Introduction
Glioblastoma is the most common and malignant type
of brain tumor in humans, accounting for 50–60% of gliomas and 20%
of all intracranial tumors (1).
Traditionally, the optimal treatment strategy for glioblastoma
treatment was surgical resection (2).
However, due to their highly infiltrative and invasive growth
pattern, as well as the existence of multifocal and subclinical
lesions, it is difficult to totally eradicate such tumors by
surgical resection (2). Concomitant
chemotherapy and adjuvant radiotherapy have been used
conventionally in the treatment of glioblastoma patients, but the
median survival time of these patients remains low (3). One of the major factors contributing to
the failures of chemotherapy is the highly infiltrative nature of
glioblastoma in normal brain tissue (4). Invasion and metastasis are defining
properties of cancer malignancy (5).
Due to high invasiveness and metastasis, glioblastoma cells
frequently invade the surrounding normal brain tissue, leading to
an indistinct boundary between normal brain tissue and glioma
tissue (6).
The epithelial-mesenchymal transition (EMT) is a
critical cellular process required in normal organogenesis and
cellular responses to stress, inflammation and hypoxia. Cancer
cells also make use of the mechanisms involved in EMT to undergo
invasion and metastasis. The biological processes of EMT include
enhanced migratory capacity and invasiveness, elevated resistance
to apoptosis, and greatly increased production of extracellular
matrix (7–9). EMT is modulated by the repression of
E-cadherin, and upregulation of mesenchymal markers, including
N-cadherin, β-catenin and zinc finger protein SNAI2 (Slug)
(10–12). Furthermore, transition to a
mesenchymal gene expression pattern is associated with the
simultaneous acquisition of cancer stem cell-like properties
(13–15).
BIX01294 (Bix; a diazepin-quinazolinamine derivate)
is a euchromatic histone-lysine N-methyltransferase 2 (G9a)
inhibitor, and it has been reported that treatment with Bix
efficiently blocks cell proliferation and induces
autophagy-associated cell death (16,17).
However, the effect of persistent low-dose Bix treatment on
glioblastoma cell migration and metastasis remains to be
elucidated. In the present study, it was shown that sequential
treatment with low-dose Bix significantly enhances in vitro
U251 cell migration, invasion and EMT-associated gene expression.
Consistent with the in vitro results, following nude mouse
experimentation, Bix-treated cells formed larger and more numerous
lung tumor metastasis nodules in vivo. Furthermore,
neurosphere formation was increased in U251 cells multi-treated
with low-dose Bix when compared with untreated and single-treated
cells. The results of the present study suggest that characteristic
changes were exhibited in the glioma cells exposed to continuous
treatment with low-dose Bix.
Materials and methods
Cell and culture conditions
Human glioblastoma cell line U251 was purchased from
the American Type Culture Collection (Manassas, VA, USA). Cells
were maintained in Dulbecco's Modified Eagle Medium (Welgene,
Daegu, South Korea) supplemented with 10% fetal bovine serum (FBS;
HyClone; GE Healthcare Life Sciences, Logan, UT, USA) and 5%
antibiotic-antimycotic (Gibco; Thermo Fisher Scientific, Inc.,
Waltham, MA, USA). Cells were cultured at 37°C in a humidified
atmosphere of 5% CO2.
Drug treatment
Stock solutions of 10 mM Bix (Tocris Bioscience,
Bristol, UK) were dissolved in dimethyl sulfoxide (DMSO) and
diluted in culture medium to 1, 2, 5 and 10 µM for cell treatment.
For a single treatment of Bix (SBT), the cells were incubated at
37°C overnight, and subsequently treated with 1, 2, 5 and 10 µM for
1 day (24 h). For the sequential treatment of Bix (SeBT), the cells
were incubated at 37°C overnight, and treated with 1 µM Bix. Bix
and medium was replaced every 2 days for 2 weeks.
Cell proliferation assay
Cell proliferation was assessed using the methyl
thiazolyl tetrazolium (MTT) colorimetric assay. U251 cells
(2×105 cells/well) were seeded in 6-well plates and
incubated at 37°C overnight. Following 24 h of incubation, the
medium was removed and replaced with the experimental medium. Cells
were treated with 1, 2, 5 and 10 µM of Bix for SBT or 1 µM of Bix
for SeBT. Subsequently, cells were washed twice with
phosphate-buffered saline (PBS), and 5 mg/ml MTT diluted in PBS was
added to each well for a total of 4 h. Following removal of the MTT
solution, solubilization solution (DMSO/ethanol, 1:1 ratio) was
added to each well to dissolve the formazan crystals. The
absorbance at 570 nm was measured using a Paradigm™ Detection
Platform (Beckman Coulter, Inc., Brea, CA, USA) and analyzed using
Multimode Analysis Software version 3.3.0.9 (Beckman Coulter,
Inc.).
Chromatin isolation by small-scale
biochemical fractionation
Chromatin was isolated from the nuclei of U251
cells. Briefly, 5×106 cells were harvested and washed
with PBS. The pellet was resuspended in buffer A [10 mM
4-(2-hydroxyethyl)-1-piperazineethanesulfonic acid (pH 7.9), 10 mM
KCl, 1.5 mM MgCl2, 0.34 M sucrose, 10% glycerol, 1 mM
dithiothreitol and protease inhibitor cocktail (Roche Diagnostics,
Indianapolis, IN, USA)] with 0.1% Triton X-100. Following an 8-min
incubation on ice, nuclei (fraction P1) were collected by
centrifugation (1,300 × g for 5 min at 4°C). The P1 nuclei
were washed once with buffer A and lysed in buffer B [3 mM
ethylenediaminetetraacetic acid, 0.2 mM ethylene glycol-bis
(β-aminoethyl ether)-N,N,N',N'-tetraacetic acid, 1 mM
dithiothreitol and protease inhibitor cocktail (Roche Diagnostics)]
for 30 min. Following lysis, the insoluble chromatin (fraction P2)
and soluble fractions were separated by centrifugation (1,700 ×
g for 5 min at 4°C). The insoluble P2 fraction was washed
once with buffer B and re-suspended in sodium dodecyl sulfate
(SDS)-Laemmli buffer and boiled for 10 min. Subsequently, the
chromatin samples were quantified by Coomassie Brilliant Blue
staining.
Reverse transcription-quantitative
polymerase chain reaction (RT-qPCR) analysis
RT-qPCR was performed using the SYBR-Green method
(18). Total RNA was isolated using
the RNeasy Mini kit (Qiagen, Inc., Valencia, CA, USA). The quantity
of RNA was measured using a NanoDrop spectrophotometer (Thermo
Fisher Scientific, Inc.), and 1 µg of RNA was reverse-transcribed
using the iScript™ cDNA synthesis kit (Bio-Rad Laboratories, Inc.,
Hercules, CA, USA). The reaction was performed at 25°C for 5 min,
42°C for 30 min and terminated by heating at 85°C for 5 min.
