Introduction
The ATPases associated with various cellular
activities (AAA+) family of proteins comprises a functionally
different group of enzymes that are involved in an array of
cellular processes, including protein degradation, protein-complex
disassembly and DNA replication (1).
The AAA+ proteins are present in all kingdoms, and are
characterized by the presence of conserved AAA+ domains that
contain Walker A and Walker B motifs, followed by a conserved
region called second region of homology (2). The majority of AAA+ proteins form
hexamers to exert their biological functions. The chemical energy
generated by adenosine triphosphate (ATP) hydrolysis by this family
of proteins induces conformational changes in the substrate
proteins to modulate their functions (3,4).
A previous study reported that certain AAA+ enzymes
are associated with tumor progression (5). For example, reptin [also known as
RuvB-like (RUVBL) 2] and pontin (RUVBL1) have been demonstrated to
be overexpressed in several tumors (5). These proteins interact with c-Myc or
β-catenin, thus modulating their transcriptional activities to
promote tumor progression (6). AAA+
proteins hydrolyze ATP, and thus, chemical inhibitors that disrupt
the activities of cancer-associated AAA+ proteins may represent
promising anti-cancer drugs.
Thyroid hormone receptor interactor 13 (TRIP13, also
known as 16E1BP and pachytene checkpoint 2) is a member of the AAA+
family proteins and is conserved in a wide range of species
(7). It was first identified as a
protein that interacts with human papilloma virus E1 proteins by a
yeast two-hybrid analysis, but the physiological function of the
interaction remains unknown (7).
Accumulating studies have demonstrated that TRIP13 plays pivotal
roles in meiotic recombination and DNA repair in plants, yeast,
worms and mice (8–12). TRIP13 forms a stable hexameric ring,
and ATP binding, as well as ATP hydrolysis, are critical for the
function of the protein (13).
Previous studies have revealed that TRIP13 is a novel component of
the spindle assembly checkpoint (SAC) pathway (14–17), which
is crucial for the accurate distribution of duplicated chromosomes
(18). Defects in the SAC pathway
induce failure in chromosome separation and result in aneuploidy,
which eventually leads to cellular apoptosis or transformation
(19). TRIP13 was also reported to
promote nonhomologous end-joining, and its overexpression resulted
in cellular transformation and resistance to chemotherapeutic
agents, indicating that aberrant expression of TRIP13 may be
associated with tumor progression (20).
The present study examined whether TRIP13 has a
tumor-promoting function in colorectal cancer (CRC) using human CRC
tissue samples and cell lines. The results demonstrated that TRIP13
is highly expressed in CRC tissues and that its depletion
suppresses the malignant characteristics of CRC cells.
Materials and methods
Cells and antibodies
HCT116 and DLD1 cells were purchased from the
American Type Culture Collection (Manassas, VA, USA) and were
cultured in Dulbecco's modified Eagle medium (DMEM; Wako Pure
Chemical Industries, Ltd., Osaka, Japan) and RPMI 1640 medium (Wako
Pure Chemical Industries, Ltd.), respectively, supplemented with
10% fetal bovine serum (FBS; Biowest, Nuaille, France) and
antibiotics. Cells were authenticated by short tandem repeat
analysis using GenePrint® 10 System (Promega
Corporation, Madison, WI, USA) in 2014. HEK293T cells (RIKEN
BioResource Center, Tsukuba, Japan) were used to produce
recombinant retroviruses and were maintained in DMEM with 10% FBS.
The anti-TRIP13 antibody (cat. no. A303-605A) was obtained from
Bethyl Laboratories (Montgomery, TX, USA) and the anti-Flag
antibody (cat. no. 1E6) from Wako Pure Chemical Industries, Ltd.
Cells were incubated with the antibodies for 1 h at room
temperature for immunoblot analysis.
Generation of stable cell lines
Full-length TRIP13 RNA was extracted from HCT116
cells using the RNeasy Mini Kit (Qiagen, Venlo, The Netherlands).
RNA was amplified by polymerase chain reaction (PCR) following
reverse-transcription using PrimeScript Reverse Transcriptase
(Takara Bio, Inc., Otsu Japan) and was cloned into the pQCXIP
retroviral vector (Clontech Laboratories, Inc., Mountainview, CA,
USA) with an N-terminal Flag tag. The following primer sequences
were used to clone TRIP13: Forward,
5′-ACTATCTCGAGATGGACGAGGCCGTGGGCGAC-3′ and reverse,
5′-TCGATAGCGGCCGCTCAGATGTAAGCTGCAAGCTTC−3′. PCR was performed using
PrimeSTAR Max DNA Polymerase (Takara Bio, Inc.). PCR was performed
under the following conditions: Denaturation at 94°C for 10 sec,
annealing at 55°C for 30 sec and extension at 72°C for 1 min. To
generate a recombinant retrovirus, HEK293T cells were transfected
with pQCXIP-Flag-TRIP13 together with pVPack-GP and pVPack-Ampho
vectors (Promega Corporation) using Lipofectamine 2000 (Invitrogen;
Thermo Fisher Scientific, Inc., Waltham, MA, USA). Following 48 h,
the supernatants were added to HCT116 or DLD1 cells in the presence
of 2 µg/ml polybrene (Sigma-Aldrich; Merck Millipore, Darmstadt,
Germany), and the infected cells were selected with 1 µg/ml
puromycin for 3 days.
