Introduction
Liver cancer is the second leading cause of
cancer-associated mortality, and hepatocellular carcinoma (HCC)
accounts for ≥90% of liver cancer cases (1). To date, liver resection and liver
transplantation remain the most effective treatments for patients
with HCC (2). However, the majority
of patients do not have the opportunity for surgery due to late
diagnosis and tumor progression (3).
Despite the emergence of novel therapies for HCC, the prognosis of
patients with HCC remains poor (3,4). The
carcinogenesis of HCC is complex and involves numerous biological
processes (5). The development of
novel interventions for HCC requires improved understanding of the
disease (6). Therefore, further
research to reveal the mechanisms underlying HCC carcinogenesis is
urgently required in order to improve the management of this type
of cancer.
Dynamins (DNMs) are a family of guanylate
triphosphatases (GTPases) that participate in vesicle budding and
membrane severing via the hydrolysis of GTPs (7). DNM3 is a member of the DNM family that
is essential for endocytosis and possesses mechanochemical
properties that are important for actin-membrane processes
(8). The multiple domains of DNMs
facilitate their interaction with numerous proteins, resulting in
their diverse functions (9). An
important function of DNMs is their ability to activate the
production of nitric oxide synthases (NOS) by protein-protein
interaction (10). Cao et al
(11) demonstrated that the
proline-rich carboxylic acid terminal domain of DNM2 can interact
with the flavin adenine dinucleotide-binding region of endothelial
(e) NOS, activating eNOS and promoting nitric oxide (NO)
production. Hyndman et al (10) revealed that DNM1, DNM2 and DNM3
increased the production of NO by their interaction with neuronal
NOS (nNOS). Conversely, Kang-Decker et al (12) identified that NO could also directly
promote DNM function by cysteine residue nitrosylation.
The role of DNM3 in malignant tumors is largely
unknown. Shen et al (13)
demonstrated in 2012 that the promoter of DNM3 was hypermethylated
in HCC tissues. To the best of our knowledge, the association
between DNM3 and HCC was first described by Inokawa et al
(14) in 2013 following a triple
combination array analysis. The authors revealed that DNM3 was a
candidate tumor suppressor gene against HCC. Hypermethylation of
the DNM3 promoter was detected more frequently in HCC tissues
compared with in normal tissues, and the relative mRNA expression
level of DNM3 in HCC tissues tended to be decreased compared with
that in normal tissues. Furthermore, HCC growth was associated with
DNM3 expression level, and patients with HCC and decreased DNM3
expression levels had poorer prognosis (14). However, the precise role of DNM3 as a
tumor suppressor in HCC has not yet been revealed.
The present study aimed to evaluate the
tumor-suppressive effect of DNM3 on HCC cells and to elucidate the
mechanism underlying the DNM3-mediated inhibition of HCC
growth.
Materials and methods
Reagents, cell lines and culture
conditions
The primary antibody against DNM3 (cat. no. ab3458)
was obtained from Abcam (Cambridge, UK). Primary antibodies against
inducible (i)NOS (cat. no. sc-7271), nNOS (cat. no. sc-136006) and
β-actin (cat. no. sc-130300) were purchased from Santa Cruz
Biotechnology, Inc. (Dallas, TX, USA). Primary antibodies against
caspase-3 (cat. no. 9662), cleaved caspase-3 (cat no. 9664),
caspase-9 (cat. no. 9508), cleaved caspase-9 (cat. no. 7237), poly
ADP-ribose polymerase (PARP; cat. no. 9532), cleaved PARP (cat. no.
