Introduction
Colorectal carcinoma (CRC) is one of the most
frequent and serious digestive tract malignancies worldwide, and
the second leading cause of cancer-associated mortality (1,2). CRC is
the fourth leading cause of cancer-associated mortality in China
(3,4).
The high mortality rate of CRC may be due to its high propensity
for recurrence and metastasis (5).
The liver is the most frequent site of CRC metastasis and ~50% of
patients with CRC in China are diagnosed with liver metastases,
either in the initial diagnosis or following primary tumor surgery,
which makes treatment difficult (6,7).
Identification of the cytokines regulating the proliferation and
metastasis of CRC is required to understand the nature of CRC. In
addition, investigation of the underlying mechanisms of CRC tumor
progression may enable the development of potential therapeutic
intervention strategies.
Ras-GTPase is an important regulator of the Ras
signaling pathway, which is involved in a number of cellular
activities. Aberrant activation of the Ras signaling pathway is
common in the majority of human cancers, and is due to mutations in
RAS genes, upstream inducers and downstream effectors (8,9). Ras
proteins (H-Ras, N-Ras and K-Ras) are activated by GDP-GTP exchange
(10).
RAS protein activator like 2 (RASAL2) functions as a
GTPase activating protein (GAP) and was recently reported to be a
tumor suppressor, and to mediate the growth and metastasis of
various types of cancer (11). RASAL2
expression is suppressed by hypermethylation of its promoter, which
leads to Ras signaling, and subsequent tumor growth and metastasis
in human breast cancer (12,13). Decreased RASAL2 expression is
correlated with metastasis, recurrence and poor disease outcome in
luminal B tumors (14). However,
RASAL2 is upregulated in a subset of triple-negative breast cancer
(TNBC) and estrogen receptor (ER)-negative tumors, and is a direct
target of microRNA 203, which has anti-invasive functions (13). RASAL2 is oncogenic in TNBC, where it
drives mesenchymal invasion and metastasis, but not in luminal B
ER-positive breast cancers. High RASAL2 expression is a predictor
of poor disease outcome (13). In
ovarian cancer, downregulation of RASAL2 promotes metastasis by
increasing epithelial-mesenchymal transition (EMT) (15). Inactivation of RASAL2 promotes lung
cancer metastasis through the induction of EMT via
mitogen-activated protein kinase 1 (ERK) activation (16). RASAL2 was reported to be
hypomethylated in hepatocellular carcinoma, suggesting that it
functions as an oncogene in this cancer (12). However, to the best of our knowledge,
no research focusing on the role of RASAL2 in CRC has been
performed.
In the present study, tissue microarray
immunochemistry was used to evaluate RASAL2 expression in CRC
tissue samples at various stages of tumor development. In addition,
a RASAL2 loss-of-function cell model was established to clarify the
functional role of RASAL2 in CRC progression.
Materials and methods
Tumor tissue sample preparation and
immunohistochemical (IHC) analysis
A total of 10 CRC samples were obtained during
initial surgery in patients at the Hepatobiliary and Enteric
Surgery Research Center of Xiangya Hospital (Central South
University, Changsha, China) between December 2014 and June 2015.
Samples included tumor, wild-type and metastatic tissue. The
patients included 6 men and 4 women, with an age range of 32–72
years. All patients showed lymph node metastasis. The reverse
transcription-quantitative polymerase chain reaction (RT-qPCR) was
used to analyze the expression of RASAL2 mRNA in the tissue
samples. All experiments were approved by the Ethics Committee of
Xiangya Hospital.
A tissue microarray containing 80 CRC specimens was
obtained from Auragene Bioscience (cat no. TC0217; Changsha,
China). Tissues for microarray analysis were fixed using 3%
methanol for 15 min and subsequently incubated at 104°C for 32 min
in citrate buffer (pH 6.0). The microarray was left to cool for 10
min at room temperature prior to blocking with 10% goat serum
(Auragene Bioscience) for 15 min at room temperature. The
microarray was incubated with the RASAL2 primary antibody (1:200;
cat. no. sc-390605; Santa Cruz Biotechnology, Inc., Dallas, TX,
USA) overnight at 4°C, prior to a 15 min incubation with
horseradish peroxidase-labeled chain mildew avidin (cat. no.
P003H-1; Auragene Bioscience) at room temperature. The
antibody-labeled microarray was subsequently incubated with
3,3′-diaminobenzidine for 5 min at room temperature.
Cell culture and transfection
Human LoVo, HCT116 and SW620 CRC cell lines, and the
wild-type colon cell line NCM460, were purchased from the American
Type Culture Collection (Manassas, VA, USA). All cells were
maintained in Dulbecco's modified Eagle's medium (DMEM)
supplemented with 10% fetal bovine serum (FBS) (both Thermo Fisher
Scientific, Inc., Waltham, MA, USA), 100 U/ml penicillin and 100
µg/ml streptomycin, and incubated at 37°C with 5% CO2.
Transfection was carried out using Lipofectamine® 2000
Reagent (Invitrogen; Thermo Fisher Scientific, Inc.), according to
the manufacturer's protocol. RASAL2-short hairpin (sh)RNA
(GCCTTCCACCTCTTCATAGTA) and control (Ct)-shRNA plasmids
(pRNAT-U6.1-RASAL2) were obtained from Auragene Bioscience. The
Ct-shRNA sequence was synthesized by scrambling the RASAL2-shRNA
sequence. All subsequent experiments were carried out using
transfected cells.
