Introduction
Lung cancer is the leading cause of
cancer-associated mortality in the United States (1). Common molecular properties of lung
cancer include hyperactivation of receptor tyrosine kinases (RTKs)
(2). Phosphatidylinositol 3-kinase
(PI3K) is a potent downstream RTK signaling target, which is able
to phosphorylate phosphatidylinositol (4,5)-bisphosphate (PIP2) to generate
phosphatidylinositol (3–5)-trisphosphate (PIP3) (3). PIP3 recruits and activates
phosphoinositide-dependent kinase 1 and Akt serine/threonine kinase
1 (Akt) leading to a pro-growth, pro-survival phenotype (4). The plasma membrane associated protein
myristoylated alanine rich C-kinase substrate (MARCKS) is able to
bind to PIP2, regulating the conversion of PIP2 to PIP3 and
subsequently influencing Akt activation (5,6).
MARCKS contains three domains: An N-terminal
myristoylation domain, an MH2 domain, and an effector domain (ED)
(7). The ED of MARCKS is comprised of
a 25 amino acid sequence containing four phosphorylatable-serine
residues and 13 positively-charged lysine residues, which allow
MARCKS to bind to the plasma membrane via electrostatic
interactions and sequester PIP2 (8–11). The
phosphorylation status of MARCKS ED is essential in regulating the
subcellular localization of MARCKS (12). Following MARCKS ED phosphorylation,
the electrostatic interaction with the plasma membrane is lost,
allowing MARCKS to migrate into the cytoplasm, and subsequently
release PIP2 (13). Following
dephosphorylation, MARCKS is able to reattach to the plasma
membrane (12).
Previously, lentiviral manipulation of A549 lung
cancer cell lines was used to overexpress a wild-type (WT) or
non-phosphorylatable (NP) MARCKS protein under the regulation of a
tetracycline promoter. The NP-MARCKS construct was generated by
substituting the serine residues of the ED for alanine residues.
Overexpression of NP-MARCKS led to increased sensitivity to
radiation and prolonged double-strand DNA breaks following
treatment with ionizing radiation (14). MARCKS does not possess endogenous
enzymatic activity and therefore a 25 amino acid peptide mimetic of
MARCKS ED was engineered in order to modulate MARCKS
phosphorylation (14). In the present
study, the ability of MARCKS targeted therapy to influence MARCKS
phosphorylation levels and increase the sensitivity of lung cancer
cells to radiation was investigated.
Materials and methods
Cell culture
Human lung cancer cell lines A549, H1792 and H1975
(American Type Culture Collection, Manassas, VA, USA) were cultured
in filtered (22 µm vacuum filtration; cat no. 431097; Corning,
Inc., Corning, MA, USA) RPMI-1640 (Thermo Fisher Scientific, Inc.,
Waltham, MA, USA) with 10% fetal bovine serum (Sigma-Aldrich; Merck
Millipore, Darmstadt, Germany), 1% penicillin-streptomycin and 1%
GlutaMAX™ (Thermo Fisher Scientific, Inc.). All cells were
maintained at 37°C in 5% CO2.
MARCKS plasmid production
A549 lung cancer cell lines were engineered to
overexpress MARCKS in a tetracycline-dependent manner as described
previously (14). WT-MARCKS and
NP-MARCKS sequences (GenScript USA, Inc., Piscataway, NJ, USA) were
cloned into a pLenti6.3/TO/V5 (ViraPower HiPerform T-REx Gateway
Expression System; cat no. A11141; Invitrogen; Thermo Fisher
Scientific, Inc.) lentiviral plasmid as described previously
(5). The NP-MARCKS was engineered by
substituting the four serine residues in the ED to alanine
residues. Exogenous MARCKS was distinguishable from endogenous
MARCKS by the addition of a V-5 epitope tag on the C-terminus of
WT- and NP-MARCKS.
Lentiviral particle production
Lentiviral particles were produced as described
previously (14). A total of 4 µg
lentiviral packaging plasmid psPAX2 (plasmid no. 12260), lentiviral
envelope plasmid PCMV-VSV-G (plasmid no. 8454; both Addgene,
Cambridge, Inc., MA, USA) and lentiviral vector plasmid, were mixed
with Lipofectamine® 2000 (cat no. 11668) and Opti-MEM™
media (cat no. 11058; both Invitrogen; Thermo Fisher Scientific,
Inc.). The lentiviral mixture was added to the culture media of
293FT cells (Invitrogen; Thermo Fisher Scientific, Inc.) and the
media was subsequently replaced the following morning. Lentiviral
supernatant was collected after 48 h and filtered through a 0.45 µm
filter, aliquoted and stored at −80°C until required. Lentiviral
particles were quantified using QuickTiter p24 ELISA (Cell Biolabs,
San Diego, CA, USA) (5).
Stable cell line selection
A549 cells were transduced with equal amounts (~30
ng/ml) of p24 quantified tetracycline-repressor (Tet-R) packaged
lentiviral particles along with 8 µg/ml polybrene (EMD Millipore,
Billerica, MA, USA). A total of 500 µg/ml Geneticin®
(cat no. G418; Thermo Fisher Scientific, Inc.) was used to select
for Tet-R positive cells. Tet-R cells were subsequently transduced
with equal amounts (~30 ng/ml) of p24 quantified WT- and NP-MARCKS
lentiviral particles. A total of 1 µg/ml Blastacidin (Thermo Fisher
Scientific, Inc.) was used to select for WT- and
NP-MARCKS-expressing cells. MARCKS expression was induced by
culturing with 2 µg/ml of the tetracycline homologue doxycycline
overnight at 37°C (14).
MARCKS peptide
MARCKS peptide was used as described previously
(14). MARCKS-ED tyrosine
aminotransferase (TAT) peptide was engineered by conjugating the
cell permeable human immunodeficiency virus (HIV) TAT peptide via
cysteine bonds to the 25 amino acid ED sequence
(KKKKKRFSFKKSFKLSGFSFKKNKK) (15).