Immunoblot analysis
When the cells had reached 80% confluence, they were
lysed using Laemmli sample buffer and boiled for 5 min. The protein
concentrations of the lysates were measured using the RC-DC Protein
Assay (Bio-Rad Laboratories, Hercules, CA, USA). Equal protein
quantities of the lysates were then separated on 0.01% SDS-PAGE
gels and transferred to polyvinylidene fluoride membranes (Merck
Millipore). Next, the membranes were blocked with 0.5% skim milk
followed by incubation with anti-TRIP13 (1:1,000 dilution) and
anti-β-actin antibody (1:3,000 dilution; cat. no. A1978;
Sigma-Aldrich; Merck Millipore) primary antibodies for 1 h at room
temperature. Subsequently, the membranes were incubated with
horseradish peroxidase-conjugated secondary antibodies (1:3,000
dilution; cat. nos. 7074 and 7076; Cell Signaling Technology, Inc.,
Danvers, MA, USA) for 1 h at room temperature. Blots were
visualized using Chemi-Lumi One Super (Nacalai Tesque, Inc., Kyoto,
Japan).
Small interfering RNA (siRNA)
transfection
The siRNA sequences used to deplete TRIP13 were
5′-GCUGGUAACCAAGAUGUUUTT-3′ (siTRIP13-1),
5′-CCCAUCGAUUUGAGUGCAUTT-3′ (siTRIP13-2) and
5′-GGAUGCAUAAUGCCAGCAATT-3′ (siTRIP13-3). The sequence of the
control siRNA targeting luciferase was 5′-CUUACGCUGAGUACUUCGATT-3′.
A total of 20 nM of siRNAs were transfected into the cells using
RNAiMAX (Invitrogen; Thermo Fisher Scientific, Inc.) according to
the manufacturer's protocol. All siRNAs were purchased from
Hokkaido System Science Co., Ltd. (Sapporo, Japan).
Cell invasion assay
To measure cell invasion using Boyden chambers, a
filter was pre-coated with Matrigel, and 2×105 HCT116 or
DLD1 cells transfected with the aforementioned siRNAs were seeded
onto the upper surface of the chamber. Cells were fixed with 70%
methanol 24 h later and stained with 0.5% crystal violet. The cells
that had invaded the lower surface of the filters were counted in
five randomly selected fields. Three independent experiments were
performed, and the data were represented as the mean ± standard
deviation (SD).
Cell migration assay
Cell migration was evaluated using Boyden chambers.
siRNA-transfected cells (5.0×104) were seeded onto the
upper surface of the chamber. The lower surface of the filter was
coated with fibronectin (Sigma-Aldrich; Merck Millipore). Cells
were fixed with 70% methanol 6 h later and stained with 0.5%
crystal violet. The cells that had migrated to the lower surface of
the filters were counted in five randomly selected fields in three
independent experiments.
Patients and ethics statement
CRC tissue samples and normal colorectal tissue
sections were obtained from patients who had undergone surgery at
Nagoya University Hospital (Nagoya, Japan) between May 2010 and
October 2014. The study was approved by the institutional review
board of Nagoya University Hospital and conformed to the standards
set by the Declaration of Helsinki. All participants provided
written informed consent to participate in the study.
Reverse transcription-quantitative PCR
(RT-qPCR) analysis
RNA was extracted from CRC samples and cells using
the RNeasy Mini kit (Qiagen GmbH, Hilden, Germany), and cDNA was
generated using PrimeScript Reverse Transcriptase (Takara Bio,
Inc.). The CRC tissue samples were obtained from patients at Nagoya
University Hospital who had provided informed consent. qPCR was
performed using the SYBR Premix Ex Taq™ II (Takara Bio, Inc.), and
the Thermal Cycler Dice Real Time System TP800 (Takara Bio, Inc.)
was used for the analysis. PCR was performed under the following
conditions: 95°C for 10 sec and 60°C for 30 sec. The relative
messenger RNA (mRNA) expression levels were normalized to the level
of glyceraldehyde 3-phosphate dehydrogenase (GAPDH) using
LightCyclerR® Nano software 1.0 (Roche Diagnostics,
Tokyo, Japan). The sequences of the primers used to amplify each
gene were 5′-AGGTGGAGGAGTGGGTGTCGCTGTT-3′ (forward) and
5′-CCGGGAAACTGTGGCGTGATGG-3′ (reverse) for GAPDH, and
5′-CTGTCTCTGGCAGTGGACAAG-3′ (forward) and
5′-TTGGTTTGCAGAAGGGATTC-3′ (reverse) for TRIP13.
Cell proliferation assay
Cells were cultured in 96-well plates, and the
number of viable cells at the indicated times was evaluated using
Cell Counting Kit-8 (Dojindo Molecular Technologies, Inc.,
Kumamoto, Japan).
5-ethynyl-2′-deoxyuridine (EdU)
incorporation assay
CRC cells were transfected with siRNAs, and EdU
incorporation assays were performed 48 h later using a
Click-iT® Plus EdU Alexa Fluor® 594 Imaging
kit (Thermo Fisher Scientific, Inc.). Briefly, siRNA-transfected
cells were incubated with 20 mM EdU for 24 h and fixed with
formaldehyde. The cells were permeabilized with 0.5% Triton X-100
and stained with Hoechst in a reaction cocktail prepared according
to the manufacturer's protocol. The cells were then imaged by
fluorescence microscopy (BX60; Olympus Corporation, Tokyo, Japan),
and the percentage of EdU-positive cells was evaluated.
