Introduction
Pharmaceutical approaches are important in the
treatment of head and neck squamous cell carcinoma (HNSCC). Current
therapeutic options include surgery, radiotherapy, chemotherapy and
immunotherapy; however, the 5-year survival rate has not improved
significantly in previous decades (1,2). Tobacco
and alcohol abuse remain the primary risk factors of HNSCC
incidence (3,4). In contrast to a decreasing incidence of
laryngeal cancer, the incidence of oropharyngeal cancer is
increasing (5). This increase may be
due to an overall human papilloma virus (HPV) infection prevalence
of >20% (6,7). HPV subtypes 16 and 18 are key regulators
in the formation of several tumor entities, including carcinoma of
the uterine cervix and oropharyngeal squamous cell carcinoma
(8). Within this group HPV 16 may be
detected in >90% of HPV-associated tumors (9). The mechanism of HPV-induced cancerous
lesions involves an increased risk of viral DNA integration into
the host genome (10). This genomic
alteration leads to the overexpression of viral oncogenes E6 and
E7, and subsequently results in a disruptive viral infection with
an abrogation of cell cycle checkpoints (11). However, HPV-associated HNSCC is
associated with an improved outcome following current treatment
options (12,13).
The extracellular matrix (ECM) is important to
nutritive cellular support, and functions as a physical barrier to
cellular migration and a regulator of intercellular communication
(14). The ECM consists of
proteoglycans, non-proteoglycan polysaccharides, including
hyaluronic acid, fibers and other components, including fibronectin
and laminin (14,15). During the process of tumor formation,
the basement membrane (BM) is essential as it connects the
epithelium to the subepithelial connective tissue and therefore
must be penetrated for invasive tumor growth. Two major components
of the basement membrane, type IV collagen and fibronectin, have
been demonstrated to be disregulated in HNSCC (16). ECM degradation occurs through the
secretion of several proteases, which leads to local tumor invasion
following pentration of the BM and ultimately, the occurrence of
lymphonodal and distant metastasis (17). Under normal conditions matrix
metalloproteinases (MMP) are important components in tissue
remodeling of the ECM, and participate in the regulation of
angiogenesis, tissue repair and morphogenesis (18). Currently, the MMP family consists of
>20 distinct zinc-dependent endopeptidases (19,20). MMPs
occur as either membrane-bound or soluble as collagenases,
gelatinases and stromelysins, and are synthesized as inactive
proenzymes by tumor cells and surrounding tumor stromal cells
(21). Among MMPs, the catalytic
gelatinase MMP-9 has been previously investigated due to its
ability to degrade type IV and V collagen in the BM (22). The degradation of type IV collagen is
associated with increased levels of MMP-9 in HNSCC (23). In HNSCC increased levels of MMP-9 are
also associated with increased lymphonodal metastasis (24).
Angiogenesis and neovascularization are essential in
tumor cell formation (25). Vascular
endothelial growth factor (VEGF), and the VEGF receptors (VEGFR)-1,
−2 and −3, serve an important role in the proliferation and
differentiation of endothelial cells (25). Increased expression of VEGF and VEGFR
has been reported in various tumor entities, including HNSCC
(26,27). Tumor growth and supporting processes,
including angiogenesis, are directly associated with VEGF in HNSCC
(28). The importance of molecular
indicators, including VEGF, in the microenvironment of tumor cells
is increasing as previous studies have revealed that plasma levels
of VEGF can be used as prognostic markers in HNSCC (27,29).
Increased expression of VEGFR has also been identified in
HPV-positive SCC cell lines (30,31). The
role of VEGFR-2 is well understood and it is known to be
overexpressed by tumor endothelial cells, and promotes cell
proliferation and migration (25). By
contrast, the role of VEGFR-1 in tumor formation is poorly
understood; it may serve a role in the process of VEGF
sequestration or stimulation of hematopoietic stem cell migration
(32). Furthermore, the expression of
VEGFR-1 appears to be associated with cell survival and
radiosensitivity (33). In
HPV-associated tumor disease, several HPV-dependent oncoproteins
have been reported to alter VEGFR-1 expression in vitro
(34,35). Intracellular VEGF signaling is
mediated by the activation and transphosphorylation of its tyrosine
kinase receptors, VEGFR-1, −2 and −3 (25). A major principle of targeted therapy
in tumor disease involves the selective inhibition of tyrosine
kinase receptors, to inhibit the process of subsequent
intracellular signaling cascades. Small molecule targeted therapies
have been established in multiple types of cancer (36–45).
Erlotinib and gefitinib are orally available selective tyrosine
kinase inhibitors of epidermal growth factor receptor (EGFR) and
are approved for the therapy of non-small cell lung cancer (NSCLC)
(36–38). Gefitinib functions through the
competitive inhibition of ATP binding to EGFR and consecutive
inhibition of receptor autophosphorylation, leading to a subsequent
decrease in proangiogenic proteins, including VEGF (39,40). It
has also been reported that gefitinib affects the synthesis of MMPs
and other extracellular matrix proteins in tumor tissues (41). BCR-ABL fusion protein (BCR-ABL)
inhibitors were designed for the treatment of chronic myeloid
leukemia (42). A reciprocal
translocation between chromosomes 9 and 22, known as the
Philadelphia chromosome, forms the BCR-ABL oncogene (42). Furthermore, the BCR-ABL inhibitors
nilotinib and dasatinib also function by inhibiting the
platelet-derived growth factor receptor (PDGFR) and mast/stem cell
growth factor receptor Kit (c-KIT) (43,44). The
inhibitory effects of dasatinib are mediated through the inhibition
of proto-oncogene tyrosine-protein kinase Src (Src), a process
associated with tumor proliferation and angiogenesis (45). To the best of our knowledge, the
present study is the first to investigate the alteration of VEGFR-1
and MMP-9 expression in HPV-associated SCC cells in vitro,
following treatment with the small molecule inhibitors erlotinib,
gefitinib, nilotinib and dasatinib.