RT-qPCR
Total RNA was extracted using TRIzol®
reagent (Thermo Fisher Scientific, Inc.) according to the
manufacturer's protocol. The RevertAid H Minus First Strand cDNA
Synthesis kit (Thermo Fisher Scientific, Inc.) was used to convert
RNA into cDNA. cDNA was quantified via qPCR using the Power SYBR
Green kit (Toyobo Co., Ltd., Osaka, Japan) and the Applied
Biosystems® 7500 Real-Time PCR system (Thermo Fisher
Scientific, Inc.). PCR thermocyling conditions were as follows:
Denaturation at 95°C for 3 min, followed by 35 cycles of 95°C for
10 sec and 58°C for 30 sec. Primer sequences are illustrated in
Table I. β-actin was used as an
endogenous control and the 2−ΔΔCq method was used to
quantify relative mRNA expression (17).
 | Table I.PCR primer sequences. |
Table I.
PCR primer sequences.
| Gene | Primer sequences |
|---|
| RASAL2 | F:
TCAACAAGGAGAAGGAGATACC |
|
| R:
GGAAACTGACCCTCGGAAG |
| E-cadherin | F:
CTCGGCCTGAAGTGACTCGTAAC |
|
| R:
CAGCAACGTGATTTCTGCATTTC |
| Vimentin | F:
GACGCCATCAACACCGAGTT |
|
| R:
CTTTGTCGTTGGTTAGCTGGT |
| N-Cadherin | F:
CAGTATCCGGTCCGATCTGC |
|
| R:
GTCCTGCTCACCACCACTAC |
| β-actin | F:
AGGGGCCGGACTCGTCATACT |
|
| R:
GGCGGCACCACCATGTACCCT |
Western blot analysis
The cells were lysed using 2X lysis buffer (cat. no.
P002A; Auragene Bioscience) on ice for 15 min. Protein (20 µg) was
separated by 10% SDS-PAGE and transferred onto a polyvinylidene
difluoride membrane. After blocking with 5% milk at room
temperature for 40 min, the membrane was incubated with primary
antibodies against Raf-1 serine/threonine kinase proto-oncogene
(Raf-1; cat no. ab32025; 1:250), phosphorylated (p)-Raf-1
(ab130572; 1:250) (both Abcam, Cambridge, UK), ERK (cat no.
Sc-292838; 1:500), p-ERK (cat no. sc-101761; 1:500), E-cadherin
(cat no. sc-31020; 1:500), N-cadherin (cat no. sc-7939; 1:500),
vimentin (YT4480; 1:1,000) (all Santa Cruz Biotechnology, Inc.) and
β-actin (cat no. LCA01; 1:2,000; Auragene Biosciences) at 4°C
overnight. Subsequently, the membrane was incubated with
horseradish peroxidase-conjugated goat anti-rabbit (cat. no.
111-035-003) or goat anti-mouse (cat. no. 111-035-008) secondary
antibodies (both 1:5,000; Jackson ImmunoResearch Laboratories,
Inc., West Grove, PA, USA), which was detected using an enhanced
chemiluminescence kit (Auragene Bioscience). β-actin was used as
the endogenous control. Image-Pro Plus software (version 6.0; Media
Cybernetics, Inc., Rockville, MD, USA) was used to quantify the
intensity of the bands.
MTT assay
The proliferation ability of cells was measured
using the MTT cell viability assay. RASAL2-shRNA and Ct-shRNA
plasmids were transfected into LoVo and HCT116 cells, and
subsequently seeded into 96-well plates at a density of
3×103 cells/well and incubated for 3 days at 37°C. Every
24 h, 10 µl 0.5 mg/ml MTT solution (Sigma-Aldrich; Merck Millipore,
Darmstadt, Germany) was added to each well and incubated for 4 h at
37°C. The absorbance values were measured at 490 nm using a
microplate reader.
Wound healing assay
Scratch wounds were made using sterile 20 µl pipette
tips when the cells had reached between 80 and 90% confluence.
Cells were washed with serum-free PBS twice and cultured in fresh
DMEM supplemented with 10% FBS for 24 h at 37°C. The medium was
subsequently changed to DMEM supplemented with 3% FBS. Images were
captured at 0 and 48 h using an inverted microscope.
Colony formation assay
A total of 48 h following transfection, 300 cells
were plated into 6-well plates. After 10 days, visible colonies
were fixed using 4% methanol for 15 min and stained with 1 ml
Giemsa. The number of colonies was counted manually and normalized
to the control group. Experiments were performed in triplicate.
Cell invasion assay
Cell invasion assays were performed in 24-well
plates using an 8.0 µm BD Biocoat™ Matrigel™ Invasion Chamber (BD
Biosciences, Franklin Lakes, NJ, USA). A total of 0.5 ml DMEM
containing 10% FBS was added to the lower chamber at 37°C for 2 h
and 0.2 ml (1×106 cells/ml) of cell solution was seeded
into the upper chamber in serum-free culture medium. Cells were
incubated for 24 h at 37°C. Cells were subsequently stained using
crystal violet for 20 min and dissolved in 10% acetic acid.