RT-qPCR was performed using a C1000™ thermal cycler (Bio-Rad
Laboratories, Inc.,) with the Maxima™ SYBR™ Green qPCR master mix
(Thermo Fisher Scientific, Inc.). PCR amplification was performed
at 94°C for 5 min, followed by 40 cycles of 94°C for 30 sec, 55°C
for 30 sec and 72°C for 30 sec. β-actin was used as an internal
control (19). To normalize the
various RNA samples, the Ct values of β-actin were subtracted from
the Ct values of the gene of interest (ΔCt). Then, the ΔΔCt values
were determined by comparing the ΔCt values of the control and the
experimental RNA samples. The following RT-qPCR primers were used:
E-cadherin forward, 5′-GTC ACT GAC ACC AAC GAT AAT CCT-3′ and
reverse, 5′-TTT CAG TGT GGT GAT TAC GAC CTT A-3′; N-cadherin
forward, 5′-CAC TGC TCA GGA CCC AGA T-3′ and reverse, 5′-TAA GCC
GAG TGA TGG TCC-3′; β-catenin forward, 5′-GCC ATT TTA AGC CTC TCG
GT-3′ and reverse, 5′-ATT GTC CAC GCT GGA TTT TC-3′; Slug forward,
5′-AGA TGC ATA TTC GGA CCC AC-3′ and reverse, 5′-CCT CAT GTT TGT
GCA GGA GA-3′; Kruppel-like factor 4 (KLF4) forward, 5′-GCT GCC GAG
GAC CTT CTG-3′ and reverse, 5′-AAG TCG CTT CAT GTG GGA GA-3′;
cluster of differentiation 133 (CD133) forward, 5′-ACA TGA AAA GAC
CTG GGG G-3′ and reverse, 5′-GAT CTG GTG TCC CAG CAT G-3′; sex
determining region Y-box 2 (SOX2) forward, 5′-TTG CTG CCT CTT TAA
GAC TAG GA-3′ and reverse, 5′-CTG GGG CTC AAA CTT CTC TC-3′;
octamer-binding transcription factor 4 (OCT4) forward, 5′-GTA TTC
AGC CAA ACG ACC ATC and reverse, 5′-CTG GTT CGC TTT CTC TTT CG; and
β-actin forward, 5′-AGC GAG CAT CCC CCA AAG TT-3′ and reverse,
5′-GGG CAC GAA GGC TCA TCA TT-3′.
Western blot analysis
Cells were harvested by trypsinization and washed
twice with PBS. Subsequently, cell pellets were collected by
centrifugation (2,500 × g for 5 min at 4°C) and then lysed
by lysis buffer [150 mM NaCl, 20 mM Tris-HCl (pH 7.5), 1% NP40, 5
mM EDTA, 10 mM NAF, 1 mM Na3VO4, 1 mM DTT and
1X PIC]. The protein concentrations were determined using a Bio-Rad
protein assay kit (Bio-Rad Laboratories, Inc.). Cell extracts were
boiled in SDS sample buffer (0.5 M Tris-HCl, pH 6.8, 4% SDS, 20%
glycerol, 0.1% bromophenol blue and 10% β-mercaptoethanol) and 20
µg of protein was loaded onto 4–15% Mini-Protean TGX™ precast gels
(Bio-Rad Laboratories, Inc.) and underwent electrophoresis,
followed by transferral to a polyvinylidene difluoride membrane (GE
Healthcare Life Sciences, Chalfont, UK). The membranes were blocked
with 5% skim milk (BD Biosciences, San Jose, CA, USA) at room
temperature for 30 min, and incubated overnight at 4°C with the
primary antibodies. On the following day, the membranes were washed
with TBS-T (20 mM Tris, 137 mM NaCl, pH 7.6, 0.2% Tween 20) for 10
min, 3 times each, and incubated with the secondary antibody at
37°C for 1 h: goat-anti-rabbit horseradish-peroxidase conjugated
antibody (dilution, 1:5,000; catalog no., ab6721, Invitrogen;
Thermo Fisher Scientific, Inc.) or rabbit-anti-mouse
horseradish-peroxidase conjugated antibody (dilution 1:5,000;
catalog no., ab97046, Invitrogen; Thermo Fisher Scientific, Inc.).
The immunoreactive proteins were detected using enhanced
chemiluminescence (Thermo Fisher Scientific, Inc.). Immunoblots
were quantified using the Fusion FX5 system (Vilber Lourmat, Marne,
France). The following primary antibodies (dilution, 1:1,000) were
used for immunoblot analysis: anti-poly (ADP-ribose) polymerase
(PARP) (catalog no. sc-7150; Santa Cruz Biotechnology, Inc.,
Dallas, TX, USA), anti-caspase-3 (catalog no. 9662; Cell Signaling
Technology, Inc., Danvers, MA, USA), anti-microtubule-associated
protein light chain 3 (LC3) B (catalog no. #3868; Cell Signaling
Technology, Inc.), anti-histone H3 (catalog no. ab1791; Abcam,
Cambridge, UK), anti-dimethyl-histone H3-Lys9 (catalog no. 07–212;
Merck Millipore, Darmstadt, Germany), anti-E-cadherin (catalog no.
sc-7870; Santa Cruz Biotechnology, Inc.), anti-N-cadherin (catalog
no. 610921; BD Biosciences), anti-β-catenin (catalog no. 610153; BD
Biosciences), anti-Slug (catalog no. sc-166476; Santa Cruz
Biotechnology, Inc.), anti-KLF4 (catalog no. ab72543; Abcam),
anti-CD133 (catalog no. ab19898; Abcam), anti-SOX2 (catalog no.
sc-20088; Santa Cruz Biotechnology, Inc.), anti-OCT4 (catalog no.
611203; BD Biosciences) and anti-β-actin (dilution, 1:5,000;
catalog no. A5441; Sigma-Aldrich; Merck Millipore).
Migration and invasion assays
Cell migration and invasion assays were performed
using Transwell® chambers (8-µm pore size; Corning Life
Sciences, Lowell, MA, USA). A total of 2×104 cells were
resuspended in 0.2 ml of serum-free growth medium for both the cell
migration and invasion assays. For the migration assay, the cells
were added to the interior of the inserts. Growth medium (0.8 ml)
supplemented with 10% (v/v) FBS was added to the lower chamber.