Materials and methods
Cell lines
A total of two distinct HPV-negative cell lines
originating from oropharyngeal and laryngeal SCC (HNSCC 11A and
HNSCC 14C) were donated by Dr T. E. Carey (University of Michigan,
Ann Arbor, MI, USA). The p16 positive CERV196 cell line was
obtained from poorly differentiated SCC cells of the uterine cervix
(Cell Line Service GmbH, Eppelheim, Germany). The CERV196 tumor
cells were cultured in Eagle's minimum essential medium (Thermo
Fisher Scientific, Inc., Waltham, MA, USA) containing 2 mM
L-glutamine and Earle's balanced salt solution (Thermo Fisher
Scientific, Inc.), adjusted to contain 1.0 g/l sodium bicarbonate,
0.1 mM non-essential amino acids, 1.0 mM sodium pyruvate and 10%
fetal bovine serum (Thermo Fisher Scientific, Inc.). The HNSCC 11A
and 14C tumor cells were cultured in Dulbecco's Eagle's minimum
essential medium (Thermo Fisher Scientific, Inc.), supplement with
10% fetal calf serum (Thermo Fisher Scientific, Inc.), 2 mM
L-glutamine and an antibiotic/antimycotic solution
(penicillin-streptomycin, 10,000 U/ml; working dilution, 1/100;
Thermo Fisher Scientific, Inc.). Cell cultures were incubated at
37°C and 5% CO2 for 24, 48, 72 or 96 h. Orally available
nilotinib, dasatinib, gefitinib and erlotinib were donated by Dr
Hofheinz (Department of Oncology, Medical Faculty Mannheim,
University of Heidelberg, Mannheim, Germany). The drugs were stored
at room temperature and dissolved in dimethyl sulfoxide. The tumor
cells were incubated at 37°C with 20 µmol/l of each of the four
substances for 24, 48, 72 and 96 h and compared with the negative
control (untreated cells). The alamarBlue (AbD Serotec, Raleigh,
NC, USA) cell proliferation assay was used to quantify
proliferating HNSCC tumor cells and establish the relative
cytotoxicity of the tyrosine kinase inhibitors according to the
manufacturer's protocol.
VEGFR-1 and MMP-9 ELISA
Determination of protein concentrations was
performed using the ELISA technique. Subcultures of the cells were
generated by diluting and dissolving the cells from the culture. A
PBS solution supplemented with a combination of 0.05% trypsin and
0.02% EDTA (Sigma-Aldrich; Merck KGaA, Darmstadt, Germany) was
added at 37°C for 5 min to passage the cells. Subcultures were
transferred to microplates for further analysis of proliferation at
a 70% confluence. Protein expression was analyzed following
centrifugation at 8,050 × g for 10 min at room temperature and
collection of the medium supernatant. The DuoSet IC Human Total
VEGFR-1 (catalog no. DY321B; R&D Systems, Inc., Minneapolis,
MN, USA) and DuoSet IC Human Total MMP-9 (catalog no. DY911;
R&D Systems, Inc.) kits were used. For the sandwich ELISA, a
solid-phase capture antibody specific for VEGFR-1 or MMP-9 was
used, as well as a specific detection antibody [standard
streptavidin-horseradish peroxidase (HRP) format]. The capture
antibody was diluted to the working concentration in PBS (VEGFR-1,
2.0 µg/ml; MMP-9, 1.0 µg/ml) and incubated overnight at room
temperature according to the manufacturer's protocol. Three washing
steps with the washing buffer containing Tween 20 were performed.
The ELISA plates were blocked by adding 300 µl of the Reagent
Diluent (Reagent Diluent, DY995; R&D Systems, Inc.) to each
well and were incubated for 1 h at room temperature and were washed
again with the washing buffer for three times. The detection
antibody was diluted to its working concentration (VEGFR-1, 0.5
µg/ml; MMP-9, 0.1 µg/ml) and incubated with the ELISA plate for 2 h
at room temperature. The ELISA plate was washed three times with
Tween 20 and incubated with streptavidin-HRP (diluted according to
the manufacturer's protocol) for 20 min at room temperature. The
wells were subsequently washed with Tween 20. The visualization
reaction was initiated by adding the substrate solution for 20 min
followed by 50 µl stop solution at room temperature according to
the manufacturer's protocol. Each ELISA was performed according to
the manufacturer's protocol. Each experiment was performed for
three times. The calibrations on the microtiter plates included
recombinant human VEGFR-1 and MMP-9 standards that were provided in
the kits. Optical density was measured using a microplate reader
(MRX ELISA Reader; Dynatech, El Paso, TX, USA) and a wavelength of
450 nm. Wavelength correction was set to 540 nm and concentrations
were reported in pg/ml. The range of detection was between 28.4 and
281.5 pg/ml for VEGFR-1, and between 0.02 and 1021.9 pg/ml for
MMP-9. Inter-assay coefficient of variation reported by the
manufacturer was <10%.
Statistical analysis
Statistical analysis was performed using the mean
values from each experiment. Each experiment was performed for at
least three times (n=3). Comparisons were made with the negative
control to evaluate statistical significance. Values are presented
as the mean ± standard deviation. P<0.05 was considered to
indicate a statistically significant difference. The
two-coefficient variance test (SAS Statistics software, version
9.3; SAS Institute, Inc., Cary, NC, USA) and Dunnett's test were
used.