The control peptide sequence was designed using the ExPAsy random
protein sequence generator (www.expasy.org) using the average amino acid
composition computed from Swiss-Prot (www.uniprot.org) (CEIEEHAWNTVEMFSSFPGTQLYNDA) to
control for peptide size. To eliminate the effect of the positive
charges, lysine and arginine residues were changed to glutamates.
Peptide was added to cultures 1 h prior to performance of in
vivo assays, including immunoblotting, RNA expression analysis,
cellular survival and DNA double-strand damage quantification. A
dose establishment study identified 6.25 µM as the lowest effective
dose (tested, 1–25 µM), which was selected for subsequent
studies.
Immunoblotting
Immunoblotting was performed as previously described
by Jarboe et al (5). Mammalian
protein extraction reagent lysis buffer supplemented with protease
(cat no. P8340; Sigma-Aldrich; Merck Millipore) and phosphatase
inhibitors (cat nos., P0044 and P5726; Sigma-Aldrich; Merck
Millipore) was used to lyse cells. Protein concentration was
determined by using Pierce Bicinchoninic Protein Assay kit (Thermo
Fisher Scientific, Inc.) and ~10 µg of protein were separated via
electrophoresis on an 8% SDS-PAGE gel and transferred to a
polyvinylidene difluoride membrane (Immobilon; Merck Millipore).
Blots were blocked in 5% bovine serum albumin (BSA) (Sigma-Aldrich;
Merck Millipore) at room temperature for 1 h and probed with the
following primary antibodies: Phosphorylated-MARCKS (cat no.,
ab81295), MARCKS (cat no., ab52616; both Abcam, Cambridge, UK),
phosphorylated-Akt (Ser473) (cat no., D9E, 4060), Phospho-Akt
(Thr308) (cat no., C31E5E, 2965), Akt (cat no., C67E7, 4691; all
Cell Signaling Technology. Inc., Danvers, MA, USA). Antibodies were
used at a dilution of 1:1,000 with overnight 4°C incubation.
Additionally, actin antibody (cat no. sc-1616; Santa Cruz
Biotechnology, Inc., Dallas, TX, USA) was used at a concentration
of 1:5,000 and incubated overnight at 4°C. Blots were washed 3
times for 5 min in TBST. Peroxidase-conjugated Affini Pure Goat
Anti-Mouse IgG (cat no., 115-035-166) and Affini-Pure Donkey
Anti-Rabbit IgG (cat no. 711-035-152) secondary antibodies (Jackson
ImmunoResearch Laboratories, Inc., West Grove, PA, USA) was added
at a dilution of 1:5,000 in 5% BSA-TBST, and incubated for 1 h at
room temperature. Enhanced chemiluminescence (ECL) using Western
Lighting-Plus ECL substrate (PerkinElmer, Inc., Waltham, MA, USA)
was used to visualize the western blots as described previously
(14).
RNA expression analysis
RNA expression analysis was performed at the UAB
Nanostring Laboratory (Birmingham, AL, USA) (www.uab.edu/medicine/radonc/en/nanostring). The
nCounter® GX Human Cancer Reference kit (NanoString
Technologies, Inc., Seattle, WA, USA) was used to analyze RNA
expression of 230 human cancer-associated genes. MARCKS expression
in A549 NP-MARCKS and A549 WT-MARCKS cells was induced by overnight
incubation at 37°C with doxycycline, and total RNA was collected
the following day. A total of 100 ng RNA was prepared in a 30 µl
reaction volume and run on the automated nCounter system. Quality
control and normalization were performed on the raw data using
positive and negative control spots on the included GX Human Cancer
Reference Panel chip and five housekeeping reference genes
(16,17).
Cell viability and survival assay
Cells were plated in black 96-well plates (cat no.
3603; Costar; Thermo Fisher Scientific, Inc.) and allowed to adhere
overnight. The following morning the control or ED peptides were
added. A total of 1 h following peptide treatment, cells in the
irradiation group were subjected to 0, 5 or 8 Gy irradiation using
a 320 kV X-ray irradiator (Kimtron Inc., Woodbury, CT, USA). A
total of 4 days following peptide addition or irradiation, ATP
levels were measured via the ATPlite™ Luminescence Assay System
(PerkinElmer, Inc., Waltham, MA, USA) using a Synergy H1 Multi-Mode
Reader (BioTek Instruments, Inc., Winooski, VT, USA).
In vivo studies
The animal protocol was provided by the University
of Alabama at Birmingham's Institutional Animal Care and Use
Committee. Mice were housed at ~24°C and given free access to food
and water. A 12 h light/dark schedule was provided for the mice. A
total of 1 million WT- or NP-MARCKS-expressing A549 lung cancer
cells, were injected into the flanks of ~7 week old female athymic
nude mice (~17 g) (Charles River Laboratories, Hartford, CT, USA).
Cells were mixed in a 1:1 mixture of PBS:Matrigel® (BD
Biosciences, San Jose, CA, USA) to a final volume of 50 µl and
loaded into a 1 ml syringe with a 25 gx 5/8” needle (Monoject™
Tuberculin Syringe; cat no. #8881501640; Covidien, Dublin, Ireland)
(18). When tumors were palpable,
mouse food was subsequently supplemented with doxycycline (cat no.
TD.05125; Harlan Laboratories, Madison, WI, USA) (19). WT- and NP-MARCKS expression was
induced with doxycycline-supplemented mouse food for 7 days prior
to tumor volume calculation. Tumor volume was measured using
Vernier calipers using the formula: (Length × width2)/2
(20,21). Values are presented as the mean fold
change in tumor volume ± standard error of the mean (SEM). Mice
were euthanized with CO2 inhalation as set forth by
institutional guidelines.