Following 24 h incubation at 37°C, the cells attached to the upper
surface of the filter were removed using a cotton swab, and
migratory cells on the lower surface of the filter were fixed and
stained for 15 min with 0.25% crystal violet (Merck Millipore), 10%
formaldehyde, and 80% methanol. Subsequently, the inserts were
washed 5 times with double-distilled H2O and
photographed (magnification, ×200). Migrated cells were determined
by counting cells in 5 microscopic fields (randomly selected) per
well and the extent of migration was expressed as the mean number
of cells per microscopic field (20).
Cells were imaged using phase contrast microscopy (Nikon Eclipse
80i; Nikon Corporation). For the invasion assay, cells were added
to the interior of the inserts pre-coated with 10 mg/ml growth
factor-reduced Matrigel™ (BD Biosciences). Growth medium (0.8 ml)
supplemented with 10% (v/v) FBS was added to the lower chamber.
Following 24 h incubation at 37°C, the inserts were processed as
described above for the migration assay.
Sphere formation assay
U251 cells were suspended at a density of 5,000
cells/well in complete NeuroCult™ NS-A Basal media (Stemcell
Technologies, Inc., Vancouver, BC, Canada) and plated in triplicate
wells on a 24-well ultra-low attachment culture plate (Corning Life
Sciences). Cells were incubated at 37°C for 7–10 days. The number
of spheres was imaged using inverted microscopy (Nikon Eclipse TS
100, Nikon Corporation) and sphere diameters were determined using
Image-Pro Plus version 7.0 software (Media Cybernetics, Rockville,
MD, USA).
In vivo experiments
Female 6-week-old nude BALB/c mice were obtained
from Japan SLC., Inc. (Hamamatsu, Japan), and were raised under
following conditions (temperature, 22±2°C; humidity, 50±10%; access
to food, ad libitum; light/dark cycle, 12/12 h). All animal
protocols used in this study were approved by the Institutional
Animal Care and Use Committee at Dongnam Institute of Radiological
& Medical Sciences (DIRAMS; Busan, Republic of Korea). Female
BALB/c nude mice (average weight, 22 g; n=27) were randomly divided
into three groups (control, SBT and SeBT). Control- or
single/sequential-Bix treated U251 cells (2×106 cells)
were inoculated intravenously into BALB/c nude mice. After 4 weeks,
the mice were sacrificed using anesthesia and exsanguination, and
lungs were corrected by dissection.
Statistical analysis
All statistical analyses were performed using
Microsoft Excel 2007 (Microsoft Corporation, Redmond, WA, USA).
Student's t-test was used for statistical comparisons. P<0.05
was considered to indicate a statistically significant
difference.
Results
Sequential treatment with Bix does not
affect cell viability in human U251 glioblastoma cells
Bix has been reported to have antiproliferative
effects in various types of cancer cell (16,17).
Therefore it was initially examined whether Bix treatment inhibited
the proliferation of U251 glioblastoma cells. U251 cells were
prepared according to the experimental scheme shown in Fig. 1A. Subsequently, viability of cells was
assessed by the MTT assay. When cells were treated with 1, 2, 5 and
10 µM Bix, the viability of U251 cells decreased in a
dose-dependent manner (P=0.143, P=0.001, P=0.002 and P=0.001,
respectively) (Fig. 1B). To
investigate whether accumulation of non-lethal doses of Bix led to
changes in cell survival rates, U251 cells were exposed to
sequential low-dose (1 µM) Bix treatment. Notably, U251 cell
viability was unaffected by multiple low-dose treatments of Bix
(Fig. 1B). As Bix has been observed
to attenuate G9a activity (17), the
effect of Bix on H3K9 dimethylation levels was initially examined.
The results revealed that the single treatment of Bix decreased the