Results
VEGFR-1 expression levels in HNSCC
11A, HNSCC 14C and CERV196 cells
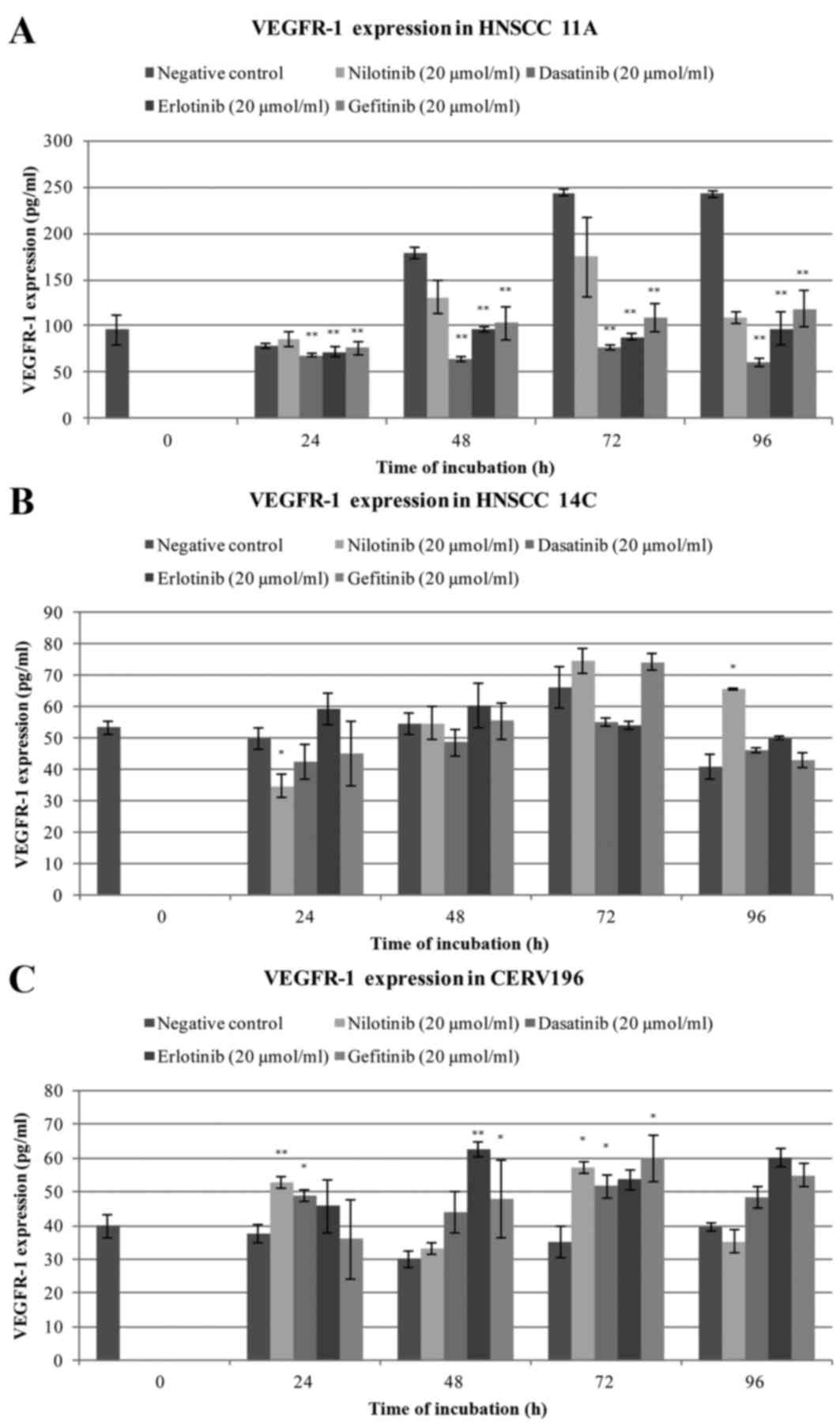
VEGFR-1 expression was detected in all three cell
lines. Treatment with 20 µmol/l dasatinib, gefitinib and erlotinib
between 48 and 96 h significantly reduced VEGFR-1 expression in
HNSCC 11A cells compared with the negative control (all P<0.001;
Fig. 1A). Treatment with 20 µmol/l
nilotinib for 48 h significantly reduced VEGFR-1 expression in
HNSCC 11A cells compared with the negative control (P=0.002;
Fig. 1A). Treatment with nilotinib
for 96 h significantly reduced VEGFR-1 expression in HNSCC 11A
cells compared with the negative control (P=0.003; Fig. 1A). There was no significant decrease
in VEGFR-1 protein expression after 24 h treatment with any of the
drugs (Fig. 1A). Treatment with
nilotinib exhibited a significant decrease in VEGFR-1 expression
after 24 h in the HNSCC 14C cells compared with the negative
control cells (P=0.040; Fig. 1B). A
significant increase in VEGRF-1 expression in HNSCC 14C cells was
observed following treatment with nilotinib for 96 h compared with
the negative control cells (P=0.038; Fig.
1B). Following treatment with nilotinib for 48 and 72 h VEGFR-1
expression appeared to increase. Treatment with gefitinib also
markedly increased VEGFR-1 expression in HNSCC 14C cells after
between 48 and 96 h compared with the negative control cells. In
the HNSCC 14C cells, treatment with dasatinib markedly decreased
VEGFR-1 expression after between 24 and 72 h compared with the
negative control cells. No significant alteration was observed
following treatment with erlotinib (Fig.
1B). In the CERV196 cells increased VEGFR-1 protein expression
was typically observed following treatment compared with the
negative control cells (Fig. 1C).
Treatment with nilotinib for 24 and 72 h significantly increased
VEGFR-1 expression compared with the negative control cells
(P=0.008 and P=0.023, respectively; Fig.
1C). Treatment with dasatinib exhibited a significant increase
in VEGFR-1 expression in the CERV196 cells after 24 and 72 h
compared with the negative controls (P=0.037 and P=0.040,
respectively; Fig. 1C). Treatment
with erlotinib significantly increased VEGFR-1 expression after 48
h compared with the negative control cells (P=0.001; Fig. 1C). Treatment with gefitinib
significantly increased protein levels of VEGFR-1 after 48 and 72 h
compared with the negative control cells (P=0.018 and P=0.041,
respectively; Fig. 1C). Decreased
VEGFR-1 expression was observed following treatment with gefitinib
for 24 h and nilotinib for 96 h (Fig.
1C). The quantified VEGFR-1 expression levels are presented in
Table I.
 | Table I.Quantification of vascular
endothelial growth factor receptor 1 expression levels in HNSCC
11A, 14C and CERV196 cells following incubation with nilotinib,
dasatinib, erlotinib and gefitinib at a concentration of 20
µmol/l. |
Table I.