Absorbance at 570 nm was measured using a microplate reader. All
experiments were performed independently in triplicate.
Cell adhesion assay
Cells were transfected with the RASAL2-shRNA or
Ct-shRNA plasmids for 48 h at 37°C. A total of 100 µl cell solution
was seeded into fibronectin-coated 96-well plates (5×105
cells/well) and incubated at 37°C for 2 h. Plates were washed with
PBS four times to remove non-adherent cells, and the remaining
cells were stained using crystal violet for 20 min and dissolved in
10% acetic acid. Absorbance at 570 nm was measured using a
microplate reader. The percentage of cell adhesion was calculated
relative to the original seeded cell solution's fluorescence at 570
nm (set to 100%).
Statistical analysis
Statistical analyses were performed using SPSS 17.0
software (SPSS, Inc., Chicago, IL, USA). Differences were evaluated
using the Student t-test or one-way analysis of variance, followed
by Fisher's least significant difference and Student-Newman-Keuls
post hoc tests. Values are presented as the mean ± standard
deviation. P<0.05 was considered to indicate a statistically
significant difference.
Results
Downregulated RASAL2 expression is
associated with CRC tumor grade and International Federation of
Gynecology and Obstetrics (FIGO) stage
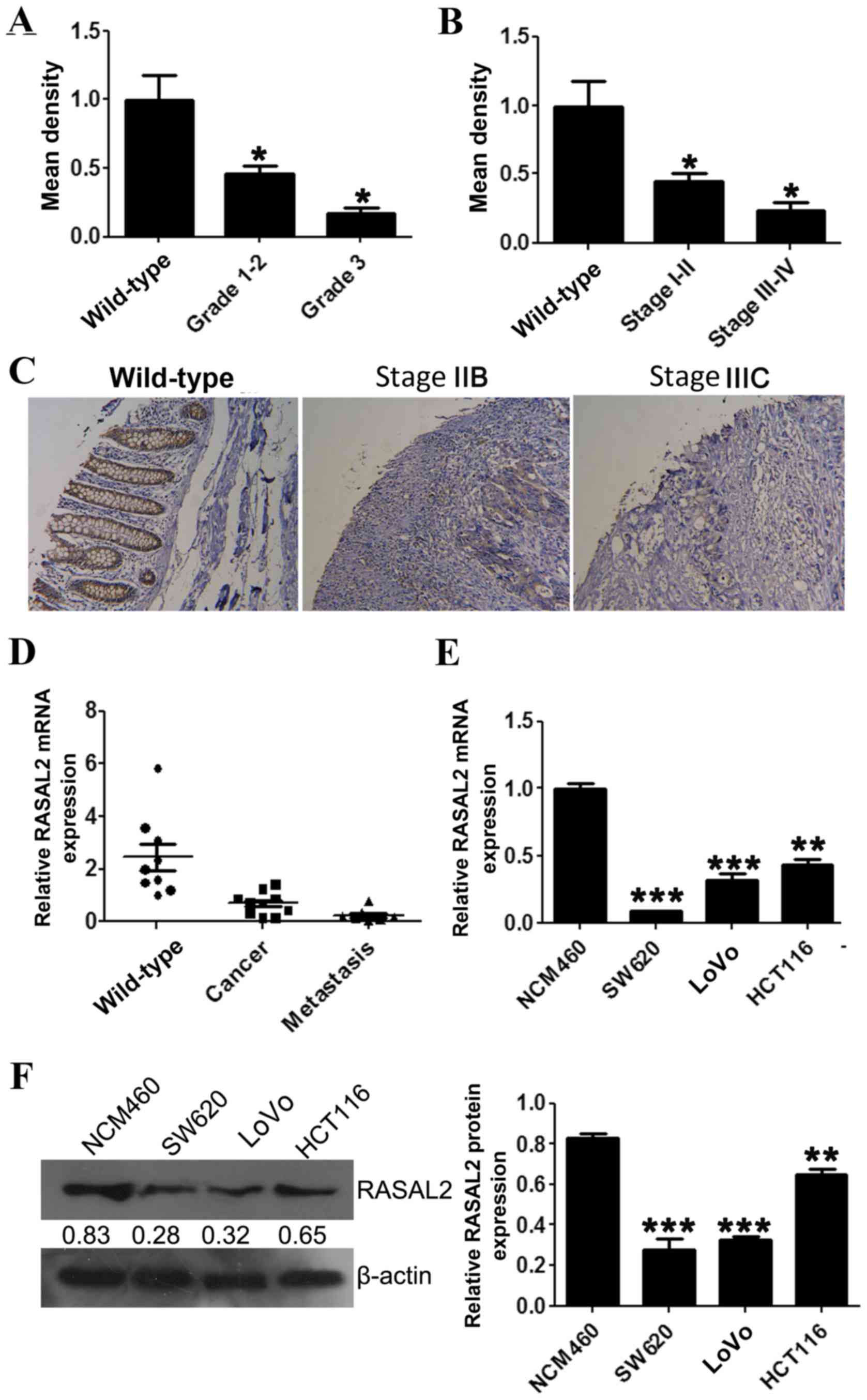
RASAL2 protein and mRNA expression levels in CRC
tissues were evaluated using an IHC microarray and RT-qPCR,
respectively. The IHC and RT-qPCR results confirmed that RASAL2
protein and mRNA levels were decreased in tumor tissue samples
compared with wild-type colorectal tissues (Fig. 1A-D). Furthermore, significantly
decreased RASAL2 protein expression was correlated with increased
CRC tumor grade (P<0.05; Fig. 1A).