levels of H3K9 demethylation, whereas sequential treatment of Bix
had no such effect (Fig. 1C).
Expression levels of apoptosis- and autophagy-associated proteins
were also measured by western blot analysis following treatment
with various concentrations of Bix. The results of the present
study demonstrated that a single treatment with 10 µM Bix in U251
cells induced PARP1 and caspase-3 cleavage, whereas the cleavage of
PARP1 and caspase-3 was not stimulated with sequential treatment
with 1 µM Bix (Fig. 1C). LC3B is
widely used as a biomarker of autophagy, during which, LC3B-I is
converted to LC3B-II through lipidation by a ubiquitin-like system
that allows LC3B to become associated with autophagic vesicles
(21). Similar to PARP1, the
conversion of LC3B-I to LC3B-II was only induced with high doses of
Bix (5 and 10 µM), and not with the sequential treatment of
low-dose Bix (Fig. 1C). These results
indicate that accumulation of low doses of Bix does not influence
U251 cell proliferation and apoptosis.
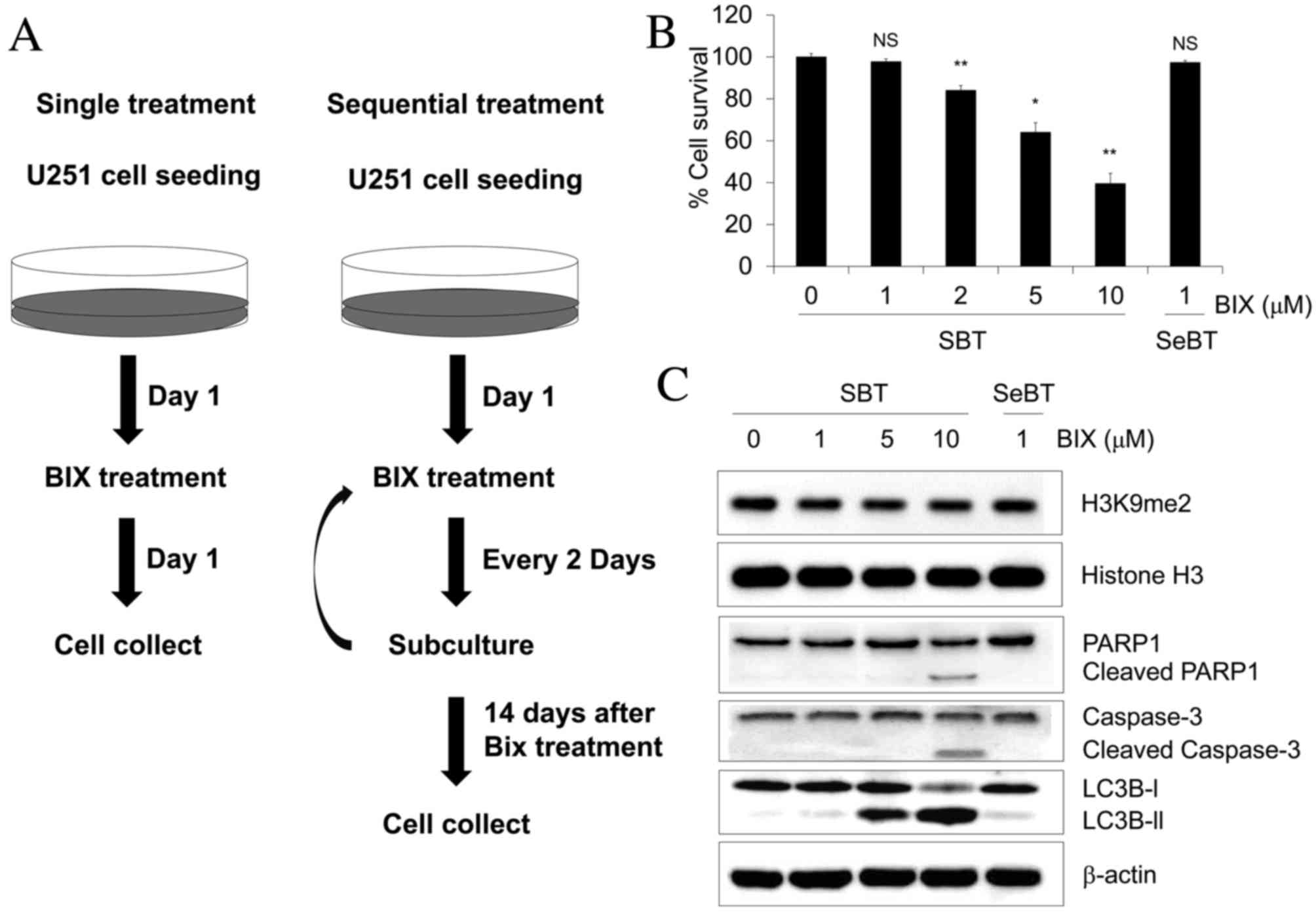 | Figure 1.Cell survival effect of Bix treatment
following the indicated dosing schedule in human U251 glioblastoma
cells. (A) Experimental scheme for Bix treatment. (B) The cells
were treated with 1, 2, 5 or 10 µM Bix and the methyl thiazolyl
tetrazolium assay was performed. (C) Levels of dimethylated H3K9
and apoptosis/autophagy-associated genes investigated by western
blot analysis. Relative optical densities of dimethylated H3K9
level were normalized to total histone H3 (1, 0.88±0.01, 0.80±0.05,
0.91±0.03, 0.92±0.1, respectively, mean ± standard error of the
mean). β-actin was used as the loading control. Western blotting
was performed at the indicated dosages (0, 1, 5 and 10 µM). Bix,
BIX01294; SBT, single treatment of Bix; SeBT, sequential treatment
of Bix; PARP, poly (ADP-ribose) polymerase. P-values were
calculated using the Student's t-test. *P<0.01 vs. the control;
**P<0.001 vs. the control; N.S. no significance. |
Sequential treatment with low-dose Bix
enhances migration and invasion of U251 cells
Subsequently, the effect of sequential treatment
with low-dose Bix (1 µM) on the migration and invasion capability
of U251 glioblastoma cells was investigated. As presented in
Fig. 2A and B, SBT cells exhibited an
approximately 3-fold increase in the number of migratory cells
(P=0.011), compared with untreated control cells, whereas SeBT
cells showed an approximately 12-fold increase (P=0.044).
Similarly, SeBT cells exhibited a 2.5-fold increase in the number
of invasive U251 cells, compared with control cells (P=0.001)
(Fig. 2A and B). As the ability to
invade and migrate is correlated with EMT capacity (22), the expression levels of EMT-associated
factors such as E-cadherin, N-cadherin, Slug and β-catenin were
evaluated by RT-qPCR (Fig. 2C) and
western blotting (Fig. 2D) in SBT and
SeBT cells. The expression of E-cadherin was demonstrably decreased
following SeBT, compared with the control (P=0.008) and SBT
(P=0.002), whereas the expression of N-cadherin and β-catenin was
significantly increased following SeBT compared with the control
(N-cadherin, P<0.001; β-catenin, P<0.001) and SBT
(N-cadherin, P=0.004; β-catenin, P=0.007). In the case of Slug, the
expression was marginally increased by SeBT, compared with the
control (Fig. 2C and D). The results
of the present study suggest that accumulation of low-dose Bix
induces invasion and migration, as well as EMT, in U251 cells.
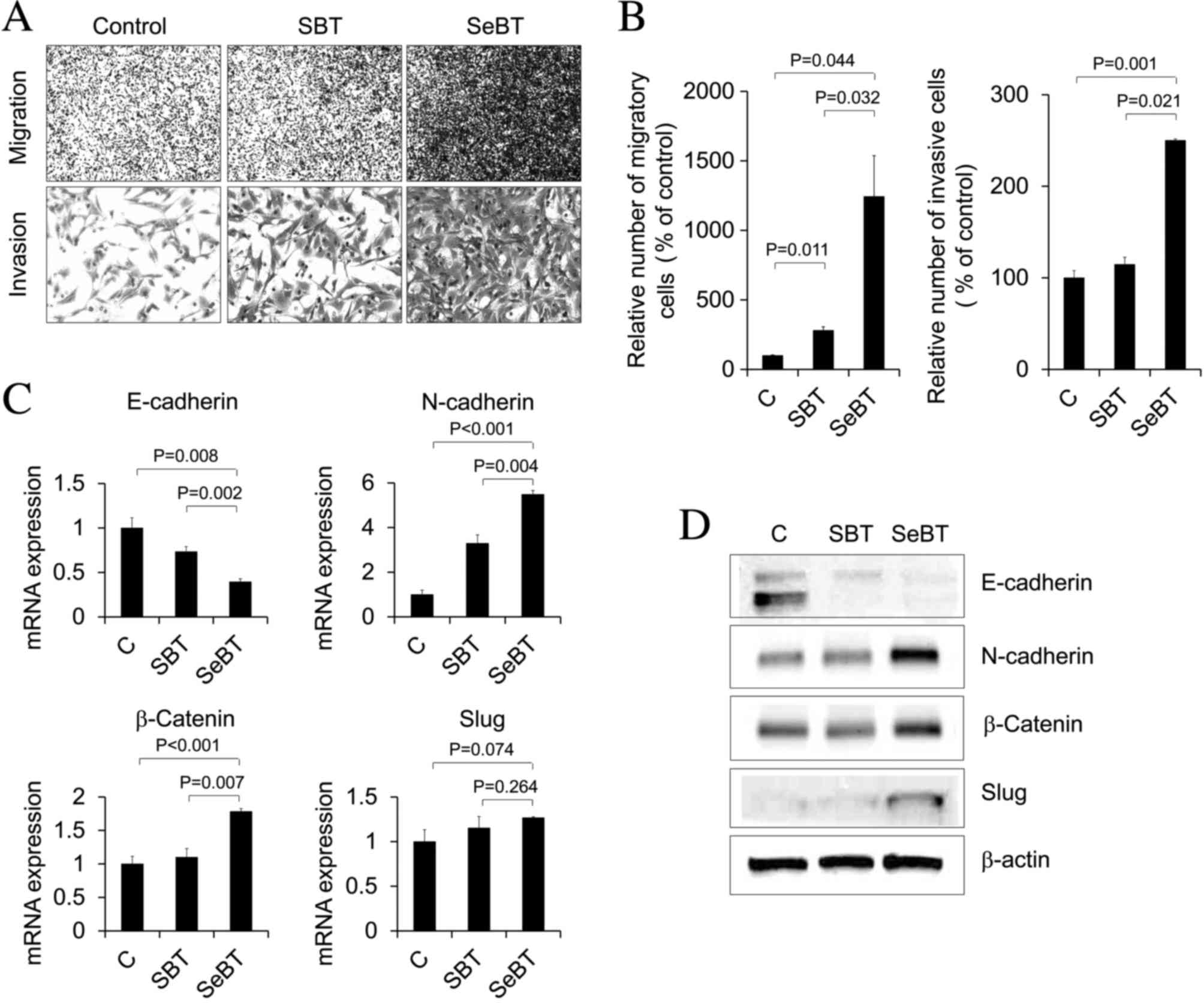 | Figure 2.Enhanced cellular migration and
invasion following sequential treatment with Bix in U251 cells. (A)
Images of migratory (upper panel) and invasive (bottom panel) cells
under phase contrast microscopy (magnification, ×200) are shown.