Double-strand DNA damage
quantification
Following overnight induction of MARCKS expression,
lung cancer cells were treated with the control or ED peptides 1 h
prior to 8 Gy irradiation. As described previously, cells were
fixed and stained with an anti-phosphorylated-γH2AX-S139 antibody
and counterstained with DAPI (14).
γH2AX staining foci marking DNA double-strand breaks were analyzed
using an EVOS FL digital inverted fluorescence microscope (Thermo
Fisher Scientific, Inc.). Positive events were defined as ≥10 foci
per cell, and positive and negative controls were included in all
experiments (14,22–24).
Statistical analysis
Statistics calculations and data graphing was
performed using GraphPad Prism (GraphPad Software, Inc., La Jolla,
CA, USA). Analysis of variance followed by the Bonferroni
correction post hoc test was used to quantify DNA damage, and a
Student's t-test was performed to analyze survival assays. All
statistics were two-sided and P<0.05 was considered to indicate
a statistically significant difference. Values are presented as the
mean ± SEM.
Results
Phosphorylation status of MARCKS ED
influences lung cancer biology
MARCKS ED is known to have multiple biological
functions; however, limited information is available regarding the
involvement of MARCKS in lung cancer biology (25). Reversible phosphorylation of MARCKS ED
is important in regulating the function and subcellular
localization of MARCKS (10,26). In the present study, A549 lung cancer
cells were engineered to express WT- or NP-MARCKS under the
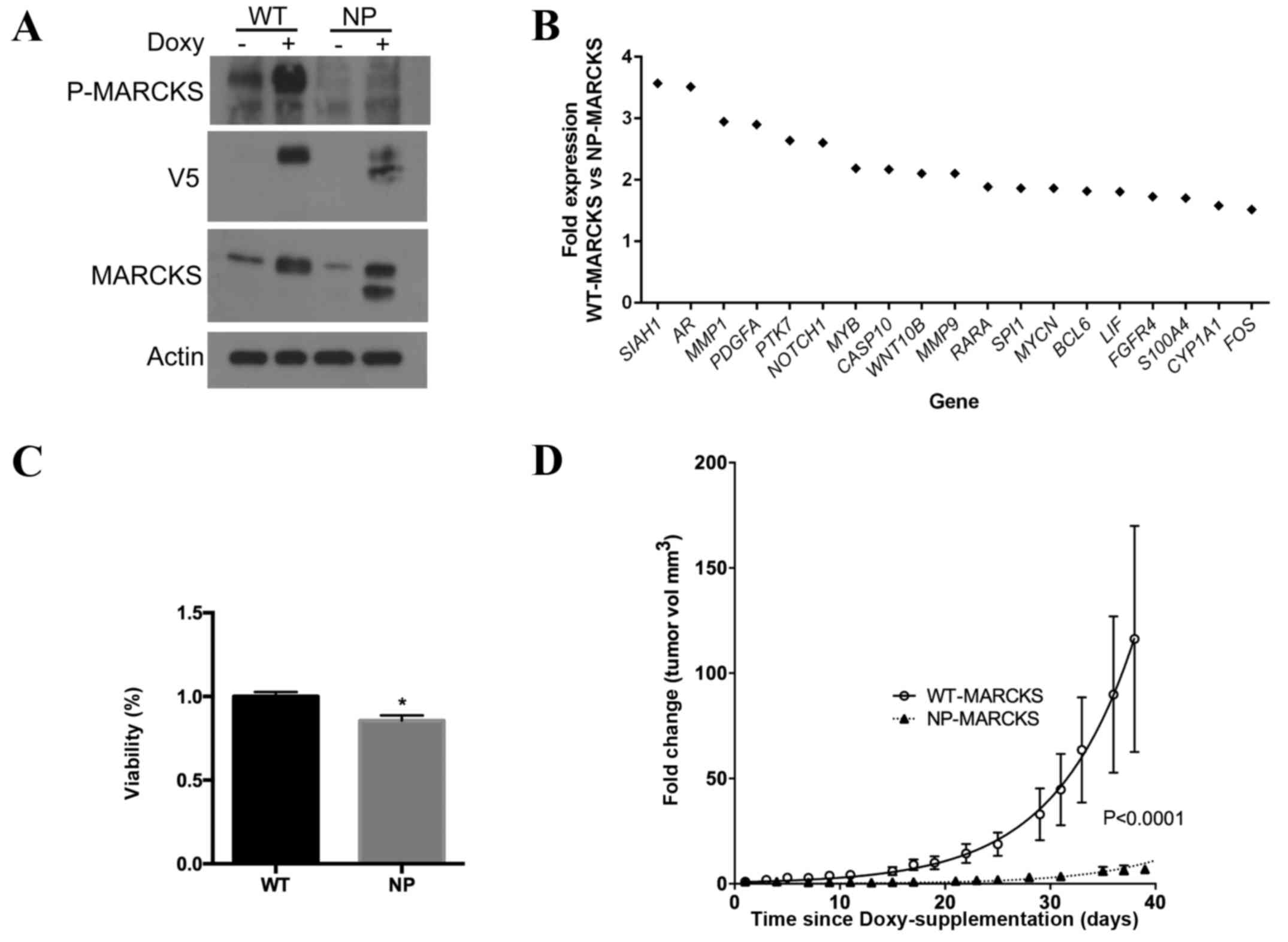
regulation of a tetracycline inducible promoter. Western blotting
(Fig. 1A) confirmed that A549 cells
were able to overexpress WT- and NP-MARCKS in a
tetracycline/doxycycline-inducible manner. Due to the ability of
MARCKS to regulate multiple signaling cascades (25), the effect of MARCKS phosphorylation on
gene expression was analyzed. Using the nCounter® GX
Human Cancer Reference kit, differences in gene expression between
A549 cells overexpressing WT- and NP-MARCKS were investigated. A
total of 19 genes exhibited a >1.5 fold increase in expression
in the WT-MARCKS cell line compared to the NP-MARCKS cell line,
including siah E3 ubiquitin protein ligase 1, androgen receptor,
matrix metalloproteinase (MMP) 1, platelet derived growth factor
subunit A, protein tyrosine kinase 7, notch 1, MYB proto-oncogene,
transcription factor, caspase 10, Wnt family member 10B, MMP9,
retinoic acid receptor alpha, WD repeat domain 87, v-myc avian
myelocytomatosis viral oncogene neuroblastoma derived homolog,
B-cell CLL/lymphoma 6, leukemia inhibitory factor, fibroblast
growth factor receptor 4, S100 calcium binding protein A4,
cytochrome P450 family 1 subfamily A member 1 and Fos
proto-oncogene, AP-1 transcription factor subunit (Fig. 1B). Several of these genes are
associated with tumorigenesis and cancer progression (27–31). Cell
viability assays were performed to investigate if the ED
phosphorylation status was important in lung cancer proliferation.