Quantification of vascular
endothelial growth factor receptor 1 expression levels in HNSCC
11A, 14C and CERV196 cells following incubation with nilotinib,
dasatinib, erlotinib and gefitinib at a concentration of 20
µmol/l.
|
|
| Treatment |
|---|
|
|
|
|
|---|
|
|
| NC | Nilotinib | Dasatinib | Erlotinib | Gefitinib |
|---|
|
|
|
|
|
|
|
|
|---|
| Cell line | Incubation time,
h | Mean | Mean | P-value | Mean | P-value | Mean | P-value | Mean | P-value |
|---|
| HNSCC 11A |
|
| 24 | 78.6 | 86.3 | 0.358 | 68.2 | 0.340 | 72.2 | 1.00 | 76.3 | 0.818 |
|
| 48 | 179.3 | 131.1 | 0.003a | 64.4 |
<0.001a | 96.5 |
<0.001a | 102.7 |
<0.001a |
|
| 72 | 243.3 | 174.2 |
<0.001a | 77.1 |
<0.001a | 88.2 |
<0.001a | 109.7 |
<0.001a |
|
| 96 | 242.2 | 109.4 | 0.002a | 60.6 |
<0.001a | 97.1 |
<0.001a | 118.6 |
<0.001a |
| HNSCC 14C |
|
| 24 | 49.8 | 34.8 | 0.040a | 42.5 | 0.999 | 59.2 | 0.637 | 45.2 | 0.999 |
|
| 48 | 54.6 | 54.7 | 0.995 | 48.5 | 0.513 | 60.2 | 0.974 | 55.6 | 0.570 |
|
| 72 | 66.3 | 74.4 | 0.997 | 55.1 | 0.172 | 54.0 | 0.071 | 74.2 | 0.962 |
|
| 96 | 40.9 | 65.8 | 0.038a | 46.1 | 0.949 | 50.0 | 0.461 | 43.1 | 0.995 |
| CERV196 |
|
| 24 | 37.5 | 52.8 | 0.008a | 48.9 | 0.037a | 45.8 | 0.218 | 36.0 | 0.753 |
|
| 48 | 30.1 | 33.0 | 0.783 | 44.1 | 0.076 | 62.4 | 0.001a | 47.9 | 0.018a |
|
| 72 | 35.2 | 57.1 | 0.023a | 51.8 | 0.040a | 53.6 | 0.141 | 60.1 | 0.041a |
|
| 96 | 39.5 | 35.4 | 0.760 | 48.3 | 0.966 | 60.1 | 0.147 | 54.8 | 0.279 |
MMP-9 expression levels in HNSCC 11A,
HNSCC 14C and CERV196 cells
MMP-9 expression was evaluated in all three cell
lines. Treatment with 20 µmol/l dasatinib, gefitinib and erlotinib
between 24 and 96 h significantly decreased MMP-9 expression in the
HNSCC 11A cells compared with the negative control cells (all
P<0.001; Fig. 2A). Treatment with
20 µmol/l nilotinib for 24 and 48 h also significantly decreased
MMP-9 expression compared with the negative control cells
(P<0.001 and P=0.014, respectively; Fig. 2A). In addition, a marked decrease in
MMP-9 expression in HNSCCC 11A cells was observed following
treatment nilotinib for 72 and 96 h. In the HNSCC 14C cells a
significant decrease in MMP-9 protein expression was observed
following treatment with dasatinib, gefitinib and erlotinib across
all time points compared with the negative control cells (all
P<0.001; Fig. 2B). Treatment with
nilotinib for between 24 and 72 h also significantly decreased
MMP-9 expression compared with the negative control cells (all
P<0.001; Fig. 2B). Treatment with
dasatinib and erlotinib for 72 h led to a significant increase in
MMP-9 expression in the CERV196 cells compared with the negative
control cells (P=0.014 and P=0.007, respectively; Fig. 2C). The majority of treatment types and
durations induced no significant alteration in MMP-9 expression in
the CERV196 cells compared with the negative control cells.
Discussion
The present study was performed in order to evaluate
the expression of VEGFR-1 and MMP-9 in HPV-positive and -negative
SCC cells and to measure the altered expression patterns of these
biomarkers following treatment with the well-established tyrosine
kinase inhibitors nilotinib, dasatinib, erlotinib and
gefitinib.
The tumor microenvironment is essential for tumor
progression, survival and the formation of metastases (25). In the context of tumor growth,
penetration of the BM is important as the BM functions as a barrier
and differentiates between non-invasive and invasive tumor growth.
MMPs are known to degrade ECM substrates and promote the invasion
of tumorous vasculature (46). The
role of MMP-9 is decisive as it dissolves the BM by degrading type
IV and V collagen (22). In the
present study, a statistically significant decrease in MMP-9
expression was observed in HPV-negative HNSCC tumor cells following
treatment with tyrosine kinase inhibitors in a time-dependent
manner, despite none of the drugs functioning as a direct inhibitor
of MMP-9. MMP-9 expression may be regulated by multiple factors
including cytokines and growth factors including EGFR and
transforming growth factors (47).
Activated EGFR may also promote cell migration through the
regulation of MMP-9 expression in an epithelial-mesenchymal
transition (EMT)-like process, which leads to the degradation of
E-cadherin. This effect has also been reported in SCC cells
(48). Additionally, multiple
signaling pathways including the mitogen-activated protein kinase
(MAPK) and protein kinase B (AKT) pathways regulate the expression
levels of MMPs including MMP-9 (49).
The process of EMT may be activated through the MAPK signaling
pathway which may lead to the migration and spread of tumor cells
(50). Furthermore, the activation of
MMPs including MMP-9 may result in long-term EMT (51).
Liu and Klominek (52)
reported a decrease in MMP-9 production in malignant mesothelioma
following treatment with erlotinib. Furthermore, a
gefitinib-induced reduction in MMP-9 and MMP-2 expression was
observed several tumor entities including oral HNSCC (41,53,54).
Erlotinib and gefitinib function as inhibitors of EGFR (37), therefore, MMP-9 regulation in HNSCC
11A and HNSCC 14C cells may be EGFR-mediated. Selective
EGFR-inhibiting proteins may therefore be used for the direct
inhibition of EGFR and the indirect inhibition of MMP-9 expression
in HPV-negative HNSCC. In the present study, treatment with
dasatinib and nilotinib also significantly reduced the expression
levels of MMP-9 in p16-negative HNSCC tumor cells. However, the
mechanism of action remains unclear as nilotinib and dasatinib
function through the inhibition of BCR-ABL, PDGFR and c-KIT, and
Src in the case of dasatinib. The mechanism of decreased MMP-9
expression following treatment with nilotinib and dasatinib remain
to be elucidated. A recent study demonstrated that MMP-9 expression
may be decreased in pituitary adenomas, suggesting that nilotinib
exerts its inhibitory effects through the inhibition of the
tyrosine kinase receptor, epithelial discoidin domain-containing
receptor 1 (DDR1) (55). The present
study hypothesizes that DDR1 may be associated with the regulation
of MMPs, including MMP-2 and MMP-9, and therefore, may be
associated with the mechanism of decreased MMP-9 levels in HNSCC,
although DDR1 expression patterns in HNSCC remain to be
elucidated.