(B) The number of migratory and invasive cells was counted and
presented. Values are presented as the mean ± standard deviation.
Representative data from three independent trials. (C) E-cadherin,
N-cadherin, β-catenin and Slug mRNA expression patterns under SBT
or SeBT were assessed by reverse transcription-quantitative
polymerase chain reaction analysis. Results represent mRNA levels
normalized to the levels of β-actin mRNA. Relative E-cadherin,
N-cadherin, β-catenin and Slug mRNA levels are presented as the
mean ± standard deviation of three independent experiments. (D)
Western blot analysis demonstrated the change in total protein
expression of E-cadherin, N-cadherin, β-catenin and Slug. The
expression level of β-actin was used as loading control. Bix,
BIX01294; SBT, single treatment of Bix; SeBT, sequential treatment
of Bix; Slug, zinc finger protein SNAI2; C, control. |
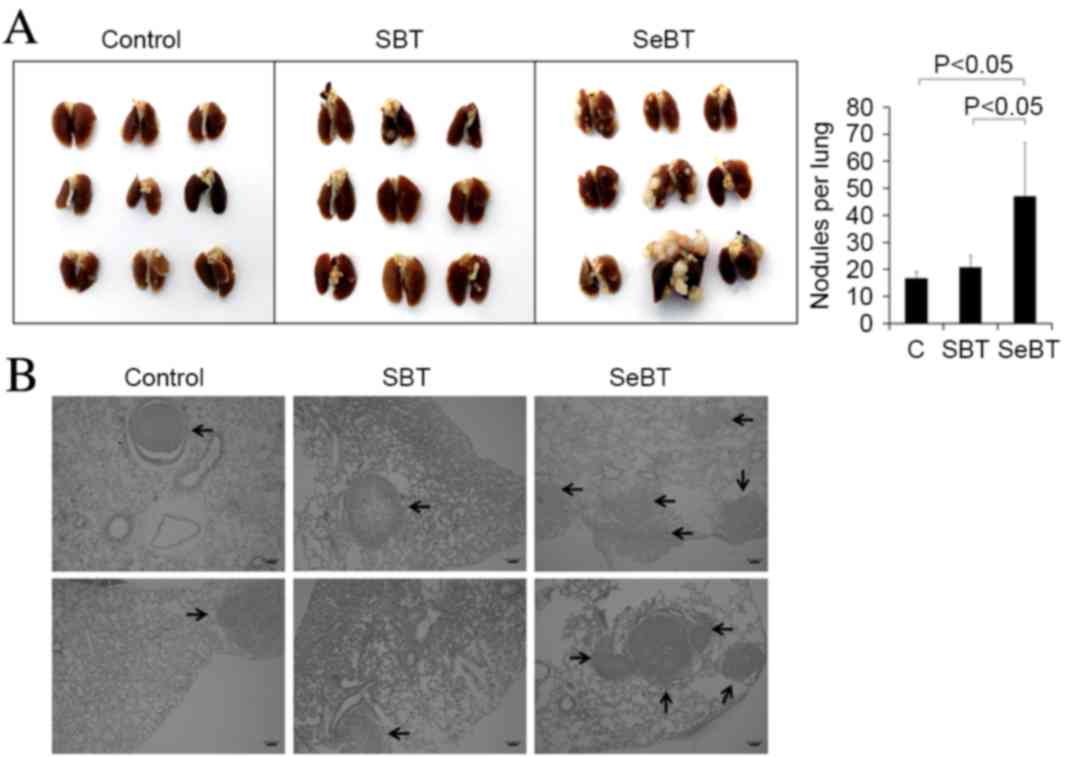
Sequential treatment with Bix enhances
U251 cell pulmonary metastasis in vivo
To investigate the impact of sequential treatment
with Bix on U251 cell metastasis, a pulmonary metastasis experiment
was performed. A total of 2×106 control, SBT or SeBT
U251 cells were intravenously injected into nude BALB/c mice. After
4 weeks, the mice were sacrificed as described in the materials and
methods section, and lung tissues were visualized (Fig. 3A, left panel). The results showed that
the mice injected with SeBT cells had larger sized nodules in lung
tissue, compared with mice injected with control (P=0.013) or SBT
(P=0.022) cells. Furthermore, the largest number of metastatic lung
nodules was observed in mice injected with SeBT cells (Fig. 3A, right panel). Induced nodule
formation by sequential treatment with Bix was also observed in
tissue sections (Fig. 3B). The
results of the present study support the idea that accumulation of
low-dose Bix enhances the ability of U251 cells to migrate and
metastasize.
Sequential treatment with Bix
increases the expression of cancer stem cell markers and
neurosphere formation
CD133 is a critical marker of self-renewal in
glioblastoma cells and its expression has been implicated in
tumorigenic potential, as well as metastatic ability (23). KLF4, SOX2 and OCT4 also have essential
roles in stem cell biology (23). To
investigate whether the SeBT-induced metastatic potential of U251
cells has any correlation with stem cell-like properties, the
expression of these markers was identified using RT-qPCR (Fig. 4A). The expression of CD133, SOX2 and
OCT4 was significantly upregulated in SeBT cells, compared with the
control (P=0.001, P=0.002 and P=0.001, respectively) and SBT
(P=0.021, P=0.096 and P=0.015, respectively) cells. By contrast,
the expression of transcription factor KLF4 was decreased in SeBT
cells, when compared with control (P=0.003) and SBT (P=0.009) cells
(Fig. 4A). Similar patterns were
observed in western blot analysis (Fig.