Significantly decreased RASAL2 protein expression was also
correlated with increased FIGO stage (18) (P<0.05; Fig. 1B).
RASAL2 mRNA and protein expression was evaluated in
the CRC cell lines SW620, LoVo and HCT116, and the wild-type colon
cell line NCM460, using RT-qPCR and western blotting, respectively.
Consistent with the results observed in the tumor tissue samples,
RASAL2 mRNA expression was significantly decreased in the CRC cell
lines compared with the NCM460 cell line (all P<0.01; Fig. 1E), which was accompanied by a similar
decrease in protein levels (all P<0.01; Fig. 1F). These results indicate that RASAL2
is downregulated in CRC tissues and cell lines.
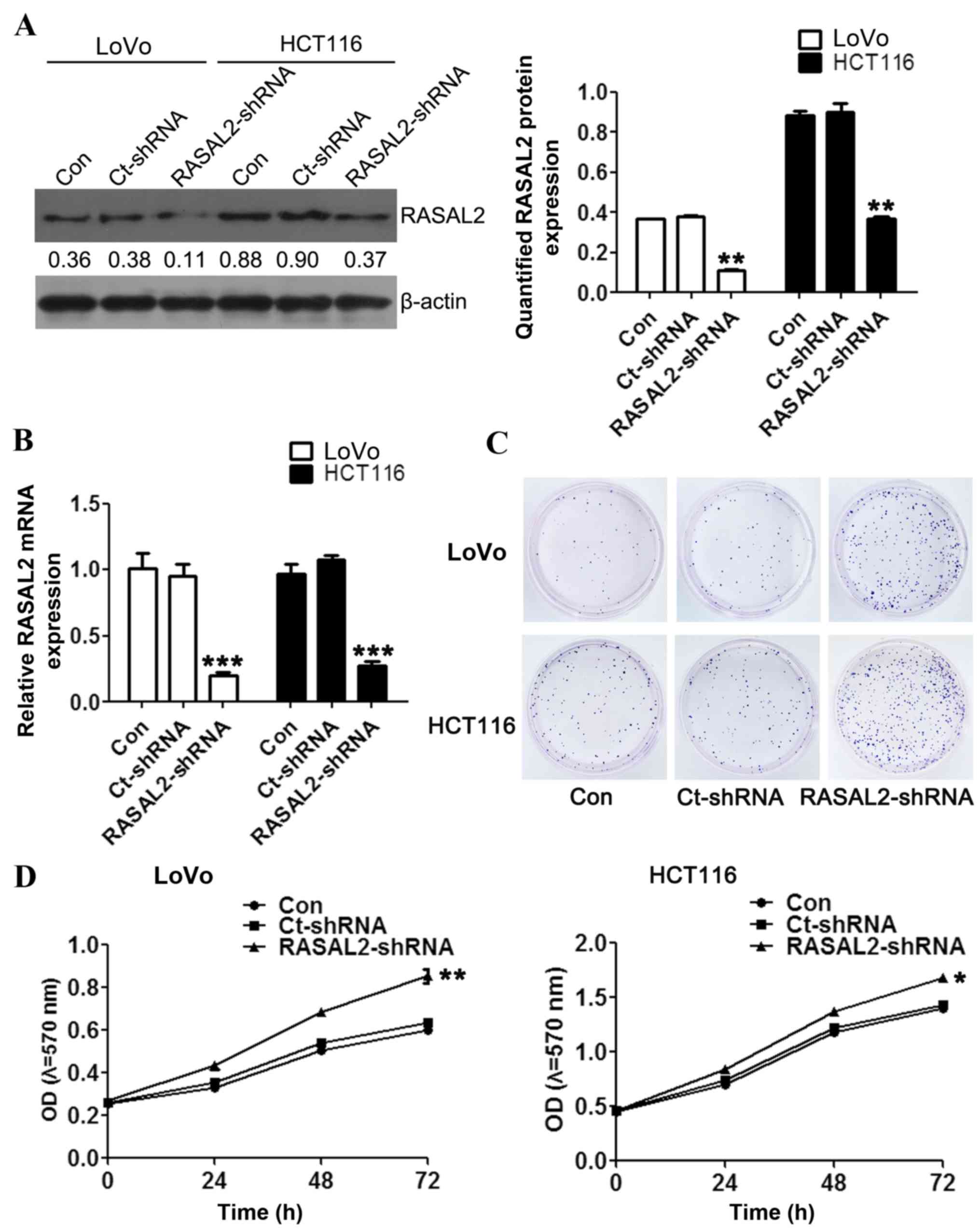
Loss of RASAL2 promotes CRC cell
proliferation
To investigate the function of RASAL2 in CRC
pathogenesis, RASAL2 loss-of-function models were established using
shRNA interference in the CRC cell lines LoVo and HCT116.