The ATPlite assay indicated significantly increased levels of ATP
in the WT-MARCKS A549 cells compared with the NP-MARCKS cell line
(Fig. 1C; P=0.002), suggesting that
non-phosphorylated MARCKS impedes cell viability. In addition,
in vivo mouse studies were used to assess the ability of
MARCKS ED phosphorylation status to influence lung cancer growth.
WT- and NP- MARCKS A549 cells were injected into the flanks of
athymic nude mice. Mouse food was supplemented with doxycycline to
induce the expression of WT- and NP- MARCKS (19). The change in tumor volume was compared
between the WT- and NP-MARCKS A549 cells over a period of 40 days.
WT-MARCKS transfected mice exhibited a greater increase in tumor
volume compared with NP-MARCKS transfected mice (Fig. 1D). By modeling the data to a nonlinear
regression fit for exponential growth, a significant difference
between the WT- and NP-MARCKS tumors (P<0.0001) was observed.
The results of the present study suggest that the phosphorylation
status of MARCKS ED influences gene expression, viability and tumor
growth.
MARCKS ED provides a potential
therapeutic intervention to alter lung cancer biology
Previously, a 25-amino acid peptide mimetic of
MARCKS ED was used to alter lung cancer biology (14). The 25-amino acid ED peptide was
conjugated to the cell permeable TAT-peptide, which allowed the ED
mimic to enter the cell. Swiss-Prot software calculated a control
peptide comprised of 25 random amino acids to control for peptide
length. The previous investigation observed changes in cell
physiology due to the ED peptide; however, the ability of the
peptide to influence MARCKS phosphorylation or proliferation was
not investigated (14). Dose
titration determined that 6.25 µM was the ideal concentration (data
not shown). A549, H1792 and H1975 lung cancer cell lines were
cultured without peptide, with 6.25 µM control peptide or with 6.25
µM ED mimetic peptide. Western blot analysis revealed that the ED
peptide mimetic was able to decrease the level of phosphorylated
MARCKS in the three lung cancer cell lines (Fig. 2A). Decreasing the level of
phosphorylated MARCKS may lead to increased plasma
membrane-associated MARCKS and decreased Akt activation. Treatment
of A549, and to a lesser extent, H1975, cells with the ED-peptide
was able to decrease levels of Akt phosphorylated at the serine 473
and threonine 308 residues (Fig. 2A).
In addition, the ED-peptide was able to reduce the total level of
Akt, which was most pronounced in the H1792 cells (Fig. 2A). Notably, minimal phosphorylated
T308 Akt was observed in the H1792 cells in all conditions, though
phosphorylated serine 473 Akt was detectable and was mildly
suppressed with the ED-peptide. The effect of MARCKS ED peptide on
lung tumor growth was investigated. ATP levels in A549, H1972 and
H1975 cell lines treated with the ED peptide mimetic were
normalized to ATP levels in lung cancer cells with no peptide
intervention. A significant decrease in ATP levels in the A549
(P=0.045), H1792 (P=0.000002) and H1975 (P=0.000003) lung cancer
cells following 4 days of treatment with the ED peptide was
observed compared with the control cells (Fig. 2B).