Src kinases are a subcategory of non-receptor
protein tyrosine kinases and contribute to tumor growth in tumor
stromal cells (56). The results of
the present study support the study by Liang et al (57), which associated the dasatinib-induced
reduction in MMP-9 expression in tumor cells with the inactivation
of Src-dependent signaling pathways. Therefore, the targeting of
the Src kinase family with dasatinib may be a promising objective
for further investigation into selective therapeutic approaches in
solid malignant tumors, including HNSCC. In the present study, a
similar effect was not observed in the p16-positive CERV196 cells.
By contrast, an increase in MMP-9 expression was detected following
treatment with dasatinib and erlotinib for 72 h in CERV196 cells.
These results indicated HPV-dependent mechanisms in SCC cells to
evade decreased MMP-9 levels. The level of MMP-9 expression was
decreased in p16-positive squamous cancer cells. p16-associated
oncoproteins E6 and E7 promote the activity of MMPs, including
MMP-9 in cervical SCC cells (58).
Therefore, a potential explanation for the intransigence or
increase in MMP-9 expression is the counter regulation of the
drug-induced decrease in MMP-9 through the activation of viral
oncoproteins. Hu et al (59)
demonstrated that activation of β-catenin, a functional protein
coordinating cell-cell adhesion and promoting the expression of ECM
components, including fibronectin, may be induced by viral
oncoproteins. It was hypothesized that other metastasis-associated
proteins may be facilitated by p16-induced oncoproteins, which is
consistent with the results of the present study of increased MMP-9
expression by nilotinib, dasatinib, erlotinib and gefitinib as it
is not affected by this type of selective tyrosine kinase
inhibition. The role of MMP-9 is complex and requires elucidation
in further studies to investigate the therapeutic potential of
targeting MMPs in HNSCC.
VEGFR-induced angiogenesis is important in local
tumor progression and the formation of distant metastases. VEGFR-1
is expressed on the surface of endothelial cells and its expression
was evaluated in all three cell lines. The expression and function
of VEGFR-1 in tumor cells as a vascular and non-vascular modulator
is less well-understood compared with VEGFR-2. The role of VEGFR-1
as a target for selective inhibition is in the early stages
(60). Currently, there are no
published data investigating the effect of the indirect inhibitors
nilotinib, dasatinib, erlotinib and gefitinib on VEGFR-1 expression
in HNSCC with respect to HPV-status. In the present study, a
decrease in VEGFR-1 expression was observed in the HNSCC 11A cells
following treatment with all the tested drugs for between 48 and 96
h. There was also a tendency towards a decrease in VEGFR-1
expression in the HPV-negative HNSCC 14C cells following treatment
with nilotinib and dasatinib. The cellular mechanism for this
effect remains unclear as nilotinib and dasatinib are not direct
inhibitors of VEGFR.
It has been reported that the activation of Src
serves an essential role in the signal transduction downstream of
various growth factor receptors including VEGFR (61). Also, Src kinase activity in tumor
cells is elevated (61,62). As previously described, Liang et
al (57) demonstrated that
dasatinib inhibits the angiogenic potential of endothelial cells
including the expression of VEGF in tumor-associated endothelial
cells, suggesting Src to be a key downstream effector of angiogenic
signaling pathways. A Src-induced stop signal in the sense of a
negative feedback mechanism for further VEGFR-expression may
therefore be a potential explanation of the nilotinib- and
dasatinib-induced effects on VEGFR-1 in HPV-negative tumor cells.
The selective tyrosine kinase inhibitors erlotinib and gefitinib
function through EGFR-inhibition. The results of the present study
support several previous studies, which suggested that EGFR
activation regulates VEGF expression (63–65). This
mechanism may provide an explanation for the decrease in VEGFR-1
expression though selective EGFR-inhibition in HNSCC 11A. In the
present study, an alteration in VEGFR-1 expression following
treatment with erlotinib and gefitinib in HNSCC 14C was not
detected. The HPV-positive CERV196 cells exhibited markedly
decreased levels of VEGFR-1 expression in comparison with the
HPV-negative tumor cells. In addition, an increase in VEGFR-1
expression in the HPV-positive CERV196 tumor cells was observed
following treatment with all of the tested drugs. It is known that
viral oncogenes may induce the expression of angiogenic factors,
including VEGF (30,34). Therefore, a potential mechanism for
the increase in VEGFR-1 expression is the drug-induced activation
or stimulation of viral oncogenic proteins, including E6 and E7. As
a result, unknown cellular autocrine mechanisms may increase the
production of angiogenic proteins. This may be a potential evasive
strategy of malignant cells following drug-induced dysregulation.
Dias et al (66) discussed a
similar mechanism in virally transformed cancer cells of the
oropharynx as a result of treatment with cetuximab. However, this
hypothesis remains to be completely elucidated.
In conclusion, the present study is one of the first
to investigate the altered expression patterns of the viable
molecular target proteins MMP-9 and VEGFR-1, in p16-positive and
-negative SCC cells, following treatment with the non-direct
selective tyrosine kinases nilotinib, dasatinib, erlotinib and
gefitinib in vitro. The results of the present study provide
an improved understanding of MMP-9 and VEGFR-1, and their
interaction with selective small molecule inhibitors. These results
may be used in further investigation into novel strategies of
targeted therapy in p16-positive and -negative HNSCC.
Acknowledgements
The authors of the present study would like to thank
Petra Prohaska for technical support (medical technical assistant;
Department of Otorhinolaryngology Head and Neck Surgery, University
Hospital Mannheim, University of Heidelberg, Germany). Statistical
analysis was performed in cooperation with Dr C. Weiss (Institute
of Biomathematics, University Hospital Mannheim, University of
Heidelberg, Germany).