Statistical analysis
Data were expressed as the mean ± SD. Comparisons
between groups were performed using unpaired t-tests.
P<0.05 was considered to indicate a statistically significant
difference.
Results
TRIP13 is expressed in CRC tissues and
cell lines
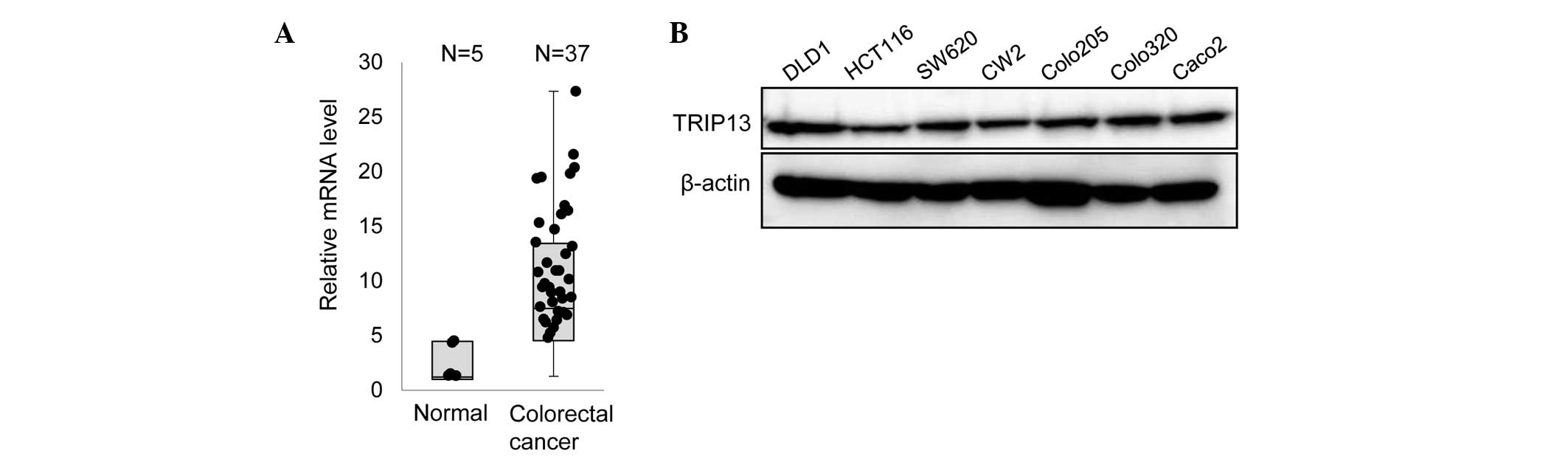
To examine whether TRIP13 has any role in CRC, the
mRNA level of TRIP13 in CRC tissue specimens was first examined by
RT-qPCR. As indicated in Fig. 1A,
higher expression of TRIP13 was observed in multiple CRC samples
compared with normal tissues. Next, the expression of TRIP13 in
several CRC cell lines was examined. Immunoblot analyses
demonstrated that all the cell lines expressed a similar amount of
TRIP13 (Fig. 1B).
Depletion of TRIP13 suppresses cell
proliferation
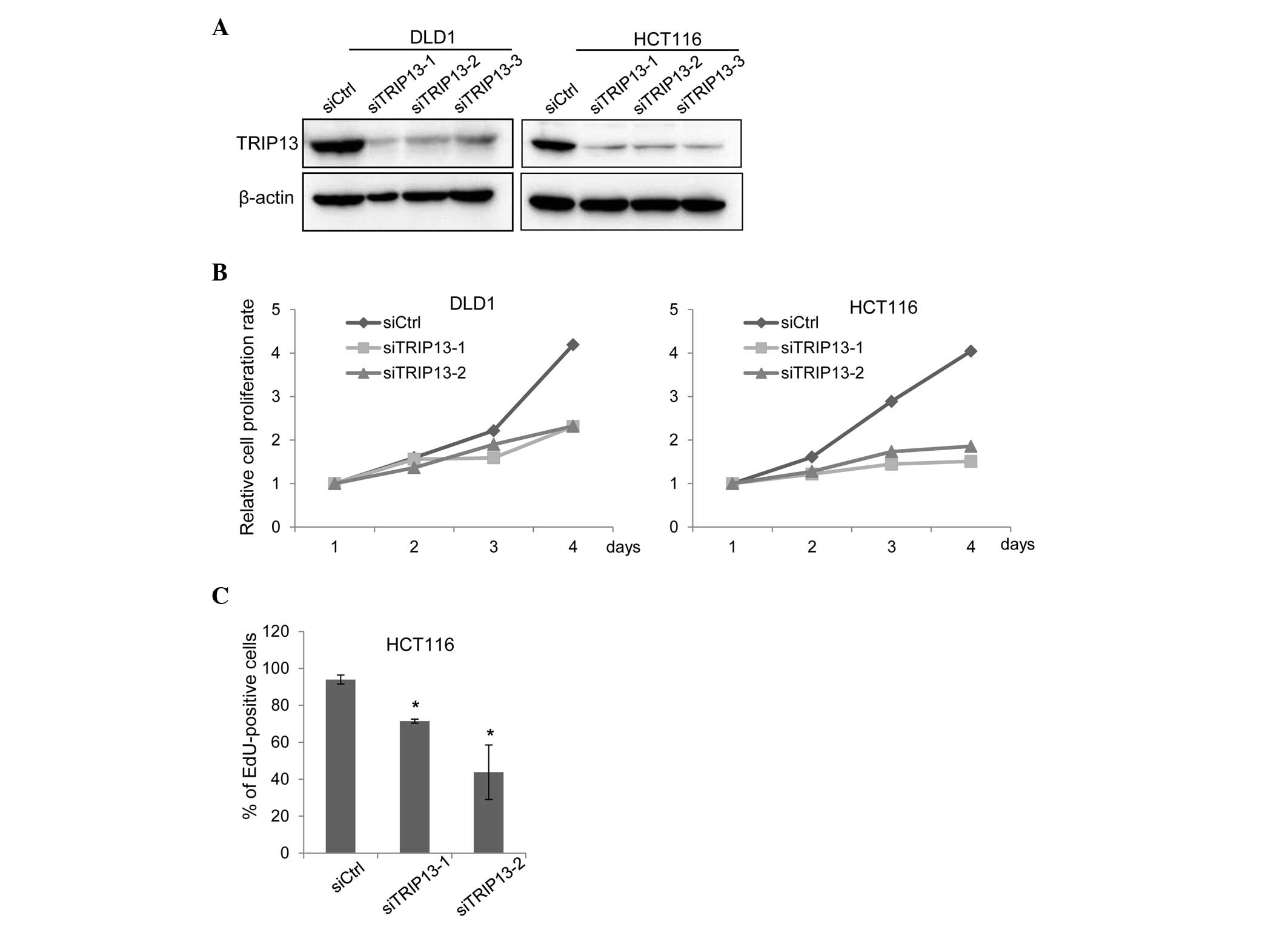
To determine whether TRIP13 has any tumor-promoting
functions, TRIP13 expression was depleted in DLD1 and HCT116 cells
using siRNAs that targeted different regions of TRIP13 mRNA.
Transfection of either siRNA sufficiently knocked down TRIP13
expression (Fig. 2A). In the absence
of TRIP13 expression, the proliferation of both cell lines was
significantly suppressed (Fig. 2B).
Reduced cell proliferation can be induced by a delay in cell cycle
progression or by the promotion of cellular apoptosis (21). To determine how TRIP13 depletion