5625), cyclin D1 (cat. no. 2978), cyclin-dependent kinase (CDK)2
(cat. no. 2546), CDK4 (cat. no. 12,790) and GAPDH (cat. no. 5174)
were obtained from Cell Signaling Technology, Inc. (Danvers, MA,
USA). The iNOS inhibitor L-canavanine was supplied by Sigma-Aldrich
(Merck KGaA, Darmstadt, Germany). L-canavanine (1 mM) was incubated
with the indicated cells at 37°C for 24 h. The L02 human hepatic
cell line and HepG2, Hep3B, SMMC-7721, PLC/PRF/5, Bel-7402 and Huh7
human HCC cell lines were obtained from the Cell Bank of the Type
Culture Collection of the Chinese Academy of Sciences (Shanghai,
China). The cells were cultured in Dulbecco's modified Eagle's
medium (Gibco; Thermo Fisher Scientific, Inc., Waltham, MA, USA)
supplemented with 10% fetal bovine serum and maintained in 5%
CO2/95% air at 37°C.
Patient tissue samples
Between January 2014 and August 2014, 10 patients
with HCC from the Department of General Surgery of Jinshan Hospital
(Fudan University, Shanghai, China) who underwent liver resection
were recruited to the present study. The patients recruited
included 7 male patients and 3 female patients aged 59–72 years
(mean age, 67.6 years). All patients were diagnosed with primary
HCC with pathological confirmation. Tumor tissues and the adjacent
non-tumor tissues from the patients obtained during resection were
used for further research. Written informed consent was obtained
from all patients prior to enrollment in the present study. The
study was approved by the Medical Ethics Committee of Fudan
University.
Animals
In total, 30 male nude mice (age, 4 weeks; weight,
15 to 18 g) were purchased from the Shanghai Institute of Material
Medicine (Shanghai, China) and housed in pathogen-free conditions
with free access to food and water. The animal procedures were
performed in accordance with the guidelines established by the
Shanghai Medical Experimental Animal Care Commission. The nude mice
were divided into 3 groups of 10. For the generation of
subcutaneous tumors, 5×106 SMMC-7721 cells suspended in
100 µl PBS were injected subcutaneously into the upper left flank
region of the nude mice. The tumor diameter was determined every 7
days. The tumor volume (V) was calculated by the following formula:
V = 1/2 × (largest diameter) × (smallest diameter)2.
Total RNA extraction and reverse
transcription-quantitative polymerase chain reaction (RT-qPCR)
Total RNA was extracted from patient tissues and
cell lines using TRIzol® reagent (Takara Bio, Inc.,
Otsu, Japan) and reverse transcribed into cDNA using PrimeScript™
RT Master Mix (Takara Bio, Inc.). The reverse transcription was
performed with 5X PrimeScript RT Master Mix (4 µl), RNA (1 µg) and
RNase-free ddH2O, to make 20 µl in total. The
thermocycler conditions were as follows: 37°C for 30 min, 85°C for
4 sec, and 4°C for 5 min. Relative mRNA expression level was
determined by qPCR with a SYBR® Premix Ex Taq™ Tli
RNaseH Plus PCR kit (Takara Bio, Inc.) and an Applied Biosystems
Prism® 7300 Sequence Detector (Thermo Fisher Scientific,
Inc.). qPCR was performed in triplicate for each sample in a 20 µl
reaction system which consisted of template cDNA (2 µl), primers (1
µM), SYBR Premix Ex Taq (10 µl), ROX Reference Dye (0.4 µl) and
ddH2O (7.6 µl). The thermocycler settings were as
follows: an initial cycle of 60 sec at 95°C, then 45 cycles of 15
sec at 95°C, 15 sec at 58°C and 30 sec at 72°C. GAPDH was used as
the internal control. The relative mRNA level was determined by the
2−ΔΔCq method (15). The
sequences (5′-3′) of the primers were as follows: DNM3 forward,
TCGAGGGTCGGG CATTGTA; DNM3 reverse, CTTCAATCTCAAGGCGAAC TTCA; GAPDH
forward, TGTGGGCATCAATGGATTTGG; and GAPDH reverse,
ACACCATGTATTCCGGGTCAAT.