4B). These data demonstrate that SeBT of cancer cells may
induce cancer stem cell expansion. As glioma stem cells have been
reported to form neurospheres at an increased efficiency compared
with differentiated cells, the present study sought to determine
the effect of SeBT cells on sphere formation, as this may be an
indicator of a cancer stem cell-like phenotype. Control, SBT or
SeBT cells were seeded on 24-well low adhesion plates at a density
of 5,000 cells per well, and after 1 week, spheres were visualized
(Fig. 4C). It was demonstrated that
SeBT cells formed a greater number of spheres, compared with
control (P=0.011) and SBT (P=0.045) cells. In addition, SeBT cells
formed larger spheres, compared with control and SBT cells. The
results of the present study suggest that prolonged exposure to
low-dose Bix induces metastatic potential and self-renewal in U251
cells.
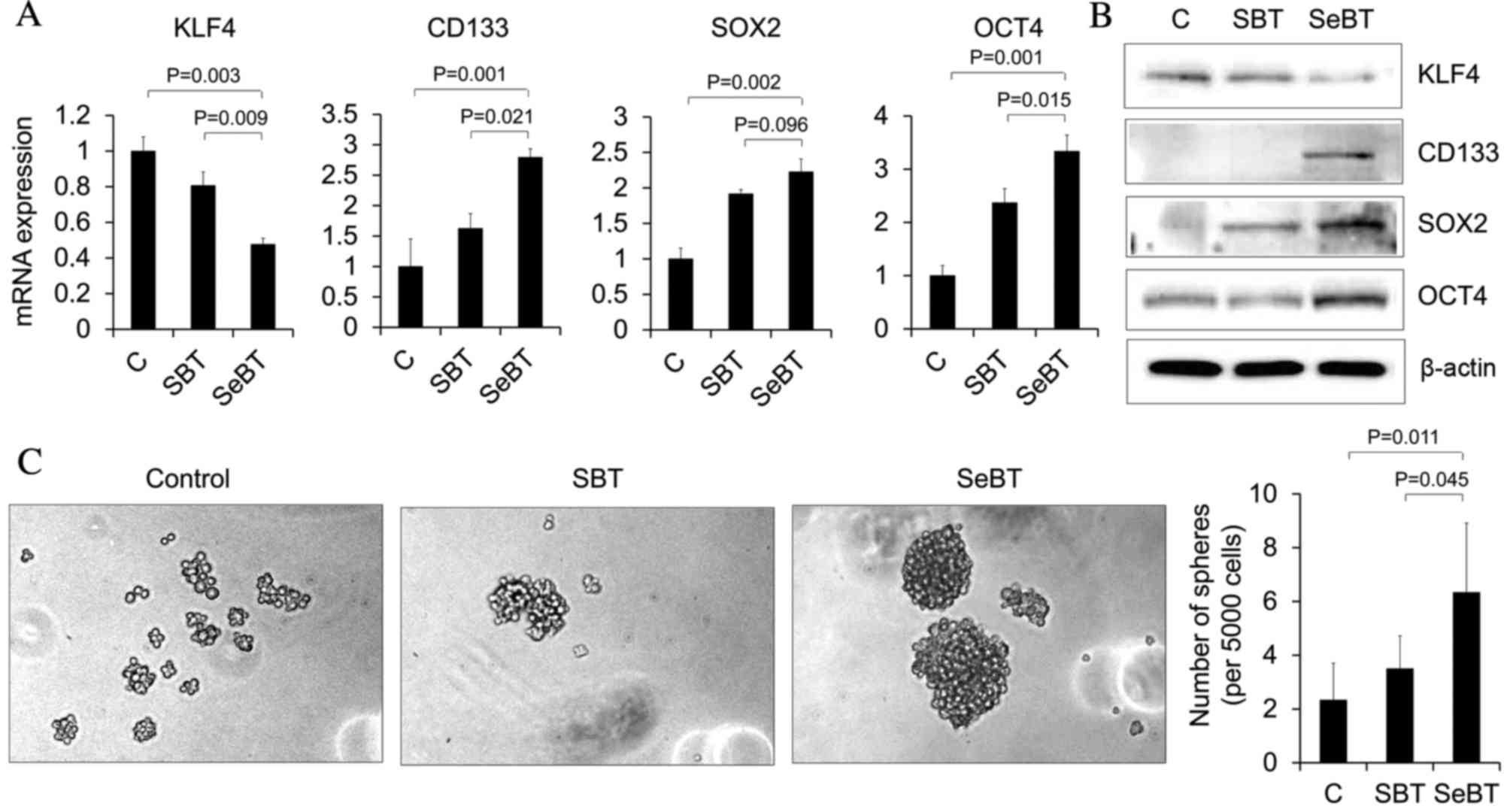 | Figure 4.Sequential treatment with Bix
increases the expression of cancer stem cell markers and
neurosphere formation in U251 cells. (A) KLF4, CD133, SOX2 and OCT4
mRNA expression patterns under single or sequential treatment with
Bix were assessed by reverse transcription-quantitative polymerase
chain reaction. Results represent mRNA levels normalized to the
levels of β-actin mRNA. Relative KLF4, CD133, SOX2 and OCT4 mRNA
levels are presented as the mean ± standard deviation of three
independent experiments. (B) Western blot analysis demonstrated the
change in total protein expression of KLF4, CD133, SOX2 and OCT4.
The expression level of β-actin was used as loading control. (C)
Representative images of neurosphere formation following single or
sequential treatment of Bix. Bix, BIX01294; KLF4, Kruppel-like
factor 4; CD133, cluster of differentiation 133; SOX2, sex
determining region Y-box 2; OCT4, octamer-binding transcription
factor 4; SBT, single treatment of Bix; SeBT, sequential treatment
of Bix; C, control. |
Discussion
Glioblastoma is characterized by rapid cell
proliferation, as well as high invasion and migration properties
(24). The median survival time of
glioblastoma is 6–14 months; in addition to this, a 2-year survival
rate is observed in only 10% of patients and 5-year survival is
observed in <5% (25). Previously,
advanced chemotherapy and adjuvant radiotherapy have been used to
treat glioblastoma patients, but the response is generally
unfavorable and the toxic chemotherapy and radiotherapy frequently
produce serious adverse side effects (26). The primary cause of treatment failure
is invasion and metastasis of glioblastoma into normal brain
tissues (4).