suppressed cell proliferation, a terminal deoxynucleotidyl
transferase dUTP nick end labeling (TUNEL) assay and an EdU
incorporation assay were performed. The TUNEL assay detects
fragmented DNA induced by apoptosis, while the EdU incorporation
assay evaluates cell cycle progression (21). Cells that progress through the S phase
incorporate the thymidine analog EdU; thus, proliferating cells can
be detected based on the level of EdU incorporated (21). As shown in Fig. 2C, TRIP knockdown reduced the ratio of
cells that incorporated EdU; however, no increase in the level of
apoptosis caused by TRIP13 depletion was observed (data not
shown).
TRIP13 knockdown inhibits cell
migration and invasion
The effects of TRIP13 knockdown on cell migration
were next studied using a modified Boyden chamber. DLD1 and HCT116
cells were transfected with siRNAs, and 72 h later, the cells were
placed on the upper surface of the filter and allowed to migrate to
the bottom surface, which was coated with fibronectin. The cells
that migrated to the bottom surface were counted to evaluate cell
migration. As shown in Fig. 3A,
TRIP13 depletion significantly suppressed the migration of both
cell lines. The invasion of TRIP13-knockdown cells was also
examined using Matrigel-coated Boyden chambers. The invasion of
both DLD1 and HCT116 cells was clearly suppressed by TRIP13
depletion (Fig. 3B).