Western blotting
Total protein was extracted using NP40 solution
(cat. no. P0013F; Beyotime Institute of Biotechnology, Haimen,
China). Western blotting was performed as described in an existing
protocol (16). Following transfer to
polyvinylidine fluoride (PVDF) membranes, the protein levels were
determined by incubation with the previously described primary
antibodies overnight at 4°C. All primary antibodies were used at a
dilution of 1:500, with the exception of GADPH, which was used at a
dilution of 1:1,000. Subsequent to washing with TBS containing
Tween 20 (TBST), the PVDF membranes were incubated with the
corresponding horseradish peroxidase-conjugated secondary
antibodies (dilution, 1:10,000) for 2 h at room temperature,
including HRP-linked anti-rabbit IgG (cat. no. 7074) or HRP-linked
anti-mouse IgG (cat. no. 7076) from Cell Signaling Technology, Inc.
(Danvers, MA, USA). Subsequent to washing with TBST, the signals
were developed using an enhanced chemiluminescence system
(ZSGB-Bio; OriGene China, Beijing, China) and visualized using the
Tanon 5200 Chemiluminescent Imaging System (Tanon Science and
Technology Co., Ltd., Shanghai, China).
Cell viability assay
SMMC-7721 cells were seeded on 96-well plates at a
density of 5×103 cells/well and incubated with Cell
Counting Kit-8 (CCK-8) reagent (Dojindo Molecular Technologies,
Inc., Kumamoto, Japan) at 37 °C for 2 h. The absorbance values at
490 nm were evaluated by using a microplate reader (Omega Bio-tek,
Inc., Norcross, GA, USA). All experiments were performed in
triplicate.
Cell cycle analysis
Cell cycle analysis was performed using a cell cycle
kit (cat. no. C1052; Beyotime Institute of Biotechnology, Haimen,
China). SMMC-7721 cells were washed with cold PBS three times and
then fixed in ice-cold 70% ethanol at −20°C for 12 h. Following
fixation, cells were resuspended in cold PBS supplemented with 200
mg/ml ribonuclease (cat. no. C1052-3; Beyotime Institute of
Biotechnology) and stained with 0.5 ml propidium iodide (PI)
staining buffer at 37°C for 30 min in the dark. Cell cycle analysis
was determined using a BD FACSVerse flow cytometer (BD Biosciences,
Franklin Lakes, NJ, USA). The populations of cells distributed in
the G0/G1, S and G2/M cell cycle
phases were evaluated by FlowJo software (version 7.6.1; Tree Star,
Inc., Ashland, OR, USA). All experiments were repeated three
times.
Annexin V-PI assay
The Annexin V-PI assay was performed using a
commercial Annexin V-FITC Apoptosis Detection kit (cat. no. C1062;
Beyotime Institute of Biotechnology). SMMC-7721 cells were
resuspended in cold PBS and harvested by centrifugation at 157 × g
for 5 min at 4°C. The cells were then washed with PBS and
resuspended in 300 µl binding buffer from the kit. Subsequently,
the cells were incubated with 5 µl Annexin V-FITC and 5 µl PI in
the dark for 15 min and subjected to flow cytometry.
Short hairpin (sh)RNA
transfection
Human DNM3 shRNA and a scrambled shRNA control were
purchased from Santa Cruz Biotechnology, Inc. and transfected into
SMMC-7721 cells using Lipofectamine 3000 (Thermo Fisher Scientific,
Inc.), according to the manufacturer's protocol.
DNM3 overexpression
The pLenti-DNM3 cDNA lentiviral vector was purchased
from Shanghai GenePharma Co., Ltd. (Shanghai, China). The vector
included an enhanced green fluorescent protein (EGFP) tag. The
pLenti-DNM3 cDNA vector and negative control (NC) vector (EGFP
only) were transfected into SMMC-7721 cells, respectively.
Determination of NO production
NO production was detected using a commercial NO
detection kit (cat. no. S0021; Beyotime Institute of Biotechnology,
Haimen, China) to evaluate nitrite accumulation in the culture
medium supernatant, using Griess reagent following conversion of
nitrate into nitrite by nitrate reductase. Absorbance values at 540
nm were evaluated using a microplate reader (Omega Bio-tek, Inc.,
Norcross, GA, USA).