Glossary
Abbreviations
Abbreviations:
|
AKT
|
protein kinase B
|
|
BCR-ABL
|
BCR-ABL fusion protein
|
|
BM
|
basement membrane
|
|
c-KIT
|
mast/stem cell growth factor receptor
Kit
|
|
DDR
|
epithelial discoidin domain-containing
receptor
|
|
ECM
|
extracellular matrix
|
|
EGFR
|
epidermal growth factor receptor
|
|
HNSCC
|
head and neck squamous cell
carcinoma
|
|
HPV
|
human papilloma virus
|
|
HRP
|
horseradish peroxidase
|
|
MAPK
|
mitogen-activated protein kinase
|
|
MMP
|
matrix metalloproteinase
|
|
NSCLC
|
non-small cell lung cancer
|
|
PDGFR
|
platelet-derived growth factor
receptor
|
|
Src
|
proto-oncogene tyrosine-protein kinase
Src
|
|
VEGFR
|
vascular endothelial growth factor
receptor
|
References
|
1
|
Siegel RL, Miller KD and Jemal A: Cancer
statistics, 2015. CA Cancer J Clin. 65:5–29. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Sepiashvili L, Hui A, Ignatchenko V, Shi
W, Su S, Xu W, Huang SH, O'Sullivan B, Waldron J, Irish JC, et al:
Potentially novel candidate biomarkers for head and neck squamous
cell carcinoma identified using an integrated cell line-based
discovery strategy. Mol Cell Proteomics. 11:1404–1415. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Hashibe M, Brennan P, Benhamou S,
Castellsague X, Chen C, Curado MP, Dal Maso L, Daudt AW, Fabianova
E, Fernandez L, et al: Alcohol drinking in never users of tobacco,
cigarette smoking in never drinkers, and the risk of head and neck
cancer: Pooled analysis in the International Head and Neck Cancer
Epidemiology Consortium. J Natl Cancer Inst. 99:777–789. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Hashibe M, Brennan P, Chuang SC, Boccia S,
Castellsague X, Chen C, Curado MP, Dal Maso L, Daudt AW, Fabianova
E, et al: Interaction between tobacco and alcohol use and the risk
of head and neck cancer: Pooled analysis in the international head
and neck cancer epidemiology consortium. Cancer Epidemiol
Biomarkers Prev. 18:541–550. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Ang KK and Sturgis EM: Human
papillomavirus as a marker of the natural history and response to
therapy of head and neck squamous cell carcinoma. Semin Radiat
Oncol. 22:128–142. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Romanitan M, Näsman A, Ramqvist T,
Dahlstrand H, Polykretis L, Vogiatzis P, Vamvakas P, Tasopoulos G,
Valavanis C, Arapantoni-Dadioti P, et al: Human papillomavirus
frequency in oral and oropharyngeal cancer in Greece. Anticancer
Res. 28:2077–2080. 2008.PubMed/NCBI
|
|
7
|
Dayyani F, Etzel CJ, Liu M, Ho CH, Lippman
SM and Tsao AS: Meta-analysis of the impact of human papillomavirus
(HPV) on cancer risk and overall survival in head and neck squamous
cell carcinomas (HNSCC). Head Neck Oncol. 2:152010. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Lopez-Ocejo O, Viloria-Petit A,
Bequet-Romero M, Mukhopadhyay D, Rak J and Kerbel RS: Oncogenes and
tumor angiogenesis: The HPV-16 E6 oncoprotein activates the
vascular endothelial growth factor (VEGF) gene promoter in a p53
independent manner. Oncogene. 19:4611–4620. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Gillison ML, Castellsagué X, Chaturvedi A,
Goodman MT, Snijders P, Tommasino M, Arbyn M and Franceschi S:
Eurogin Roadmap: Comparative epidemiology of HPV infection and
associated cancers of the head and neck and cervix. Int J Cancer.
134:497–507. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Dok R and Nuyts S: HPV positive head and
neck cancers: Molecular pathogenesis and evolving treatment
strategies. Cancers (Basel). 8:pii: E41. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Doorbar J: Molecular biology of human
papillomavirus infection and cervical cancer. Clin Sci (Lond).
110:525–541. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Sturgis EM and Ang KK: The epidemic of
HPV-associated oropharyngeal cancer is here: Is it time to change
our treatment paradigms? J Natl Compr Canc Netw. 9:665–673.
2011.PubMed/NCBI
|
|
13
|
O'Sullivan B, Huang SH, Perez-Ordonez B,
Massey C, Siu LL, Weinreb I, Hope A, Kim J, Bayley AJ, Cummings B,
et al: Outcomes of HPV-related oropharyngeal cancer patients
treated by radiotherapy alone using altered fractionation.
Radiother Oncol. 103:49–56. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Park CC, Bissell MJ and Barcellos-Hoff MH:
The influence of the microenvironment on the malignant phenotype.
Mol Med Today. 6:324–329. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Aderhold C, Umbreit C, Faber A, Birk R,
Sommer JU, Hörmann K and Schultz JD: Matrix metalloproteinase-2 and
−14 in p16-positive and -negative HNSCC after exposure To 5-FU and
docetaxel in vitro. Anticancer Res. 34:4929–4937. 2014.PubMed/NCBI
|
|
16
|
Curry JM, Sprandio J, Cognetti D,
Luginbuhl A, Bar-ad V, Pribitkin E and Tuluc M: Tumor
microenvironment in head and neck squamous cell carcinoma. Semin
Oncol. 41:217–234. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Jimenez L, Sharma VP, Condeelis J, Harris
T, Ow TJ, Prystowsky MB, Childs G and Segall JE: MicroRNA-375
suppresses extracellular matrix degradation and invadopodial
activity in head and neck squamous cell carcinoma. Arch Pathol Lab
Med. 139:1349–1361. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Rosenthal EL and Matrisian LM: Matrix
metalloproteases in head and neck cancer. Head Neck. 28:639–648.
2006. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Nelson AR, Fingleton B, Rothenberg ML and
Matrisian LM: Matrix metalloproteinases: Biologic activity and
clinical implications. J Clin Oncol. 18:1135–1149. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Ravanti L and Kähäri VM: Matrix
metalloproteinases in wound repair (review). Int J Mol Med.
6:391–407. 2000.PubMed/NCBI
|
|
21
|
Monsky WL, Kelly T, Lin CY, Yeh Y,
Stetler-Stevenson WG, Mueller SC and Chen WT: Binding and
localization of M(r) 72,000 matrix metalloproteinase at cell
surface invadopodia. Cancer Res. 53:3159–3164. 1993.PubMed/NCBI
|
|
22
|
Rundhaug JE: Matrix metalloproteinases,
angiogenesis, and cancer: Commentary re: A. C. Lockhart et
al: Reduction of wound angiogenesis in patients treated with
BMS-275291, a broad spectrum matrix metalloproteinase inhibitor.