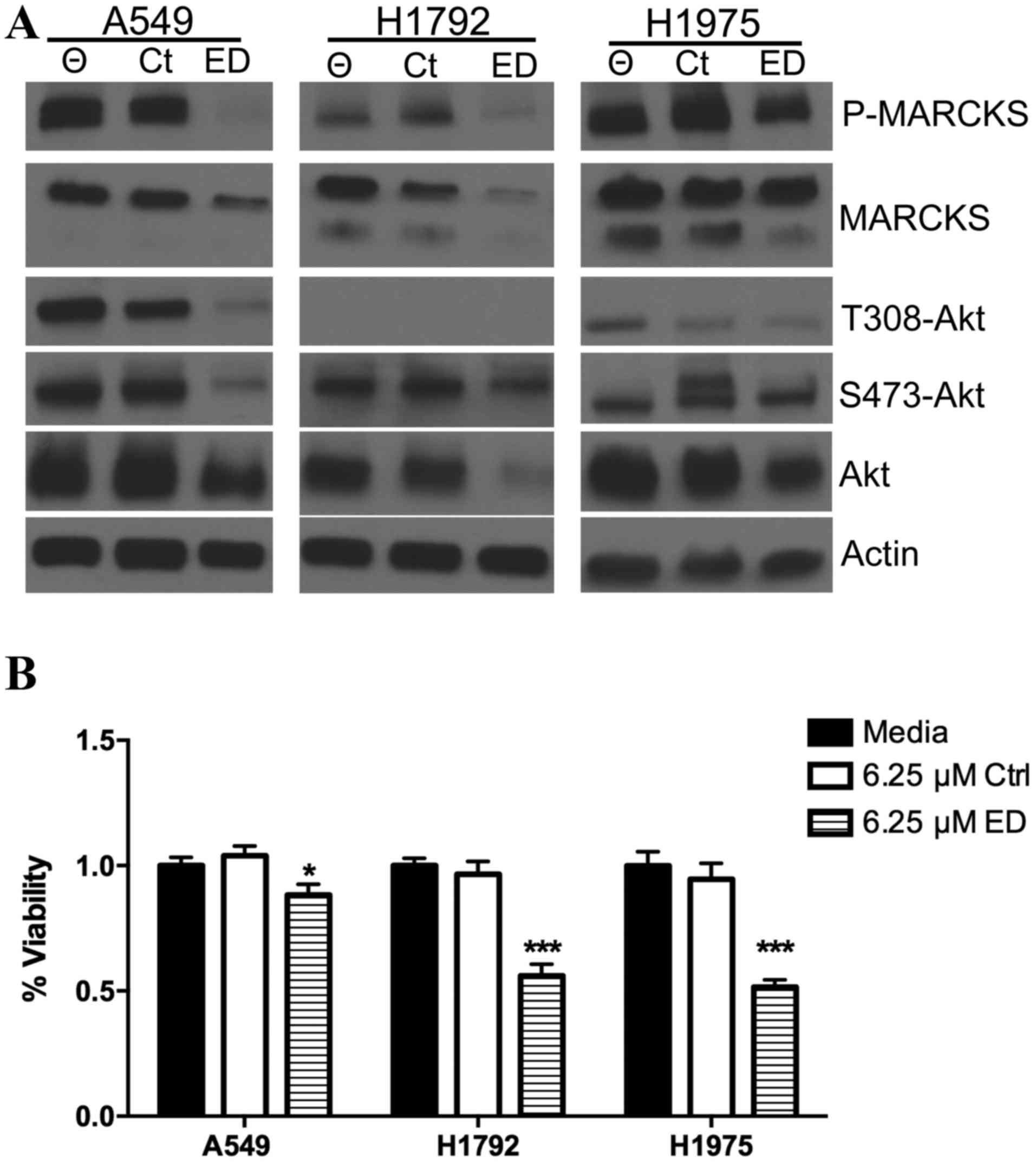 | Figure 2.MARCKS targeted peptide therapy
influences lung cancer biology. (A) A549, H1792 and H1975 lung
cancer cells were cultured with no peptide, 6.25 µM control
peptide, or 6.25 µM ED peptide. Western blot analysis was used to
probe for phosphorylated MARCKS, MARCKS, phosphorylated Akt (T308),
phosphorylated Akt (S473), Akt and broad range actin. (B) ATPlite
assay measured ATP levels 4 days following A549, H1792 and H1975
cell transfection with no peptide, 6.25 µM control peptide, or 6.25
µM ED peptide. *P<0.05 compared with Ctrl; ***P<0.0001
compared with Ctrl. Θ indicates treatment without peptide. MARCKS,
myristoylated alanine rich C-kinase substrate; Ctrl, control; ED,
effector domain; Akt, Akt serine/threonine kinase 1; P,
phosphorylated. |
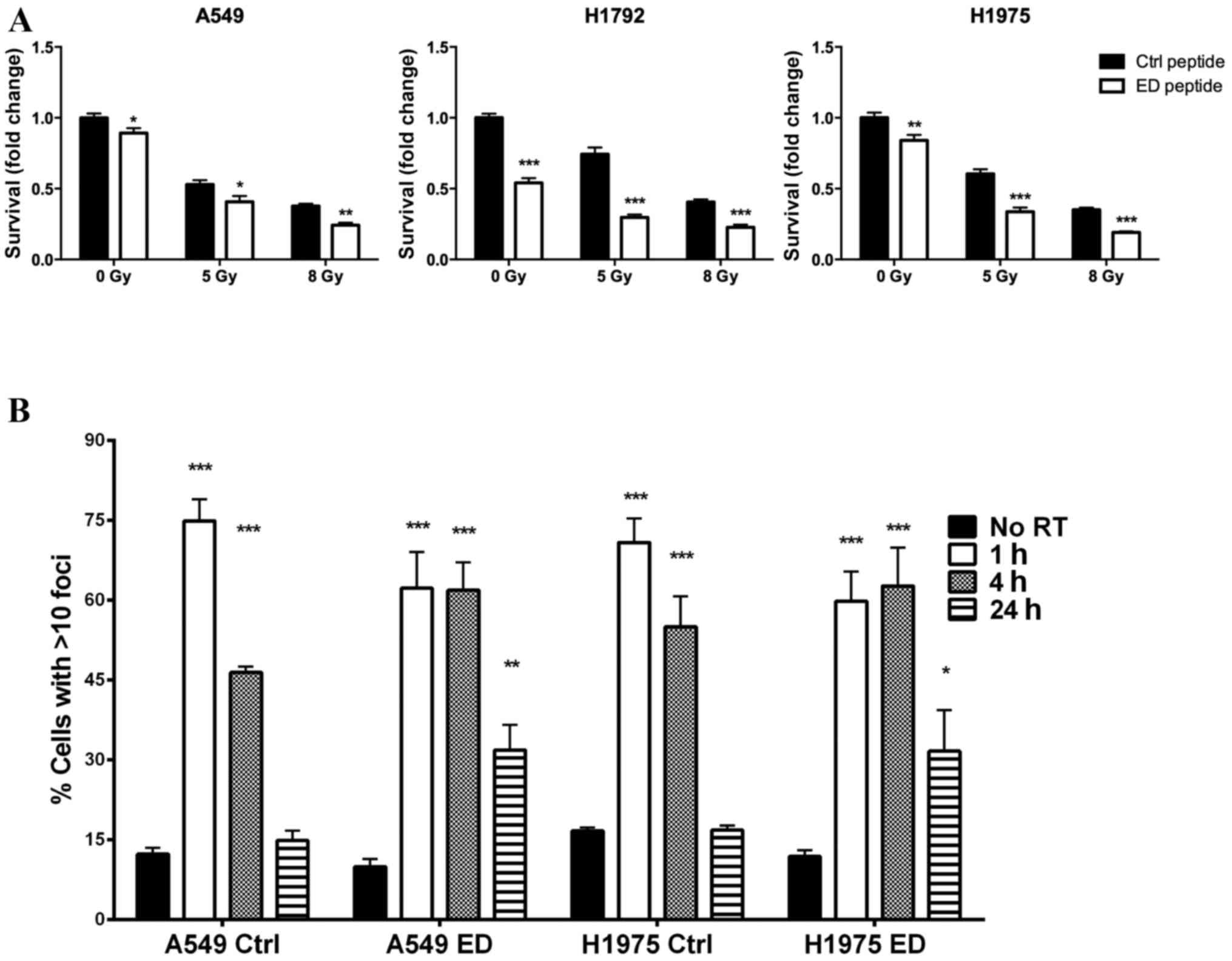
MARCKS targeted peptide increases the
sensitivity of lung cancer cells to radiation and increases γH2AX
foci staining
Previously, lentiviral manipulation was used to
overexpress NP-MARCKS in A549 cells and their sensitivity to
radiation increased (14). In the
present study, the effect of treatment with the MARCKS ED peptide
mimetic on the radiation sensitivity of lung cancer cells was
analyzed. A549, H1792 and H1975 cells were cultured with either the
control or ED peptides 1 h prior to treatment with ionizing
radiation. The three cell lines exhibited significantly decreased
survival following ED peptide treatment compared with the control
group at irradiation levels 0, 5 and 8 Gy (Fig. 3A; P<0.05). DNA double-strand breaks
in A549 and H1975 cells were examined by measuring γH2AX foci
(22). γH2AX foci increased in all
groups 1 h following irradiation; however, 24 h following
irradiation A549 and H1975 cells treated with the control peptide
exhibited basal level γH2AX foci staining. Conversely, A549 and
H1975 cells treated with the ED peptide demonstrated persistent
γH2AX foci staining (Fig. 3B;
P<0.01 compared with the control). The results of the present
study suggest that altering the phosphorylation status of MARCKS ED
increases lung cell sensitivity to radiation and prolongs DNA
damage following irradiation.
Discussion
MARCKS appears to have tumor suppressive properties
in glioblastoma and colon cancer (5,32),
however, may function as a tumor promoter in breast and
cholangiocarcinoma cancers (33,34). The
exact role MARCKS serves in cancer biology remains unclear. The
subcellular localization and phosphorylation status of MARCKS ED
are important in the normal biological and oncogenic function and
regulation of MARCKS (25,35), particularly in lung cancer (36). Hanada et al (37) reported that squamous cell carcinoma
with increased MARCKS expression indicated poor prognosis, however
subcellular localization and phosphorylation status were not
investigated. Chen et al (38)
demonstrated that MARCKS phosphorylation is involved in
invasiveness and that lung tumors with elevated and phosphorylated