Evaluation of ROS generation
Intracellular expression levels of ROS were
determined by the oxidative conversion of 2,7-dichlorofluorescein
diacetate (DCFH-DA) to the highly fluorescent compound
2,7-dichlorofluorescein (DCF). Following their harvesting and
washing in PBS, the cells were incubated with 10 µM DCFH-DA for 20
min at 37°C in the dark. DCF fluorescence was evaluated using a
GLOMAX Multidetection System (Promega Corporation, Madison, WI,
USA) with a blue filter (excitation 490 nm; emission 510–570
nm).
Mitochondrial membrane potential
assay
The mitochondrial membrane potential was detected
with a commercial
5,5,6,6-tetrachloro-1,1,3,3-tetraethyl-imidacarbocyanine iodide
(JC-1) probe kit used according to the manufacturer's protocol
(cat. no. C2006; Beyotime Institute of Biotechnology). Briefly,
cells seeded in 96-well plates were incubated with JC-1 (5 µg/ml)
at 37°C for 20 min. The mitochondrial membrane potential was
determined by the relative intensity of dual emissions from
mitochondrial JC-1 monomers or aggregates under a fluorescence
microscope with a 488 nm excitation wavelength.
Intracellular Ca2+
concentration detection
Intracellular Ca2+ concentration was
determined with Fluo-3/acetoxymethyl ester (Fluo-3/AM; cat. no.
S1056; Beyotime Institute of Biotechnology) staining conducted as
according to the manufacturer's protocol. Briefly, cells were
suspended in Tyrode's buffer and incubated with 5 µM Fluo-3/AM at
room temperature for 30 min, in the dark. The extent of Fluo-3
fluorescence in cells was then analyzed with a FACSCalibur flow
cytometer (BD Biosciences).
Immunohistochemistry (IHC)
The iNOS protein levels in the subcutaneous tumors
of nude mice were determined by IHC. The nude mice were sacrificed
42 days after the generation of the xenograft model, and the tumors
were excised, fixed in 4% paraformaldehyde and embedded in
paraffin. Subsequently, tumor tissue sections of 5-µm thickness
were obtained. IHC was performed according to a previously
published protocol (17). A primary
antibody against iNOS was used at a dilution of 1:200. Sections
were incubated with the primary antibody at 4°C overnight. After
washing with TBST, the sections were incubated with the
aforementioned HRP-linked anti-mouse IgG at room temperature for 2
h (dilution, 1:10,000). Reactivity was visualized with
3,3′-diaminobenzidine staining at room temperature for 10 min. The
sections were counterstained with hematoxylin. Pathological changes
were photographed with a BX51 light microscope (Olympus
Corporation, Tokyo, Japan).
Statistical analysis
The data are expressed as the means of 3 repetitions
± standard deviation, and differences between groups were analyzed
using a Student's t-test or a one-way analysis of variance.
P<0.05 was considered to indicate a statistically significant
difference.