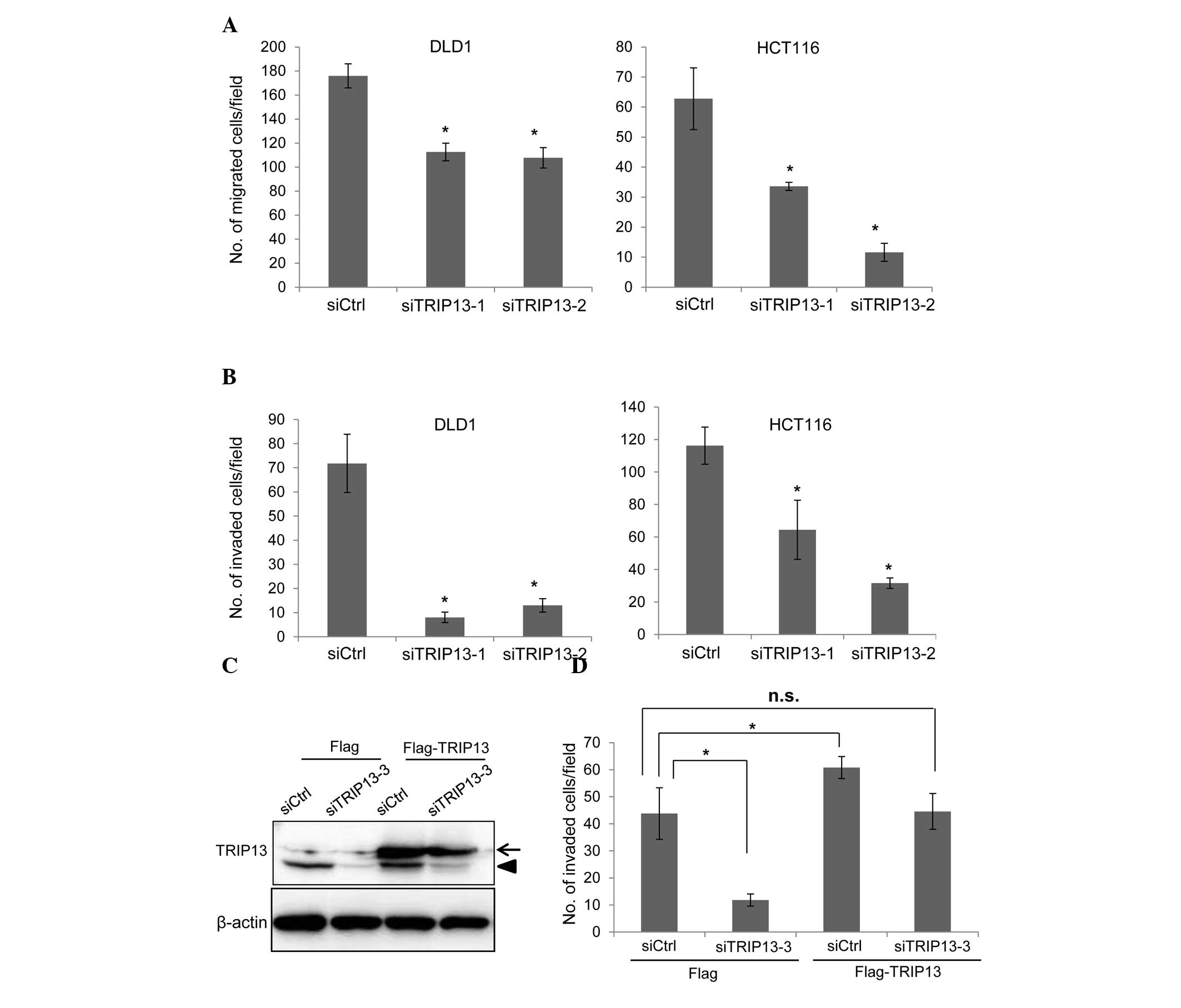 | Figure 3.Knockdown of TRIP13 inhibits cell
migration and invasion. (A) Cells were transfected with siRNAs, and
72 h later, cell migration was examined. The graph shows the
average number of migrated cells per field. Three independent
experiments were performed, and the data are shown as the mean ± SD
(*P<0.05). (B) siRNA-transfected cells were subjected to an
invasion assay. The graph shows the average number of invaded cells
per field. Three independent experiments were performed, and the
data are shown as the mean ± SD (*P<0.05). (C) HCT116 cells that
constitutively expressed Flag tag or Flag-TRIP13 were established
by retroviral infection. Cells were transfected with control siRNA
or with TRIP13 siRNA (siTRIP13-3), and the expression of TRIP13 was
examined by immunoblotting 72 h later. siTRIP13-3 targets the
3′-untranslated region of TRIP13 messenger RNA. The arrow indicates
Flag-TRIP13 while the arrowhead indicates endogenous TRIP13. (D)
Cells transfected with siRNAs were subjected to an invasion assay.
The graph shows the average number of invaded cells per field.
Three independent experiments were performed, and the data are
shown as the mean ± SD (*P<0.05). SD, standard deviation;
TRIP13, thyroid hormone receptor interactor 13; siRNA, small
interfering RNA; Ctrl, control; n.s., not significant. |
To confirm that TRIP13 is associated with the
invasion of cancer cells, a rescue experiment was performed. HCT116
cells that constitutively expressed either Flag or Flag-TRIP13 were
generated by retrovirus infection. To specifically deplete
endogenous TRIP13, an additional siRNA (siTRIP13-3) that targeted
the 3′-untranslated region of TRIP13 mRNA was used. Transfection of
the siTRIP13-3 depleted endogenous TRIP13, but the expression level
of Flag-TRIP13 was not affected by this siRNA (Fig. 3C). The invasion of siRNA-transfected
cells was then examined using Matrigel-coated Boyden chambers. The
invasion of Flag-expressing HCT116 cells was significantly reduced
by transfection with siTRIP13-3 (Fig.
3D). Exogenous expression of Flag-TRIP13 significantly promoted
cell invasion (Fig. 3D), while
depletion of endogenous TRIP13 in Flag-TRIP13-expressing cells
suppressed cell invasion to a level similar to that of control
siRNA-transfected Flag-expressing cells (Fig. 3D). These results indicate that the
reduction in cell invasion caused by the TRIP13 siRNAs was mediated
by the depletion of TRIP13.