MARCKS exhibited increased invasion in vitro and in
patients. Recently, Chen et al (36) demonstrated that increased levels of
phosphorylated MARCKS are associated with decreased patient
survival and increased tumor growth. A similar 25-amino acid
peptide was used to modulate MARCKS phosphorylation and led to
reduce migration (36). Previously,
the present authors demonstrated that the phosphorylation status of
MARCKS is important in subcellular localization, drug sensitivity
and the DNA damage response (14).
Specifically, lentivirally manipulated A549 lung cancer cell lines
were used and showed that NP-MARCKS-expressing cells exhibited
increased levels of MARCKS fluorescent staining around the plasma
membrane compared with the WT-MARCKS-expressing cells, suggesting
ED phosphorylation status may impact MARCKS location (14). In the present study, A549
NP-MARCKS-expressing cells exhibited increased sensitivity to
radiation and prolonged double-strand DNA breaks, when assessed
using γH2AX foci staining. A549 lung cancer cells are typically
inherently resistant to radiation (39). The present results demonstrated that
the phosphorylation status of MARCKS is essential to radiation
sensitivity. As MARCKS lacks endogenous enzymatic activity, a
peptide mimetic was designed to modulate MARCKS phosphorylation.
The 25-amino acid sequence of MARCKS ED was conjugated to the HIV
TAT peptide, which increased the ability of the peptide to enter
the cell.
In the present study, the importance of MARCKS ED
phosphorylation status in lung cancer biology was further
characterized and the therapeutic benefit of ED peptide treatment
in lung cancer was investigated. RNA expression was observed to be
altered dependent on the phosphorylation status of MARCKS in
lentivirally-engineered A549 cells. Furthermore, the
phosphorylation status of MARCKS ED in vivo was observed to
impact on lung tumor growth. The efficacy of the ED peptide was
tested. Parental A549, H1792 and H1975 lung cancer cells treated
with the ED peptide exhibited a decrease in MARCKS phosphorylation.
In addition, a decrease in T308 and S473 phosphorylation and total
Akt levels was observed. While it is unclear why T308
phosphorylation was so low in the H1792 cells, the ED peptide was
able to decrease A549, H1792 and H1975 proliferation, and increased
radiation sensitivity, though it caused prolonged double-strand DNA
breaks only in A549 and H1975 cells following irradiation.
Lung cancer is the leading cause of
cancer-associated mortality in the United States, and despite
targeted therapy and early detection, drug resistance frequently
develops (40). Additionally,
resistance to ionizing radiation has been observed (41). Targeting MARCKS has the potential to
provide a novel mechanism for targeting the RTK-PI3K-Akt signaling
cascade. The ED peptide may be able to modulate radiation
sensitivity and serve as a future method of avoiding drug
resistance (42).