Clin. Cancer Res., 9: 00-00, 2003. Clin Cancer Res. 9:551–554.
2003.PubMed/NCBI
|
|
23
|
Koontongkaew S, Amornphimoltham P,
Monthanpisut P, Saensuk T and Leelakriangsak M: Fibroblasts and
extracellular matrix differently modulate MMP activation by primary
and metastatic head and neck cancer cells. Med Oncol. 29:690–703.
2012. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Koontongkaew S: The tumor microenvironment
contribution to development, growth, invasion and metastasis of
head and neck squamous cell carcinomas. J Cancer. 4:66–83. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Ferrara N, Gerber HP and LeCouter J: The
biology of VEGF and its receptors. Nat Med. 9:669–676. 2003.
View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Folkman J: The role of angiogenesis in
tumor growth. Semin Cancer Biol. 3:65–71. 1992.PubMed/NCBI
|
|
27
|
Hsu HW, Wall NR, Hsueh CT, Kim S, Ferris
RL, Chen CS and Mirshahidi S: Combination antiangiogenic therapy
and radiation in head and neck cancers. Oral Oncol. 50:19–26. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Mineta H, Miura K, Ogino T, Takebayashi S,
Misawa K, Ueda Y, Suzuki I, Dictor M, Borg A and Wennerberg J:
Prognostic value of vascular endothelial growth factor (VEGF) in
head and neck squamous cell carcinomas. Br J Cancer. 83:775–781.
2000. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Argiris A, Lee SC, Feinstein T, Thomas S,
Branstetter BF IV, Seethala R, Wang L, Gooding W, Grandis JR and
Ferris RL: Serum biomarkers as potential predictors of antitumor
activity of cetuximab-containing therapy for locally advanced head
and neck cancer. Oral Oncol. 47:961–966. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Aderhold C, Faber A, Umbreit C,
Chakraborty A, Bockmayer A, Birk R, Sommer JU, Hörmann K and
Schultz JD: Small molecules alter VEGFR and PTEN expression in
HPV-positive and -negative SCC: New hope for targeted-therapy.
Anticancer Res. 35:1389–1399. 2015.PubMed/NCBI
|
|
31
|
Kramer B, Hock C, Birk R, Sauter A, Stuck
BA, Hörmann K, Schultz JD and Aderhold C: Targeted therapies in
HPV-positive and -negative HNSCC-alteration of EGFR and VEGFR-2
expression in vitro. Anticancer Res. 36:2799–2807. 2016.PubMed/NCBI
|
|
32
|
Cabebe E and Wakelee H: Role of
anti-angiogenesis agents in treating NSCLC: Focus on bevacizumab
and VEGFR tyrosine kinase inhibitors. Curr Treat Options Oncol.
8:15–27. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Van Limbergen EJ, Zabrocki P, Porcu M,
Hauben E, Cools J and Nuyts S: FLT1 kinase is a mediator of
radioresistance and survival in head and neck squamous cell
carcinoma. Acta Oncol. 53:637–645. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Le Buanec H, D'Anna R, Lachgar A, Zagury
JF, Bernard J, Ittelé D, d'Alessio P, Hallez S, Giannouli C, Burny
A, et al: HPV-16 E7 but not E6 oncogenic protein triggers both
cellular immunosuppression and angiogenic processes. Biomed
Pharmacother. 53:424–431. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
No JH, Jo H, Kim SH, Park IA, Kang D, Han
SS, Kim JW, Park NH, Kang SB and Song YS: Expression of vascular
endothelial growth factor and hypoxia inducible factor-1alpha in
cervical neoplasia. Ann N Y Acad Sci. 1171:105–110. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Ward WH, Cook PN, Slater AM, Davies DH,
Holdgate GA and Green LR: Epidermal growth factor receptor tyrosine
kinase. Investigation of catalytic mechanism, structure-based
searching and discovery of a potent inhibitor. Biochem Pharmacol.
48:659–666. 1994. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Bareschino MA, Schettino C, Troiani T,
Martinelli E, Morgillo F and Ciardiello F: Erlotinib in cancer
treatment. Ann Oncol. 18:(Suppl 6). vi35–vi41. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Gridelli C, Bareschino MA, Schettino C,
Rossi A, Maione P and Ciardiello F: Erlotinib in non-small cell
lung cancer treatment: Current status and future development. The
Oncologist. 12:840–849. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Hirata A, Ogawa S, Kometani T, Kuwano T,
Naito S, Kuwano M and Ono M: ZD1839 (Iressa) induces antiangiogenic
effects through inhibition of epidermal growth factor receptor
tyrosine kinase. Cancer Res. 62:2554–2560. 2002.PubMed/NCBI
|
|
40
|
Wakeling AE: Inhibitors of growth factor
signalling. Endocr Relat Cancer. 12:(Suppl 1). S183–S187. 2005.
View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Toda D, Ota T, Tsukuda K, Watanabe K,
Fujiyama T, Murakami M, Naito M and Shimizu N: Gefitinib decreases
the synthesis of matrix metalloproteinase and the adhesion to
extracellular matrix proteins of colon cancer cells. Anticancer
Res. 26:129–134. 2006.PubMed/NCBI
|
|
42
|
Kantarjian H, Giles F, Wunderle L, Bhalla
K, O'Brien S, Wassmann B, Tanaka C, Manley P, Rae P, Mietlowski W,
et al: Nilotinib in imatinib-resistant CML and Philadelphia
chromosome-positive ALL. N Engl J Med. 354:2542–2551. 2006.
View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Manley PW, Drueckes P, Fendrich G, Furet
P, Liebetanz J, Martiny-Baron G, Mestan J, Trappe J, Wartmann M and
Fabbro D: Extended kinase profile and properties of the protein
kinase inhibitor nilotinib. Biochim Biophys Acta. 1804:445–453.
2010. View Article : Google Scholar : PubMed/NCBI
|
|
44
|
le Coutre P, Schwarz M and Kim TD: New
developments in tyrosine kinase inhibitor therapy for newly
diagnosed chronic myeloid leukemia. Clin Cancer Res. 16:1771–1780.