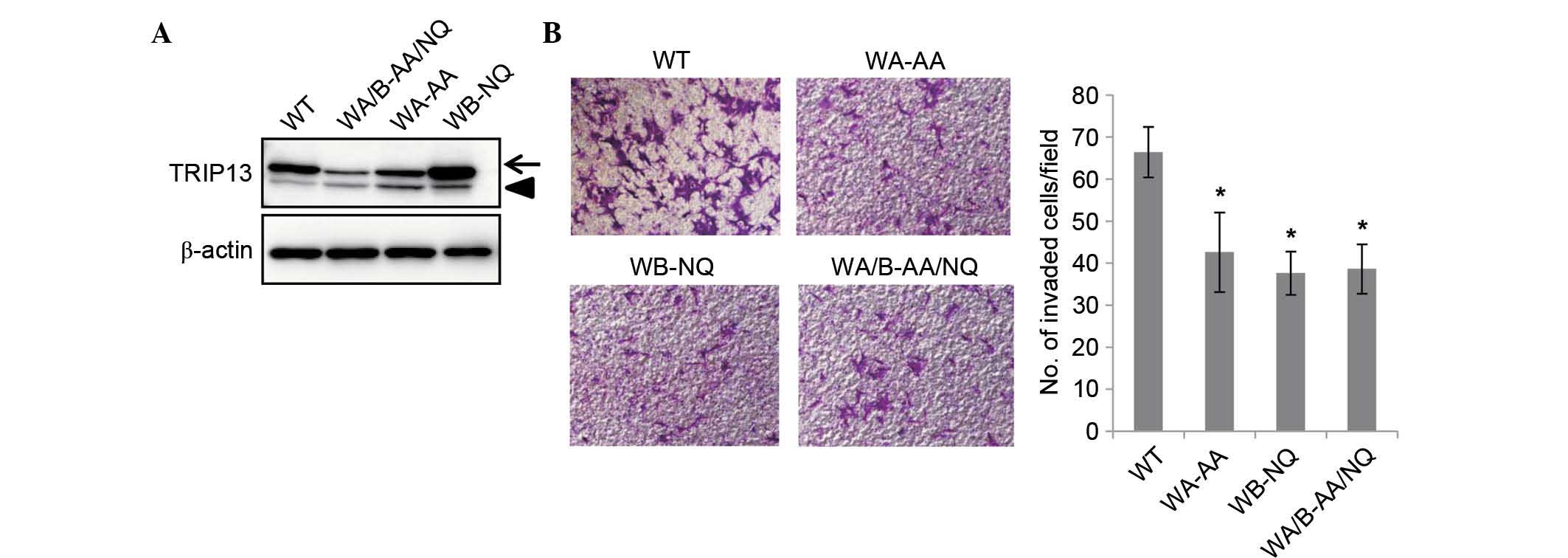
The catalytic activity of TRIP13 is
required for the promotion of cell invasion
It was next examined whether the catalytic activity
of TRIP13 was required for the promotion of cell invasion.
Mutations in the Walker A motif are known to disrupt ATP binding,
whereas Walker B motif mutations disrupt ATP hydrolysis (2). Both were shown to be essential for the
function of TRIP13 in yeast (13).
The present study generated TRIP13 mutants that have mutations
either in the Walker A or Walker B motifs, or in both motifs,
including TRIP13-GK/AA, which has glycine 184 and lysine 185 in the
Walker A motif substituted with alanine; and TRIP13-DE/NQ, which
has aspartic acid 252 and glutamic acid 253 in the Walker B motif
substituted with asparagine and glutamine, respectively; and
TRIP13-GK/AA-DE/NQ, which has both mutations. HCT116 cells that
constitutively expressed Flag-tagged versions of each mutant were
established by retrovirus infection. Although the expression of
TRIP13-GK/AA and TRIP13-GK/AA-DE/NQ was lower than that of
Flag-TRIP13, the expression level was higher than that of
endogenous TRIP13 (Fig. 4A). The
invasion of these cell lines was examined using Matrigel-coated
Boyden chambers. As shown in Fig. 4B,
the invasion of these mutant cell lines was significantly reduced
compared with that of the wild type TRIP13-expressing cells. These
results demonstrated that the catalytic activity of TRIP13 is
required for the promotion of cell invasion.
Discussion
TRIP13 has been demonstrated to be a
kinetochore-localized protein that ensures accurate progression of
cell division (8–12). A number of kinetochore-localized
proteins are highly expressed in various cancers, and their
expression is associated with the genomic instability or malignant
conversion of cancer cells (19). For
example, aurora B, which is localized to the kinetochore (as well
as to the spindle midzone) during mitosis, is a protein kinase
critical for the accurate distribution of chromosomes (22). Aurora B is highly expressed in lung,
prostate and thyroid cancers, and deregulated aurora B promotes
tumorigenesis by inducing aneuploidy (23,24). In
addition to aurora B, other kinetochore proteins such as highly
expressed in cancer 1 and monopolar spindle 1 kinase have also been
shown to be overexpressed in certain cancers, and small molecules
that inhibit the functions of these proteins are regarded as
promising candidate agents for cancer treatment (25–27). These
previous studies indicate the important functions of
kinetochore-localized proteins for cancer progression. The present
report revealed that TRIP13 was highly expressed in CRC tumor
tissues and various CRC cell lines. High expression of TRIP13 has
also been reported in head and neck cancer, as well as in prostate
cancer (20,28), indicating that TRIP13 may be
overexpressed in a variety of cancers, similar to other
kinetochore-localized proteins (25–27).