Acknowledgements
The present study was supported by a Research
Scholar Grant from the American Cancer Society (grant no.
RSG-14-071-01-TBG), a pilot grant from P20 from the National Cancer
Institute of the National Institutes of Health (grant no.
CA151129-02), the 2008 ASTRO Junior Faculty Training Research Award
and the UAB's Radiation Oncology Pilot Grant.
References
|
1
|
Siegel R, Naishadham D and Jemal A: Cancer
statistics, 2013. CA Cancer J Clin. 63:11–30. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Herbst RS, Heymach JV and Lippman SM: Lung
cancer. N Engl J Med. 359:1367–1380. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Engelman JA: Targeting PI3K signalling in
cancer: Opportunities, challenges and limitations. Nat Rev Cancer.
9:550–562. 2009. View
Article : Google Scholar : PubMed/NCBI
|
|
4
|
Davies MA: Regulation, role, and targeting
of Akt in cancer. J Clin Oncol. 29:4715–4717. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Jarboe JS, Anderson JC, Duarte CW, Mehta
T, Nowsheen S, Hicks PH, Whitley AC, Rohrbach TD, McCubrey RO, Chiu
S, et al: MARCKS regulates growth and radiation sensitivity and is
a novel prognostic factor for glioma. Clin Cancer Res.
18:3030–3041. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Glaser M, Wanaski S, Buser CA, Boguslavsky
V, Rashidzada W, Morris A, Rebecchi M, Scarlata SF, Runnels LW,
Prestwich GD, et al: Myristoylated alanine-rich C kinase substrate
(MARCKS) produces reversible inhibition of phospholipase C by
sequestering phosphatidylinositol 4,5-bisphosphate in lateral
domains. J Biol Chem. 271:26187–26193. 1996. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Aderem A: Signal transduction and the
actin cytoskeleton: The roles of MARCKS and profilin. Trends
Biochem Sci. 17:438–443. 1992. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Stumpo DJ, Graff JM, Albert KA, Greengard
P and Blackshear PJ: Nucleotide sequence of a cDNA for the bovine
myristoylated alanine-rich C kinase substrate (MARCKS). Nucleic
Acids Res. 17:3987–3988. 1989. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Heemskerk FM, Chen HC and Huang FL:
Protein kinase C phosphorylates Ser152, Ser156 and Ser163 but not
Ser160 of MARCKS in rat brain. Biochem Biophys Res Commun.
190:236–241. 1993. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Gambhir A, Hangyás-Mihályné G, Zaitseva I,
Cafiso DS, Wang J, Murray D, Pentyala SN, Smith SO and McLaughlin
S: Electrostatic sequestration of PIP2 on phospholipid membranes by
basic/aromatic regions of proteins. Biophys J. 86:2188–2207. 2004.
View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Wang J, Arbuzova A, Hangyás-Mihályné G and
McLaughlin S: The effector domain of myristoylated alanine-rich C
kinase substrate binds strongly to phosphatidylinositol
4,5-bisphosphate. J Biol Chem. 276:5012–5019. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
McLaughlin S and Aderem A: The
myristoyl-electrostatic switch: A modulator of reversible
protein-membrane interactions. Trends Biochem Sci. 20:272–276.
1995. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Manenti S, Sorokine O, Van Dorsselaer A
and Taniguchi H: Affinity purification and characterization of
myristoylated alanine-rich protein kinase C substrate (MARCKS) from
bovine brain. Comparison of the cytoplasmic and the membrane-bound
forms. J Biol Chem. 267:22310–22315. 1992.PubMed/NCBI
|
|
14
|
Rohrbach TD, Jarboe JS, Anderson JC,
Trummell HQ, Hicks PH, Weaver AN, Yang ES, Oster RA, Deshane JS,
Steele C, et al: Targeting the effector domain of the myristoylated
alanine rich C-kinase substrate enhances lung cancer radiation
sensitivity. Int J Oncol. 46:1079–1088. 2015.PubMed/NCBI
|
|
15
|
Graff JM, Rajan RR, Randall RR, Nairn AC
and Blackshear PJ: Protein kinase C substrate and inhibitor
characteristics of peptides derived from the myristoylated
alanine-rich C kinase substrate (MARCKS) protein phosphorylation
site domain. J Biol Chem. 266:14390–14398. 1991.PubMed/NCBI
|
|
16
|
Isayeva T, Xu J, Ragin C, Dai Q, Cooper T,
Carroll W, Dayan D, Vered M, Wenig B, Rosenthal E, et al: The
protective effect of p16(INK4a) in oral cavity carcinomas: p16
(Ink4A) dampens tumor invasion-integrated analysis of expression
and kinomics pathways. Mod Pathol. 28:631–653. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Malkov VA, Serikawa KA, Balantac N,
Watters J, Geiss G, Mashadi-Hossein A and Fare T: Multiplexed
measurements of gene signatures in different analytes using the
Nanostring nCounter Assay System. BMC Res Notes. 2:802009.
View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Willey CD, Xiao D, Tu T, Kim KW, Moretti
L, Niermann KJ, Tawtawy MN, Quarles CC and Lu B: Enzastaurin
(LY317615), a protein kinase C beta selective inhibitor, enhances
antiangiogenic effect of radiation. Int J Radiat Oncol Biol Phys.