Significantly decreased RASAL2 mRNA expression was detected in
cells following transfection with the RASAL2- or Ct-shRNA (all
P<0.01 vs. the control group; Fig.
2A), which was accompanied by a similar decrease in protein
levels (both P<0.01; Fig. 2B). In
order to investigate the effect of RASAL2 knockdown on
tumorigenesis, cell proliferation was measured following
transfection using the colony formation and MTT assays,
respectively. The colony formation assay demonstrated increased
colony formation in the LoVo and HCT116 cells following
RASAL2-shRNA transfection compared with the untransfected and
Ct-shRNA-transfected cells (Fig. 2C).
Furthermore, RASAL2-shRNA transfection significantly enhanced the
viability of the CRC cells compared with the untransfected cells
(both P<0.05; Fig. 2D). These
results indicate that RASAL2 suppresses CRC cell proliferation
in vitro.
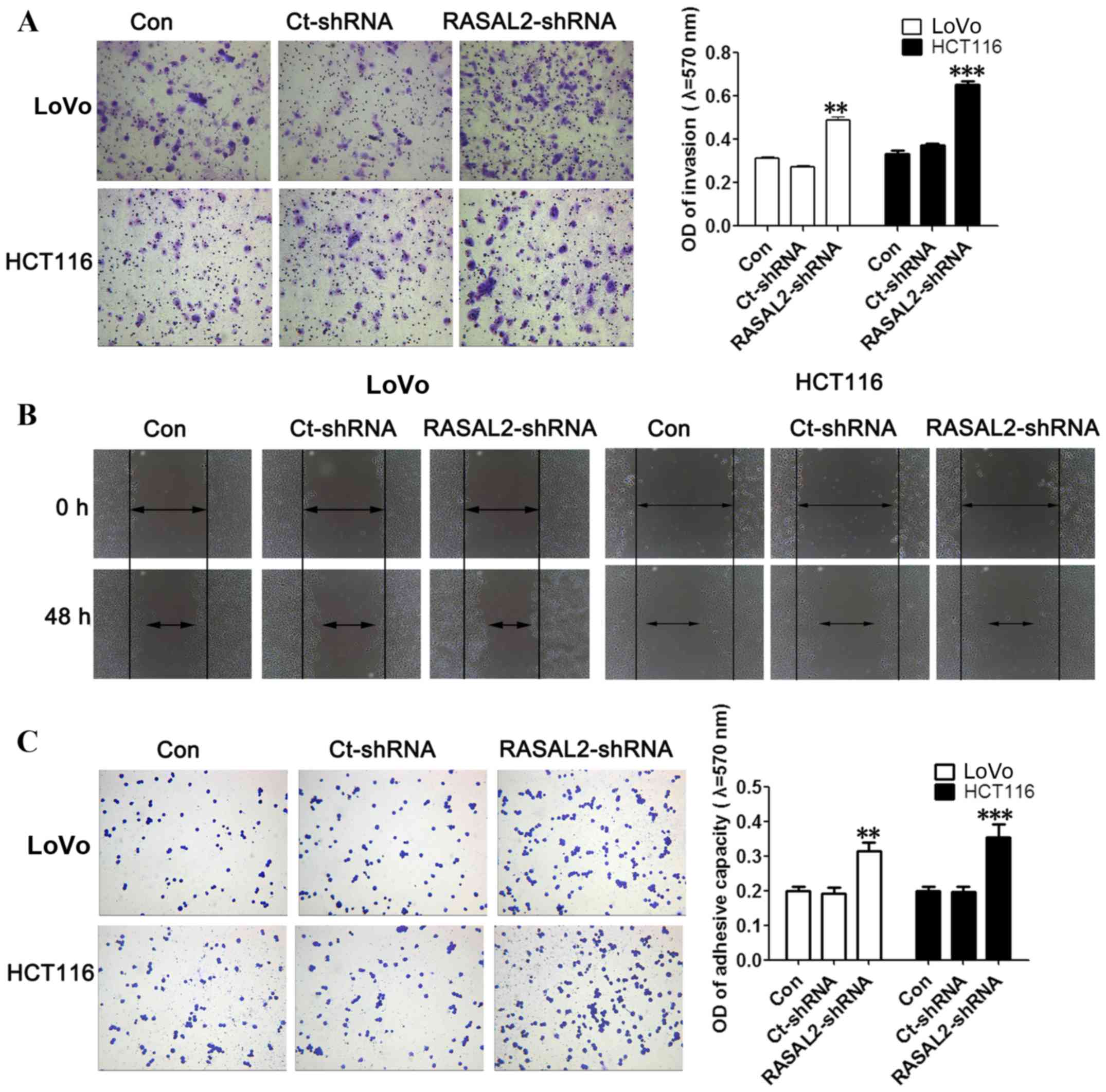
RASAL2 suppresses CRC cell migration
and invasion
The effect of RASAL2 knockdown on the invasion and
migration of LoVo and HCT116 cells was investigated using the cell
invasion and wound healing assays, respectively. RASAL2 knockdown
was demonstrated to significantly promote invasion (both P<0.01;
Fig. 3A) and markedly increase
migration (Fig. 3B) in LoVo and
HCT116 cells compared with the untransfected control cells. In
addition, the effect of RASAL2 on cell adhesion was evaluated using
a cell adherence assay. RASAL2 knockdown significantly enhanced the
adhesive ability of LoVo and HCT116 cells compared with the
untransfected control cells (both P<0.01; Fig. 3C). These results demonstrate that
RASAL2 knockdown promotes CRC cell invasion, migration and adhesion
in vitro.