Together with the current results, those studies suggest that
TRIP13 is important in the progression of multiple cancers.
Mitotic defects often lead to aneuploidy, which
promotes the genetic instability of cells and induces apoptosis or
cellular transformation (19). The
mitotic SAC ensures that all the kinetochores are properly attached
to the mitotic spindle to mediate the accurate distribution of
chromosomes (18). A critical
component of the SAC is the mitotic checkpoint complex (MCC), which
is composed of budding uninhibited by benzimidazole-related 1,
budding uninhibited by benzimidazoles 3, mitotic arrest deficient
(MAD) 2 and cell division cycle 20 (29,30).
Deregulated expression or inactivation of MCC components is
associated with numerous types of cancers. Mice with MAD2 or MAD1
heterozygous deletions were prone to develop lung cancer or various
types of cancer, respectively (31,32). The
embryonic fibroblasts of these mice were defective at maintaining
the SAC, and consequently exhibited a high frequency of aneuploidy
(32). These results clearly
demonstrate that maintenance of the SAC is critical to prevent
cancer formation. TRIP13 has been shown to promote the dissociation
of the MCC complex (15–17), which subsequently inactivates the SAC
to prevent the completion of mitosis (18). Thus, overexpression of TRIP13 and
silencing of MCC components have similar effects on cells, since
both can lead to dysregulation of the SAC. Overexpression of TRIP13
may promote chromosomal instability in cancer cells, allowing
further acquisition of malignant characteristics.
In summary, the present study has demonstrated that
TRIP13 is overexpressed in CRC, and that the suppression of TRIP13
reduces CRC cell proliferation and invasion. TRIP13 has ATPase
activity, and an inactivating mutant of TRIP13 was unable to
promote cancer cell invasion, suggesting that the catalytic
activity of TRIP13 is essential for cancer progression. Recent
studies have reported that small chemical molecules can inhibit the
activity of members of the AAA+ family (33). Thus, small chemicals that target
TRIP13 may represent a promising drug for the treatment of various
types of cancer.
Acknowledgements
The authors would like to thank the members of the
Division of Cancer Biology, Nagoya University Graduate School of
Medicine (Nagoya, Japan) for their helpful discussions and
technical assistance. The present study was funded by a grant from
The Naito Foundation (Tokyo, Japan).
References
|
1
|
Ogura T and Wilkinson AJ: AAA+ superfamily
ATPases: Common structure-diverse function. Genes Cells. 6:575–597.
2001. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Hanson PI and Whiteheart SW: AAA+
proteins: Have engine, will work. Nat Rev Mol Cell Biol. 6:519–529.
2005. View
Article : Google Scholar : PubMed/NCBI
|
|
3
|
Snider J and Houry WA: AAA+ proteins:
Diversity in function, similarity in structure. Biochem Soc Trans.
36:72–77. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Wendler P, Ciniawsky S, Kock M and Kube S:
Structure and function of the AAA+ nucleotide binding pocket.
Biochim Biophys Acta. 1823:2–14. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Grigoletto A, Lestienne P and Rosenbaum J:
The multifaceted proteins Reptin and Pontin as major players in
cancer. Biochim Biophys Acta. 1815:147–157. 2011.PubMed/NCBI
|
|
6
|
Huber O, Ménard L, Haurie V, Nicou A,
Taras D and Rosenbaum J: Pontin and reptin, two related ATPases
with multiple roles in cancer. Cancer Res. 68:6873–6876. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Lee JW, Choi HS, Gyuris J, Brent R and
Moore DD: Two classes of proteins dependent on either the presence
or absence of thyroid hormone for interaction with the thyroid
hormone receptor. Mol Endocrinol. 9:243–254. 1995. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Li XC and Schimenti JC: Mouse pachytene
checkpoint 2 (trip13) is required for completing meiotic
recombination but not synapsis. PLoS Genet. 3:e1302007. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Roig I, Dowdle JA, Toth A, de Rooij DG,
Jasin M and Keeney S: Mouse TRIP13/PCH2 is required for
recombination and normal higher-order chromosome structure during
meiosis. PLoS Genet. 6:e10010622010. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Ho HC and Burgess SM: Pch2 acts through
Xrs2 and Tel1/ATM to modulate interhomolog bias and checkpoint
function during meiosis. PLoS Genet. 7:e10023512011. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Wojtasz L, Daniel K, Roig I, Bolcun-Filas
E, Xu H, Boonsanay V, Eckmann CR, Cooke HJ, Jasin M, Keeney S, et
al: Mouse HORMAD1 and HORMAD2, two conserved meiotic chromosomal
proteins, are depleted from synapsed chromosome axes with the help
of TRIP13 AAA-ATPase. PLoS Genet. 5:e10007022009. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Farmer S, Hong EJ, Leung WK, Argunhan B,
Terentyev Y, Humphryes N, Toyoizumi H and Tsubouchi H: Budding
yeast Pch2, a widely conserved meiotic protein, is involved in the
initiation of meiotic recombination. PLoS One. 7:e397242012.