In the present study, it was demonstrated that
accumulation of low-dose Bix induces the migratory and invasive
phenotype in U251 glioblastoma cells. The results of the present
study revealed that Bix inhibited proliferation and induced
apoptosis in U251 cells in a dose-dependent manner. However,
multiple treatments of low-dose Bix did not influence cell survival
or apoptosis. Although accumulation of damage by sequential
treatment with non-lethal doses of Bix was not able to change the
cell proliferation rate, SeBT cells exhibited an increased
migratory and invasive phenotype compared with control or SBT
cells. EMT is a critical event in the development of invasive and
metastatic potential in cancer progression (6). EMT is known to be initiated through the
regulation of the Wnt/β-catenin, transforming growth factor-β,
Smad, Notch and Hedgehog signaling pathways (22,27,28). This
complex signaling network begins with the cleavage of E-cadherin,
which causes adherent junction breakdown and an indirect increase
in the expression of transcription factors, including Slug and
β-catenin. The repression of the epithelial gene E-cadherin
indirectly leads to an increase in the expression of N-cadherin and
other mesenchymal genes (29). It was
also demonstrated that enhanced migratory and invasive ability,
which is exhibited by SeBT cells, was accompanied by downregulation
of E-cadherin, and upregulation of N-cadherin and β-catenin. This
indicated that the EMT process is involved in the induced migration
and invasion of SeBT cells. Notably, the mRNA expression level of
Slug was not markedly altered between SBT and SeBT cells. However,
the protein level of Slug was upregulated in SeBT cells.
Futhermore, these gene expression changes were not directly induced
by the Bix-mediated inhibition of G9a, as H3K9 dimethylation levels
were not correspondingly altered. The results of the present study
suggest that there may be another regulatory mechanism of Bix,
leading to EMT stimulation. In order to confirm this phenomenon
in vivo, treated U251cells were injected into nude mice via
the tail vein. The results revealed that SeBT cell-injected mice
had more and larger nodules compared with control and SBT
cell-injected mice. Expansion of cancer stem cells also enhances
the invasive and metastatic potential of cancer cells (28). Cancer stem cells are tumor-initiating
cells found in the bulk of tumors, that possess the ability to
self-renew, divide and differentiate into multiple cell lineages.
They are able to initiate the formation of a new tumor, leading to
tumor recurrence and metastasis following removal of the primary
tumor (30).
The interplay between EMT and stemness signature has
become increasingly relevant to the field of cancer research.
Multiple studies have revealed that cancer stem cell properties may
contribute to tumor heterogeneity, maintenance, metastasis,
radioresistance and chemoresistance in a various types of cancer
(28,31). Such cells have the capacity to give
rise to whole tumors due to two fundamental properties: The ability
to self-renew, and the ability to differentiate into multiple cell
types (32). Critical genes for
self-renewal and pluripotency in human embryonic stem cells,
including KLF4, SOX2 and OCT4, are likewise elevated in cancer stem
cells (33). In addition, the surface
glycoprotein CD133 has been identified as a key marker of the
cancer stem cell subpopulation in glioblastoma and various types of
cancer (23). Consistent with
previous reports, SeBT cells exhibited increased expression levels
of CD133, SOX2 and OCT4 compared with control and SBT cells.
Although it remains unclear why KLF4 expression was decreased in
SeBT cells, this reduction may involve the enhanced invasive
capability of SeBT cells (34).
Cancer stem cells and normal stem cells have been reported to form
spheres at increased efficiency compared with differentiated cells
(35). The present results obtained
from a sphere formation assay demonstrated that SeBT cells were
able to form larger spheres compared with control and SBT cells,
indicating additional phenotypic changes caused by sequential
treatment with low-dose Bix. In conclusion, the results of the
present study indicate that SeBT cells induce distant invasion and
metastasis through mediation of EMT-promoting markers and that
these effects regulate cancer stem cell characteristics in
glioblastoma. These findings suggest that SeBT-induced
invasion/migration and/or cancer stem cell formation may be major
factors contributing to the failure of chemotherapy in glioblastoma
patients.
Acknowledgements
The present study was supported by the Dongnam
Institute of Radiological and Medical Science grant funded by the
Korean government (grant no. 50590-2015) and the Basic Science
Research Program through the National Research Foundation of Korea
(grant nos. NRF-2013, M2A2A, 7043665) funded by the Ministry of
Science, ICT & Future Planning.
References
|
1
|
Lehrer S, Green S, Ramanathan L,
Rosenzweig K and Labombardi V: No consistent relationship of
glioblastoma incidence and cytomegalovirus seropositivity in
whites, blacks, and Hispanics. Anticancer Res. 32:1113–1115.
2012.PubMed/NCBI
|
|
2
|
Hu Y, Lin X, Wang P, Xue YX, Li Z, Liu LB,
Yu B, Feng TD and Liu YH: CRM197 in combination with shRNA
interference of VCAM-1 displays enhanced inhibitory effects on
human glioblastoma cells. J Cell Physiol. 220:1713–1728. 2015.
View Article : Google Scholar
|
|
3
|
Stupp R, Hegi ME, Mason WP, van den Bent
MJ, Taphoorn MJ, Janzer RC, Ludwin SK, Allgeier A, Fisher B,
Belanger K, et al: Effects of radiotherapy with concomitant and
adjuvant temozolomide versus radiotherapy alone on survival in
glioblastoma in a randomised phase III study: 5-year analysis of
the EORTC-NCIC trial. Lancet Oncol. 10:459–466. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Oberoi RK, Parrish KE, Sio TT, Mittapalli
RK, Elmquist WF and Sarkaria JN: Strategies to improve delivery of
anticancer drugs across the blood-brain barrier to treat
glioblastoma. Neuro Oncol. 18:27–36. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Jiang WG, Sanders AJ, Katoh M, Ungefroren
H, Gieseler F, Prince M, Thompson SK, Zollo M, Spano D, Dhawan P,
et al: Tissue invasion and metastasis: Molecular, biological and
clinical perspectives. Semin Cancer Biol. 35:(Suppl). S244–S275.
2015. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Scott RW, Crighton D and Olson MF:
Modeling and imaging 3-dimensional collective cell invasion. J Vis
Exp pii. 35252011.
|
|
7
|
Yang J and Weinberg RA:
Epithelial-mesenchymal transition: At the crossroads of development
and tumor metastasis. Dev Cell. 14:818–829. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Kalluri R and Weinberg RA: The basics of
epithelial-mesenchymal transition. J Clin Invest. 119:1420–1428.
2009. View
Article : Google Scholar : PubMed/NCBI
|
|
9
|
Li L and Li W: Epithelial-mesenchymal
transition in human cancer: Comprehensive reprogramming of
metabolism, epigenetics, and differentiation. Pharmacol Ther.