RASAL2 functions upstream of the ERK
and EMT signaling pathways
EMT is a critical event in the progression and
metastasis of cancer (19). To
investigate whether the effect of RASAL2 knockdown on CRC cell
migration and invasion was EMT-mediated, the expression of several
EMT markers was assessed using western blotting and RT-qPCR
analyses. RASAL2 knockdown significantly increased the protein and
mRNA levels of the mesenchymal markers vimentin and N-cadherin,
while significantly decreasing the protein and mRNA expression of
the epithelial marker E-cadherin, compared with the untransfected
control cells (all P<0.05; Fig.
4). Furthermore, activation of the ERK and Raf-1 signaling
pathways was evaluated using western blot analysis. This identified
no significant difference in the protein expression of Raf-1 and
ERK between the cell lines (all P>0.05; Fig. 4A). However, RASAL2 knockdown led to
significantly increased expression of p-Raf-1 and p-ERK compared
with the untransfected control cells (all P<0.05; Fig. 4A.) These results indicate that RASAL2
knockdown activates the ERK signaling pathway and promotes EMT in
CRC cells in vitro.
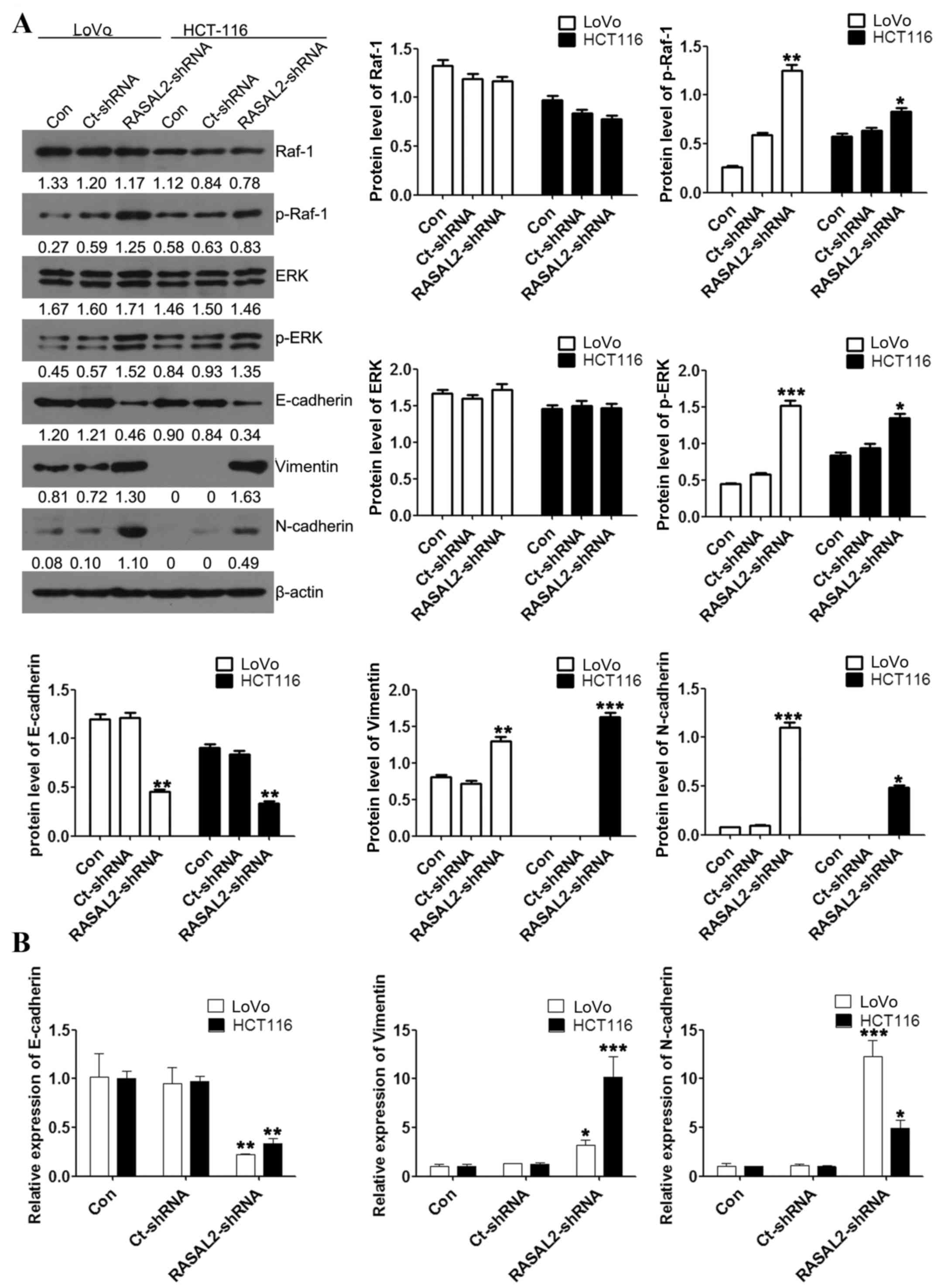 | Figure 4.RASAL2 regulates EMT by activating the
ERK signaling pathway in LoVo and HCT116 cells. (A) Raf-1, p-Raf-1,
ERK, p-ERK, E-cadherin, vimentin and N-cadherin protein expression
levels relative to β-actin were assessed using western blotting.
