poole BB, Cheng KH, et
al: Targeting Notch, a key pathway for ovarian cancer stem cells,
sensitizes tumors to platinum therapy. Proc Natl Acad Sci USA.
109:E2939–E2948. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Rose SL, Kunnimalaiyaan M, Drenzek J and
Seiler N: Notch 1 signaling is active in ovarian cancer. Gynecol
Oncol. 117:130–133. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Hopfer O, Zwahlen D, Fey MF and Aebi S:
The Notch pathway in ovarian carcinomas and adenomas. Br J Cancer.
93:709–718. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Capaccione KM and Pine SR: The Notch
signaling pathway as a mediator of tumor survival. Carcinogenesis.
34:1420–1430. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Park JT, Li M, Nakayama K, Mao TL,
Davidson B, Zhang Z, Kurman RJ, Eberhart CG, Shih IeM and Wang TL:
Notch3 gene amplification in ovarian cancer. Cancer Res.
66:6312–6387. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Park JT, Chen X, Tropè CG, Davidson B,
Shih IM and Wang TL: Notch3 overexpression is related to the
recurrence of ovarian cancer and confers resistance to carboplatin.
Am J Pathol. 177:1087–1094. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Tan DSP, Miller RE and Kaye SB: New
perspectives on molecular targeted therapy in ovarian clear cell
carcinoma. Br J Cancer. 108:1553–1559. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Jung SG, Kwon YD, Song JA, Back MJ, Lee
SY, Lee C, Hwang YY and An HJ: Prognostic significance of Notch 3
gene expression in ovarian serous carcinoma. Cancer Sci.
101:1977–1983. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Iso T, Kedes L and Hamamori Y: HES and
HERP families: Multiple effectors of the Notch signaling pathway. J
Cell Physiol. 194:237–255. 2003. View Article : Google Scholar : PubMed/NCBI
|