150:33–46. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Lim J and Thiery JP:
Epithelial-mesenchymal transitions: Insights from development.
Development. 139:3471–3486. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Lamouille S, Xu J and Derynck R: Molecular
mechanisms of epithelial-mesenchymal transition. Nat Rev Mol Cell
Biol. 15:178–196. 2014. View
Article : Google Scholar : PubMed/NCBI
|
|
12
|
Zang M, Zhang B, Zhang Y, Li J, Su L, Zhu
Z, Gu Q, Liu B and Yan M: CEACAM6 promotes gastric cancer invasion
and metastasis by inducing epithelial-mesenchymal transition via
PI3K/AKT signaling pathway. PLoS One. 9:e1129082014. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Lo JF, Yu CC, Chiou SH, Huang CY, Jan CI,
Lin SC, Liu CJ, Hu WY and Yu YH: The epithelial-mesenchymal
transition mediator S100A4 maintains cancer-initiating cells in
head and neck cancers. Cancer Res. 71:1912–1923. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Blick T, Hugo H, Widodo E, Pinto C, Mani
SA, Weinberg RA, Neve RM, Lenburg ME and Thompson EW: Epithelial
mesenchymal transition traits in human breast cancer cell lines
parallel the CD44(hi/)CD24 (lo/−) stem cell phenotype in human
breast cancer. J Mammary Gland Biol Neoplasia. 15:235–252. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Kubicek S, O'Sullivan RJ, August EM,
Hickey ER, Zhang Q, Teodoro ML, Rea S, Mechtler K, Kowalski JA,
Homon CA, et al: Reversal of H3K9me2 by a small-molecule inhibitor
for the G9a histone methyltransferase. Mol Cell. 25:473–481. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Fan JD, Lei PJ, Zheng JY, Wang X, Li S,
Liu H, He YL, Wang ZN, Wei G, Zhang X, et al: The selective
activation of p53 target genes regulated by SMYD2 in BIX-01294
induced autophagy-related cell death. PLoS One. 10:e01167822015.
View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Kim Y, Kim YS, Kim DE, Lee JS, Song JH,
Kim HG, Cho DH, Jeong SY, Jin DH, Jang SJ, et al: BIX-01294 induces
autophagy-associated cell death via EHMT2/G9a dysfunction and
intracellular reactive oxygen species production. Autophagy.
9:2126–2139. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Schmittgen TD, Zakrajsek BA, Mills AG,
Gorn V, Singer MJ and Reed MW: Quantitative reverse
transcription-polymerase chain reaction to study mRNA decay:
Comparison of endpoint and real-time methods. Anal Biochem.
285:194–204. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Dheda K, Huggett JF, Bustin SA, Johnson
MA, Rook G and Zumla A: Validation of housekeeping genes for
normalizing RNA expression in real-time PCR. Biotechniques.
37:112–114. 2004.PubMed/NCBI
|
|
20
|
Uekita T, Tanaka M, Takigahira M, Miyazawa
Y, Nakanishi Y, Kanai Y, Yanagihara K and Sakai R:
CUB-domain-containing protein 1 regulates peritoneal dissemination
of gastric scirrhous carcinoma. Am J Pathol. 172:1729–1739. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Koukourakis MI, Kalamida D, Giatromanolaki
A, Zois CE, Sivridis E, Pouliliou S, Mitrakas A, Gatter KC and
Harris AL: Autophagosome proteins LC3A, LC3B and LC3C have distinct
subcellular distribution kinetics and expression in cancer cell
lines. PLos One. 10:e01376752015. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Wu Y and Zhou BP: New insights of
epithelial-mesenchymal transition in cancer metastasis. Acta
Biochim Biophys Sin (Shanghai). 40:643–650. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Auffinger B, Tobias AL, Han Y, Lee G, Guo
D, Dey M, Lesniak MS and Ahmed AU: Conversion of differentiated
cancer cells into cancer stem-like cells in a glioblastoma model
after primary chemotherapy. Cell Death Differ. 21:1119–1131. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Chintala SK, Tonn JC and Rao JS: Matrix
metalloproteinases and their biological function in human gliomas.
Int J Dev Neurosci. 17:495–502. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Schwartzbaum JA, Fisher JL, Aldape KD and
Wrensch M: Epidemiology and molecular pathology of glioma. Nat Clin
Pract Neurol. 2:494–503. 2006.quiz 1 p following 516. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Omuro A and DeAngelis LM: Glioblastoma and
other malignant gliomas: A clinical review. Jama. 310:1842–1850.
2013. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Lee JM, Dedhar S, Kalluri R and Thompson
EW: The epithelial-mesenchymal transition: New insights in
signaling, development, and disease. J Cell Biol. 172:973–981.
2006. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Singh A and Settleman J: EMT, cancer stem
cells and drug resistance: An emerging axis of evil in the war on
cancer. Oncogene. 29:4741–4751. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Osorio LA, Farfán NM, Castellón EA and
Contreras HR: SNAIL transcription factor increases the motility and
invasive capacity of prostate cancer cells. Mol Med Rep.
13:778–786. 2016.PubMed/NCBI
|
|
30
|
Chow AK, Ng L, Lam CS, Wong SK, Wan TM,
Cheng NS, Yau TC, Poon RT and Pang RW: The Enhanced metastatic
potential of hepatocellular carcinoma (HCC) cells with sorafenib
resistance. PloS One. 8:e786752013. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Colak S and Medema JP: Cancer stem
cells-important players in tumor therapy resistance. FEBS J.
281:4779–4791. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Reya T, Morrison SJ, Clarke MF and
Weissman IL: Stem cells, cancer and cancer stem cells. Nature.
414:105–111. 2001. View
Article : Google Scholar : PubMed/NCBI
|
|
33
|
Wang XQ, Ng RK, Ming X, Zhang W, Chen L,
Chu AC, Pang R, Lo CM, Tsao SW, Liu X, et al: Epigenetic regulation
of pluripotent genes mediates stem cell features in human
hepatocellular carcinoma and cancer cell lines. PloS One.
8:e724352013. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Zhou Y, Hofstetter WL, He Y, Hu W, Pataer
A, Wang L, Wang J, Zhou Y, Yu L, Fang B and Swisher SG: KLF4
inhibition of lung cancer cell invasion by suppression of SPARC
expression. Cancer Biol Ther. 9:507–513. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Han L, Shi S, Gong T, Zhang Z and Sun X:
Cancer stem cells: Therapeutic implications and perspectives in
cancer therapy. Acta Pharmaceutica Sinica B. 3:65–75. 2013.
View Article : Google Scholar
|