(B) E-cadherin, vimentin and N-cadherin mRNA expression levels
relative to β-actin were assessed using the reverse
transcription-quantitative polymerase chain reaction. Values are
presented as the mean ± standard deviation of triplicate data.
*P<0.05, **P<0.01, ***P<0.001 vs. the Con cells. RASAL2,
RAS protein activator-like 2; EMT, epithelial-mesenchymal
transition; ERK, mitogen-activated protein kinase 1; Raf-1, Raf-1
serine/threonine kinase proto-oncogene; p, phosphorylated; Con,
untransfected; Ct, control; shRNA, short hairpin RNA. |
Discussion
The RAS gene family contains a number of
proto-oncogenes involved in the proliferation, differentiation,
apoptosis, cytoskeletal organization and motility of tumor cells.
Mutations in RAS genes frequently lead to permanently activated RAS
and thus its downstream effectors, which causes aberrant cell
proliferation and potential tumorigenesis (14). Previous studies have demonstrated that
mutation of RAS genes is the primary mechanism of aberrant RAS
activation (8,20,21).
However, other underlying mechanisms for RAS activation exist,
which lead to abnormal activation of the RAS signaling pathway,
including deregulation of RAS GTPase-activating proteins (RAS-GAPs)
or RAS guanine nucleotide exchange factors (22).
A novel mechanism for the permanent activation of
the RAS signaling pathway is the knockdown of RAS-GAPs. RASAL2 was
reported to be mutated or suppressed in RAS- and ERK-hyperactivated
breast cancer, where RASAL2 downregulation promoted tumor growth,
progression and metastasis in two distinct human xenograft tumor
models (14). Silencing of the
RAS-GAPs RASAL1, DAB2 interacting protein (DAB2IP) and
neurofibromin 1 leads to aberrant RAS activation in HCC cells
(23). In a previous study,
reactivation of RASAL1, DAB2IP and paired like homeodomain 1
inhibited cell growth, and induced apoptosis, while their silencing
increased proliferation and inhibited apoptosis (23). In the present study, RASAL2 expression
was downregulated in CRC tissue samples and cells. RASAL2 knockdown
in CRC cells led to significantly increased expression of p-Raf-1
and p-ERK, indicating hyperactivation of the RAS signaling
pathway.
Epithelial cell adhesion and communication, with the
extracellular matrix and neighbouring cells, serves a fundamental
role in tumour differentiation and progression (24,25). In
addition, EMT is a critical event in cancer metastasis, during
which differentiated epithelial cells obtain the characteristics of
mesenchymal cells and subsequently migrate (26). During this process, epithelial marker
expression is reduced and mesenchymal marker expression is
increased (26). EMT in mammary
epithelial cells typically facilitates tumor cell migration and
invasion (27). Previously, RASAL2
was identified as a mediator of EMT and metastasis in ovarian
cancer, lung adenocarcinoma and breast cancer (14–16). In
the present study, RASAL2 was demonstrated to be a mediator of EMT,
migration and invasion in CRC cells.
In conclusion, the present study confirmed that
RASAL2 knockdown promotes proliferation and metastasis in CRC
cells, through the activation of the RAS/ERK signaling pathway and
subsequent induction of EMT in vitro. Therefore, further
investigations of therapeutic strategies targeting RASAL2 for the
treatment of CRC are warranted.
Acknowledgements
The present study was supported by the National and
International Cooperation in Science and Technology Special Project
(grant no. 2013DFA32150) and the Young Teachers Booster Project
(grant no. 502010334).
References
|
1
|
Mao JD, Wu P, Huang JX, Wu J and Yang G:
Role of ERK-MAPK signaling pathway in pentagastrin-regulated growth
of large intestinal carcinoma. World J Gastroenterol.
20:12542–12550. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Shin HR, Carlos MC and Varghese C: Cancer
control in the Asia Pacific region: Current status and concerns.
Jpn J Clin Oncol. 42:867–881. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Chen W, Zheng R, Zhang S, Zhao P, Li G, Wu
L and He J: Report of incidence and mortality in China cancer
registries, 2009. Chin J Cancer Res. 25:10–21. 2013.PubMed/NCBI
|
|
4
|
Wang X, Song ZF, Xie RM, Pei J, Xiang MF
and Wang H: Analysis of death causes of in-patients with malignant
tumors in sichuan cancer hospital of China from 2002 to 2012. Asian
Pac J Cancer Prev. 14:4399–4402. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Li Y, Lv Z, He G, Wang J, Zhang X, Lu G,
Ren X, Wang F, Zhu X, Ding Y, et al: The SOX17/miR-371-5p/SOX2 axis
inhibits EMT, stem cell properties and metastasis in colorectal
cancer. Oncotarget. 6:9099–9112. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Hayashi H, Beppu T, Sakamoto Y, Miyamoto
Y, Yokoyama N, Higashi T, Nitta H, Hashimoto D, Chikamoto A and
Baba H: Prognostic value of Ki-67 expression in conversion therapy
for colorectal liver-limited metastases. Am J Cancer Res.