2010. View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Bromann PA, Korkaya H and Courtneidge SA:
The interplay between Src family kinases and receptor tyrosine
kinases. Oncogene. 23:7957–7968. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
46
|
Willis AL, Sabeh F, Li XY and Weiss SJ:
Extracellular matrix determinants and the regulation of cancer cell
invasion stratagems. J Microsc. 251:250–260. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
47
|
Yao J, Xiong S, Klos K, Nguyen N, Grijalva
R, Li P and Yu D: Multiple signaling pathways involved in
activation of matrix metalloproteinase-9 (MMP-9) by heregulin-beta1
in human breast cancer cells. Oncogene. 20:8066–8074. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
48
|
Zuo JH, Zhu W, Li MY, Li XH, Yi H, Zeng
GQ, Wan XX, He QY, Li JH, Qu JQ, et al: Activation of EGFR promotes
squamous carcinoma SCC10A cell migration and invasion via inducing
EMT-like phenotype change and MMP-9-mediated degradation of
E-cadherin. J Cell Biochem. 112:2508–2517. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
49
|
Zhang YQ, Wei XL, Liang YK, Chen WL, Zhang
F, Bai JW, Qiu SQ, Du CW, Huang WH and Zhang GJ: Over-expressed
twist associates with markers of epithelial mesenchymal transition
and predicts poor prognosis in breast cancers via ERK and Akt
activation. PLoS One. 10:e01358512015. View Article : Google Scholar : PubMed/NCBI
|
|
50
|
Zhao L, Geng H, Liang ZF, Zhang ZQ, Zhang
T, Yu DX and Zhong CY: Benzidine induces epithelial-mesenchymal
transition in human uroepithelial cells through ERK1/2 pathway.
Biochem Biophys Res Commun. 459:643–649. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
51
|
Qiao B, Johnson NW and Gao J:
Epithelial-mesenchymal transition in oral squamous cell carcinoma
triggered by transforming growth factor-beta1 is Snail
family-dependent and correlates with matrix metalloproteinase-2 and
−9 expressions. Int J Oncol. 37:663–668. 2010.PubMed/NCBI
|
|
52
|
Liu Z and Klominek J: Inhibition of
proliferation, migration, and matrix metalloprotease production in
malignant mesothelioma cells by tyrosine kinase inhibitors.
Neoplasia. 6:705–712. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
53
|
Normanno N and Gullick WJ: Epidermal
growth factor receptor tyrosine kinase inhibitors and bone
metastases: Different mechanisms of action for a novel therapeutic
application? Endocr Relat Cancer. 13:3–6. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
54
|
Lee EJ, Whang JH, Jeon NK and Kim J: The
epidermal growth factor receptor tyrosine kinase inhibitor ZD1839
(Iressa) suppresses proliferation and invasion of human oral
squamous carcinoma cells via p53 independent and MMP, uPAR
dependent mechanism. Ann N Y Acad Sci. 1095:113–128. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
55
|
Li S, Zhang Z, Xue J, Guo X, Liang S and
Liu A: Effect of hypoxia on DDR1 expression in pituitary adenomas.
Med Sci Monit. 21:2433–2438. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
56
|
Kilarski WW, Jura N and Gerwins P:
Inactivation of Src family kinases inhibits angiogenesis in vivo:
Implications for a mechanism involving organization of the actin
cytoskeleton. Exp Cell Res. 291:70–82. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
57
|
Liang W, Kujawski M, Wu J, Lu J, Herrmann
A, Loera S, Yen Y, Lee F, Yu H, Wen W and Jove R: Antitumor
activity of targeting SRC kinases in endothelial and myeloid cell
compartments of the tumor microenvironment. Clin Cancer Res.
16:924–935. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
58
|
Zhu D, Ye M and Zhang W: E6/E7
oncoproteins of high risk HPV-16 upregulate MT1-MMP, MMP-2 and
MMP-9 and promote the migration of cervical cancer cells. Int J
Clin Exp Pathol. 8:4981–4989. 2015.PubMed/NCBI
|
|
59
|
Hu Z, Müller S, Qian G, Xu J, Kim S, Chen
Z, Jiang N, Wang D, Zhang H, Saba NF, et al: Human papillomavirus
16 oncoprotein regulates the translocation of β-catenin via the
activation of epidermal growth factor receptor. Cancer.
121:214–225. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
60
|
Schwartz JD, Rowinsky EK, Youssoufian H,
Pytowski B and Wu Y: Vascular endothelial growth factor receptor-1
in human cancer: Concise review and rationale for development of
IMC-18F1 (Human antibody targeting vascular endothelial growth
factor receptor-1). Cancer. 116:(4 Suppl). S1027–S1032. 2010.
View Article : Google Scholar
|
|
61
|
Mayer EL and Krop IE: Advances in
targeting SRC in the treatment of breast cancer and other solid
malignancies. Clin Cancer Res. 16:3526–3532. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
62
|
Kanda S, Miyata Y, Kanetake H and
Smithgall TE: Non-receptor protein-tyrosine kinases as molecular
targets for antiangiogenic therapy (Review). Int J Mol Med.
20:113–121. 2007.PubMed/NCBI
|
|
63
|
Argiris A, Kotsakis AP, Hoang T, Worden
FP, Savvides P, Gibson MK, Gyanchandani R, Blumenschein GR Jr, Chen
HX, Grandis JR, et al: Cetuximab and bevacizumab: Preclinical data
and phase II trial in recurrent or metastatic squamous cell
carcinoma of the head and neck. Ann Oncol. 24:220–225. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
64
|
Cohen EE, Davis DW, Karrison TG, Seiwert
TY, Wong SJ, Nattam S, Kozloff MF, Clark JI, Yan DH, Liu W, et al:
Erlotinib and bevacizumab in patients with recurrent or metastatic
squamous-cell carcinoma of the head and neck: A phase I/II study.
Lancet Oncol. 10:247–257. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
65
|
Tabernero J: The role of VEGF and EGFR
inhibition: Implications for combining anti-VEGF and anti-EGFR
agents. Mol Cancer Res. 5:203–220. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
66
|
Dias JD, Guse K, Nokisalmi P, Eriksson M,
Chen DT, Diaconu I, Tenhunen M, Liikanen I, Grénman R, Savontaus M,
et al: Multimodal approach using oncolytic adenovirus, cetuximab,
chemotherapy and radiotherapy in HNSCC low passage tumour cell
cultures. Eur J Cancer. 46:625–635. 2010. View Article : Google Scholar : PubMed/NCBI
|