77:1518–1526. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Cawthorne C, Swindell R, Stratford IJ,
Dive C and Welman A: Comparison of doxycycline delivery methods for
Tet-inducible gene expression in a subcutaneous xenograft model. J
Biomol Tech. 18:120–123. 2007.PubMed/NCBI
|
|
20
|
Ayers GD, McKinley ET, Zhao P, Fritz JM,
Metry RE, Deal BC, Adlerz KM, Coffey RJ and Manning HC: Volume of
preclinical xenograft tumors is more accurately assessed by
ultrasound imaging than manual caliper measurements. J Ultrasound
Med. 29:891–901. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Jensen MM, Jørgensen JT, Binderup T and
Kjaer A: Tumor volume in subcutaneous mouse xenografts measured by
microCT is more accurate and reproducible than determined by
18F-FDG-microPET or external caliper. BMC Med Imaging. 8:162008.
View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Yang ES, Wang H, Jiang G, Nowsheen S, Fu
A, Hallahan DE and Xia F: Lithium-mediated protection of
hippocampal cells involves enhancement of DNA-PK-dependent repair
in mice. J Clin Invest. 119:1124–1135. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Nowsheen S, Bonner JA and Yang ES: The
poly (ADP-Ribose) polymerase inhibitor ABT-888 reduces
radiation-induced nuclear EGFR and augments head and neck tumor
response to radiotherapy. Radiother Oncol. 99:331–338. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Yang ES, Nowsheen S, Wang T, Thotala DK
and Xia F: Glycogen synthase kinase 3beta inhibition enhances
repair of DNA double-strand breaks in irradiated hippocampal
neurons. Neuro Oncol. 13:459–470. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Arbuzova A, Schmitz AA and Vergères G:
Cross-talk unfolded: MARCKS proteins. Biochem J. 362:1–12. 2002.
View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Seykora JT, Myat MM, Allen LA, Ravetch JV
and Aderem A: Molecular determinants of the myristoyl-electrostatic
switch of MARCKS. J Biol Chem. 271:18797–18802. 1996. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Gialeli C, Theocharis AD and Karamanos NK:
Roles of matrix metalloproteinases in cancer progression and their
pharmacological targeting. FEBS J. 278:16–27. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Witsch E, Sela M and Yarden Y: Roles for
growth factors in cancer progression. Physiology (Bethesda).
25:85–101. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Cascorbi I, Brockmöller J and Roots I: A
C4887A polymorphism in exon 7 of human CYP1A1: Population
frequency, mutation linkages, and impact on lung cancer
susceptibility. Cancer Res. 56:4965–4969. 1996.PubMed/NCBI
|
|
30
|
Ma L, Young J, Prabhala H, Pan E, Mestdagh
P, Muth D, Teruya-Feldstein J, Reinhardt F, Onder TT, Valastyan S,
et al: miR-9, a MYC/MYCN-activated microRNA, regulates E-cadherin
and cancer metastasis. Nat Cell Biol. 12:247–256. 2010.PubMed/NCBI
|
|
31
|
Westhoff B, Colaluca IN, D'Ario G,
Donzelli M, Tosoni D, Volorio S, Pelosi G, Spaggiari L, Mazzarol G,
Viale G, et al: Alterations of the Notch pathway in lung cancer.
Proc Natl Acad Sci USA. 106:22293–22298. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Bickeboller M, Tagscherer KE, Kloor M,
Jansen L, Chang-Claude J, Brenner H, Hoffmeister M, Toth C,
Schirmacher P, Roth W and Bläker H: Functional characterization of
the tumor-suppressor MARCKS in colorectal cancer and its
association with survival. Oncogene. 34:1150–1159. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Browne BC, Hochgräfe F, Wu J, Millar EK,
Barraclough J, Stone A, McCloy RA, Lee CS, Roberts C, Ali NA, et
al: Global characterization of signalling networks associated with
tamoxifen resistance in breast cancer. FEBS J. 280:5237–5257. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Techasen A, Loilome W, Namwat N, et al:
Myristoylated alanine-rich C kinase substrate phosphorylation
promotes cholangiocarcinoma cell migration and metastasis via the
protein kinase C-dependent pathwayCancer Sci. England: pp.
pp658–pp665. 2010, View Article : Google Scholar
|
|
35
|
Wright PE and Dyson HJ: Intrinsically
unstructured proteins: Re-assessing the protein structure-function
paradigm. J Mol Biol. 293:321–331. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Chen CH, Chiu CL, Adler KB and Wu R: A
novel predictor of cancer malignancy: Up-regulation of
myristoylated alanine-rich C kinase substrate phosphorylation in
lung cancer. Am J Respir Crit Care Med. 189:1002–1004. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Hanada S, Kakehashi A, Nishiyama N, Wei M,
Yamano S, Chung K, Komatsu H, Inoue H, Suehiro S and Wanibuchi H:
Myristoylated alanine-rich C-kinase substrate as a prognostic
biomarker in human primary lung squamous cell carcinoma. Cancer
Biomark. 13:289–298. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Chen CH, Thai P, Yoneda K, Adler KB, Yang
PC and Wu R: A peptide that inhibits function of Myristoylated
Alanine-Rich C Kinase Substrate (MARCKS) reduces lung cancer
metastasis. Oncogene. 33:3696–3706. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Yang HJ, Kim N, Seong KM, Youn H and Youn
B: Investigation of radiation-induced transcriptome profile of
radioresistant non-small cell lung cancer A549 cells using RNA-seq.
PloS One. 8:e593192013. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Shanker M, Willcutts D and Roth JA: Drug
resistance in lung cancer. Lung Cancer: Targets and Therapy.
1:23–36. 2010.
|
|
41
|
Schuurbiers OC, Kaanders JH, van der
Heijden HF, Dekhuijzen RP, Oyen WJ and Bussink J: The
PI3-K/AKT-pathway and radiation resistance mechanisms in non-small
cell lung cancer. J Thorac Oncol. 4:761–767. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Garraway LA and Jänne PA: Circumventing
cancer drug resistance in the era of personalized medicine. Cancer
Discov. 2:214–226. 2012. View Article : Google Scholar : PubMed/NCBI
|