View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Chen C, Jomaa A, Ortega J and Alani EE:
Pch2 is a hexameric ring ATPase that remodels the chromosome axis
protein Hop1. Proc Natl Acad Sci USA. 111:E44–E53. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Tipton AR, Wang K, Oladimeji P, Sufi S, Gu
Z and Liu ST: Identification of novel mitosis regulators through
data mining with human centromere/kinetochore proteins as group
queries. BMC Cell Biol. 13:152012. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Wang K, Sturt-Gillespie B, Hittle JC,
Macdonald D, Chan GK, Yen TJ and Liu ST: Thyroid hormone receptor
interacting protein 13 (TRIP13) AAA-ATPase is a novel mitotic
checkpoint-silencing protein. J Biol Chem. 289:23928–23937. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Eytan E, Wang K, Miniowitz-Shemtov S,
Sitry-Shevah D, Kaisari S, Yen TJ, Liu ST and Hershko A:
Disassembly of mitotic checkpoint complexes by the joint action of
the AAA-ATPase TRIP13 and p31 (comet). Proc Natl Acad Sci USA.
111:12019–12024. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Ye Q, Rosenberg SC, Moeller A, Speir JA,
Su TY and Corbett KD: TRIP13 is a protein-remodeling AAA+ ATPase
that catalyzes MAD2 conformation switching. Elife. 4:2015.
View Article : Google Scholar
|
|
18
|
Lara-Gonzalez P, Westhorpe FG and Taylor
SS: The spindle assembly checkpoint. Curr Biol. 22:R966–R980. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Rao CV, Yamada HY, Yao Y and Dai W:
Enhanced genomic instabilities caused by deregulated microtubule
dynamics and chromosome segregation: A perspective from genetic
studies in mice. Carcinogenesis. 30:1469–1474. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Banerjee R, Russo N, Liu M, Basrur V,
Bellile E, Palanisamy N, Scanlon CS, van Tubergen E, Inglehart RC,
Metwally T, et al: TRIP13 promotes error-prone nonhomologous end
joining and induces chemoresistance in head and neck cancer. Nat
Commun. 5:45272014. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Wong M, Hyodo T, Asano E, Funasaka K,
Miyahara R, Hirooka Y, Goto H, Hamaguchi M and Senga T: Silencing
of STRN4 suppresses the malignant characteristics of cancer cells.
Cancer Sci. 105:1526–1532. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Giet R, Petretti C and Prigent C: Aurora
kinases, aneuploidy and cancer, a coincidence or a real link?
Trends Cell Biol. 15:241–250. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Gautschi O, Heighway J, Mack PC, Purnell
PR, Lara PN Jr and Gandara DR: Aurora kinases as anticancer drug
targets. Clin Cancer Res. 14:1639–1648. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Nguyen HG, Makitalo M, Yang D, Chinnappan
D, St Hilaire C and Ravid K: Deregulated Aurora-B induced
tetraploidy promotes tumorigenesis. FASEB J. 23:2741–2748. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Huang LY, Chang CC, Lee YS, Chang JM,
Huang JJ, Chuang SH, Kao KJ, Lau GM, Tsai PY, Liu CW, et al:
Activity of a novel Hec1-targeted anticancer compound against
breast cancer cell lines in vitro and in vivo. Mol Cancer Ther.
13:1419–1430. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Huang LY, Chang CC, Lee YS, Huang JJ,
Chuang SH, Chang JM, Kao KJ, Lau GM, Tsai PY, Liu CW, et al:
Inhibition of Hec1 as a novel approach for treatment of primary
liver cancer. Cancer Chemother Pharmacol. 74:511–520. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Maachani UB, Kramp T, Hanson R, Zhao S,
Celiku O, Shankavaram U, Colombo R, Caplen NJ, Camphausen K and
Tandle A: Targeting MPS1 enhances Radiosensitization of human
Glioblastoma by modulating DNA repair proteins. Mol Cancer Res.
13:852–862. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Larkin SE, Holmes S, Cree IA, Walker T,
Basketter V, Bickers B, Harris S, Garbis SD, Townsend PA and
Aukim-Hastie C: Identification of markers of prostate cancer
progression using candidate gene expression. Br J Cancer.
106:157–165. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Sudakin V, Chan GK and Yen TJ: Checkpoint
inhibition of the APC/C in HeLa cells is mediated by a complex of
BUBR1, BUB3, CDC20, and MAD2. J Cell Biol. 154:925–936. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Tipton AR, Tipton M, Yen T and Liu ST:
Closed MAD2 (C-MAD2) is selectively incorporated into the mitotic
checkpoint complex (MCC). Cell Cycle. 10:3740–3750. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Sotillo R, Schvartzman JM, Socci ND and
Benezra R: Mad2-induced chromosome instability leads to lung tumour
relapse after oncogene withdrawal. Nature. 464:436–440. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Iwanaga Y, Chi YH, Miyazato A, Sheleg S,
Haller K, Peloponese JM Jr, Li Y, Ward JM, Benezra R and Jeang KT:
Heterozygous deletion of mitotic arrest-deficient protein 1 (MAD1)
increases the incidence of tumors in mice. Cancer Res. 67:160–166.
2007. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Chapman E, Maksim N, de la Cruz F and La
Clair JJ: Inhibitors of the AAA+ chaperone p97. Molecules.
20:3027–3049. 2015. View Article : Google Scholar : PubMed/NCBI
|