5:1225–1233. 2015.PubMed/NCBI
|
|
7
|
Ruers T and Bleichrodt RP: Treatment of
liver metastases, an update on the possibilities and results. Eur J
Cancer. 38:1023–1033. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Downward J: Targeting RAS signalling
pathways in cancer therapy. Nat Rev Cancer. 3:11–22. 2003.
View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Malumbres M and Barbacid M: RAS oncogenes:
The first 30 years. Nat Rev Cancer. 3:459–465. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Cherfils J and Zeghouf M: Regulation of
small GTPases by GEFs, GAPs, and GDIs. Physiol Rev. 93:269–309.
2013. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Xu Y, Deng Y, Ji Z, Liu H, Liu Y, Peng H,
Wu J and Fan J: Identification of thyroid carcinoma related genes
with mRMR and shortest path approaches. PLoS one. 9:e940222014.
View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Stefanska B, Cheishvili D, Suderman M,
Arakelian A, Huang J, Hallett M, Han ZG, Al-Mahtab M, Akbar SM,
Khan WA, et al: Genome-wide study of hypomethylated and induced
genes in patients with liver cancer unravels novel anticancer
targets. Clin Cancer Res. 20:3118–3132. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Feng M, Bao Y, Li Z, Li J, Gong M, Lam S,
Wang J, Marzese DM, Donovan N, Tan EY, et al: RASAL2 activates RAC1
to promote triple-negative breast cancer progression. J Clin
Invest. 124:5291–5304. 2014. View
Article : Google Scholar : PubMed/NCBI
|
|
14
|
McLaughlin SK, Olsen SN, Dake B, De Raedt
T, Lim E, Bronson RT, Beroukhim R, Polyak K, Brown M, Kuperwasser C
and Cichowski K: The RasGAP gene, RASAL2, is a tumor and metastasis
suppressor. Cancer Cell. 24:365–378. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Huang Y, Zhao M, Xu H, Wang K, Fu Z, Jiang
Y and Yao Z: RASAL2 down-regulation in ovarian cancer promotes
epithelial-mesenchymal transition and metastasis. Oncotarget.
5:6734–6745. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Li N and Li S: RASAL2 promotes lung cancer
metastasis through epithelial-mesenchymal transition. Biochem
Biophys Res Commun. 455:358–362. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Livak KJ and Schmittgen TD: Analysis of
relative gene expression data using real-time quantitative PCR and
the 2(−Delta Delta C(T)) method. Methods. 25:402–408. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Kamata S and Uchida M: Relationship
between TNM-classification and survival rate in cases of carcinoma
of the oral cavity: A comparison between UICC and AJCC TNM staging
systems. Gan No Rinsho. 32:1339–1343. 1986.(In Japanese).
PubMed/NCBI
|
|
19
|
Cao H, Xu E, Liu H, Wan L and Lai M:
Epithelial-mesenchymal transition in colorectal cancer metastasis:
A system review. Pathol Res Pract. 211:557–569. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Karnoub AE and Weinberg RA: Ras oncogenes:
Split personalities. Nat Rev Mol Cell Biol. 9:517–531. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Pylayeva-Gupta Y, Grabocka E and Bar-Sagi
D: RAS oncogenes: Weaving a tumorigenic web. Nat Rev Cancer.
11:761–774. 2011. View
Article : Google Scholar : PubMed/NCBI
|
|
22
|
Chen H, Zhao JY, Qian XC, Cheng ZY, Liu Y
and Wang Z: RASAL1 attenuates gastric carcinogenesis in nude mice
by blocking RAS/ERK signaling. Asian Pac J Cancer Prev.
16:1077–1082. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Calvisi DF, Ladu S, Conner EA, Seo D,
Hsieh JT, Factor VM and Thorgeirsson SS: Inactivation of Ras
GTPase-activating proteins promotes unrestrained activity of
wild-type Ras in human liver cancer. J Hepatol. 54:311–319. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Kalluri R and Weinberg RA: The basics of
epithelial-mesenchymal transition. J Clin Invest. 119:1420–1428.
2009. View
Article : Google Scholar : PubMed/NCBI
|
|
25
|
An J, Lv J, Li A, Qiao J, Fang L, Li Z, Li
B, Zhao W, Chen H and Wang L: Constitutive expression of Bcl-2
induces epithelial-Mesenchymal transition in mammary epithelial
cells. BMC Cancer. 15:4762015. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Zhang W, Shi X, Peng Y, Wu M, Zhang P, Xie
R, Wu Y, Yan Q, Liu S and Wang J: HIF-1α promotes
epithelial-mesenchymal transition and metastasis through direct
regulation of ZEB1 in colorectal cancer. PLoS One. 10:e01296032015.
View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Keleg S, Büchler P, Ludwig R, Büchler MW
and Friess H: Invasion and metastasis in pancreatic cancer. Mol
Cancer. 2:142003. View Article : Google Scholar : PubMed/NCBI
|