Results
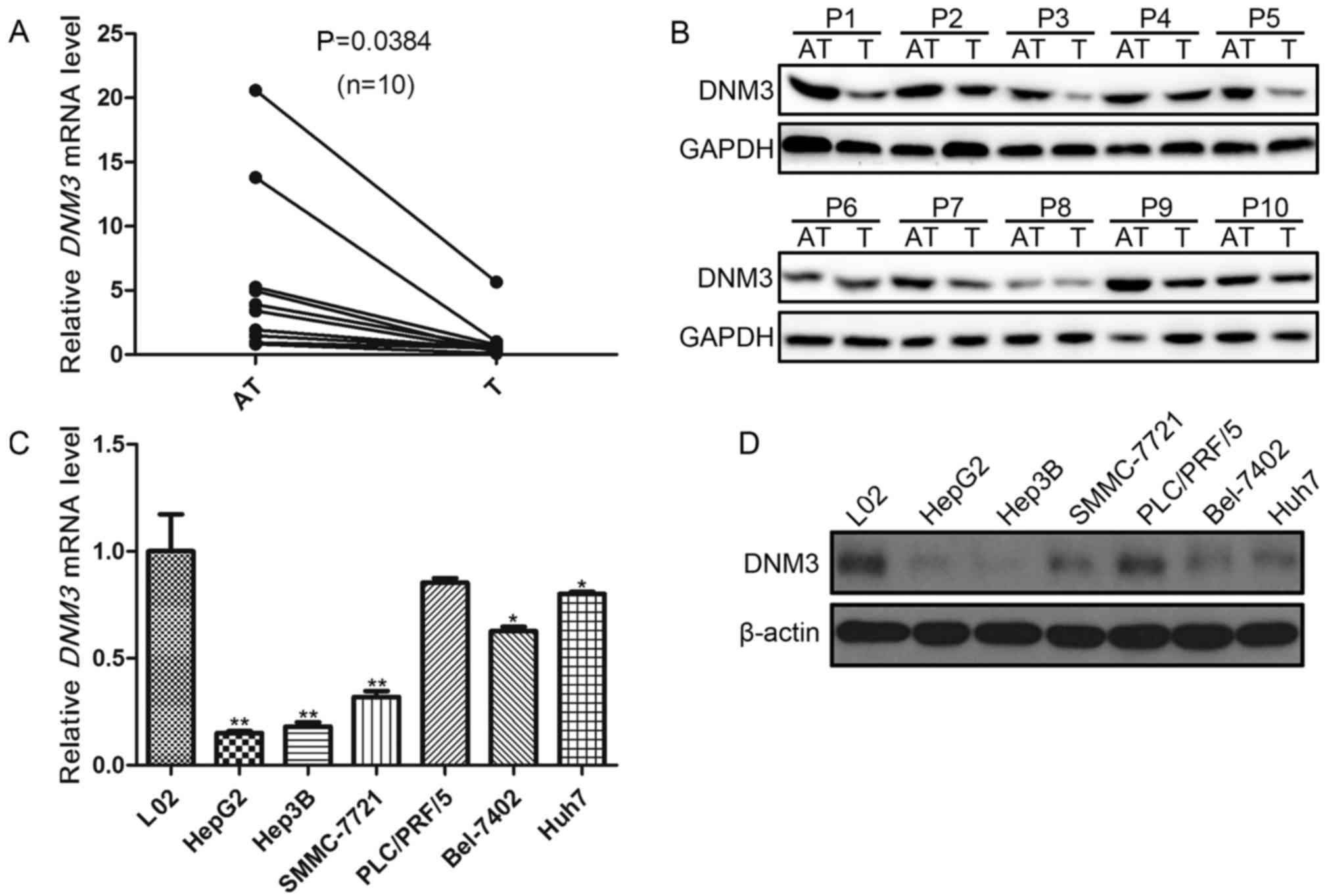
DNM3 is downregulated in human HCC
tissues and cells
In order to investigate the role of DNM3 in HCC
proliferation, the present study first evaluated DNM3 expression
levels in 10 pairs of HCC tissues and the peritumoral tissues.
RT-qPCR demonstrated a downregulation of DNM3 mRNA in the tumor
tissues compared with that in the adjacent tissues (P=0.0384;
Fig. 1A). In addition, decreased DNM3
protein expression levels in were detected in HCC tumor tissues
compared with their paired tumor-adjacent tissues (Fig. 1B).
Subsequently, DNM3 expression levels in 6
human HCC cell lines were evaluated. The DNM3 mRNA
expression levels in the L02 human hepatic cell line was used as
the control. It was revealed that DNM3 expression levels
were significantly decreased in HepG2, Hep3B, SMMC-7721, Bel-7402
and Huh7 cells compared with L02 cells (P<0.05; Fig. 1C); however, DNM3 was not
significantly decreased in PLC/PRF/5 cells. The protein expression
levels of DNM3 were also decreased in HCC cell lines compared with
L02 cells (Fig. 1D). These results
were consistent with the findings by Inokawa et al (14) that DNM3 was downregulated in human HCC
tissues.
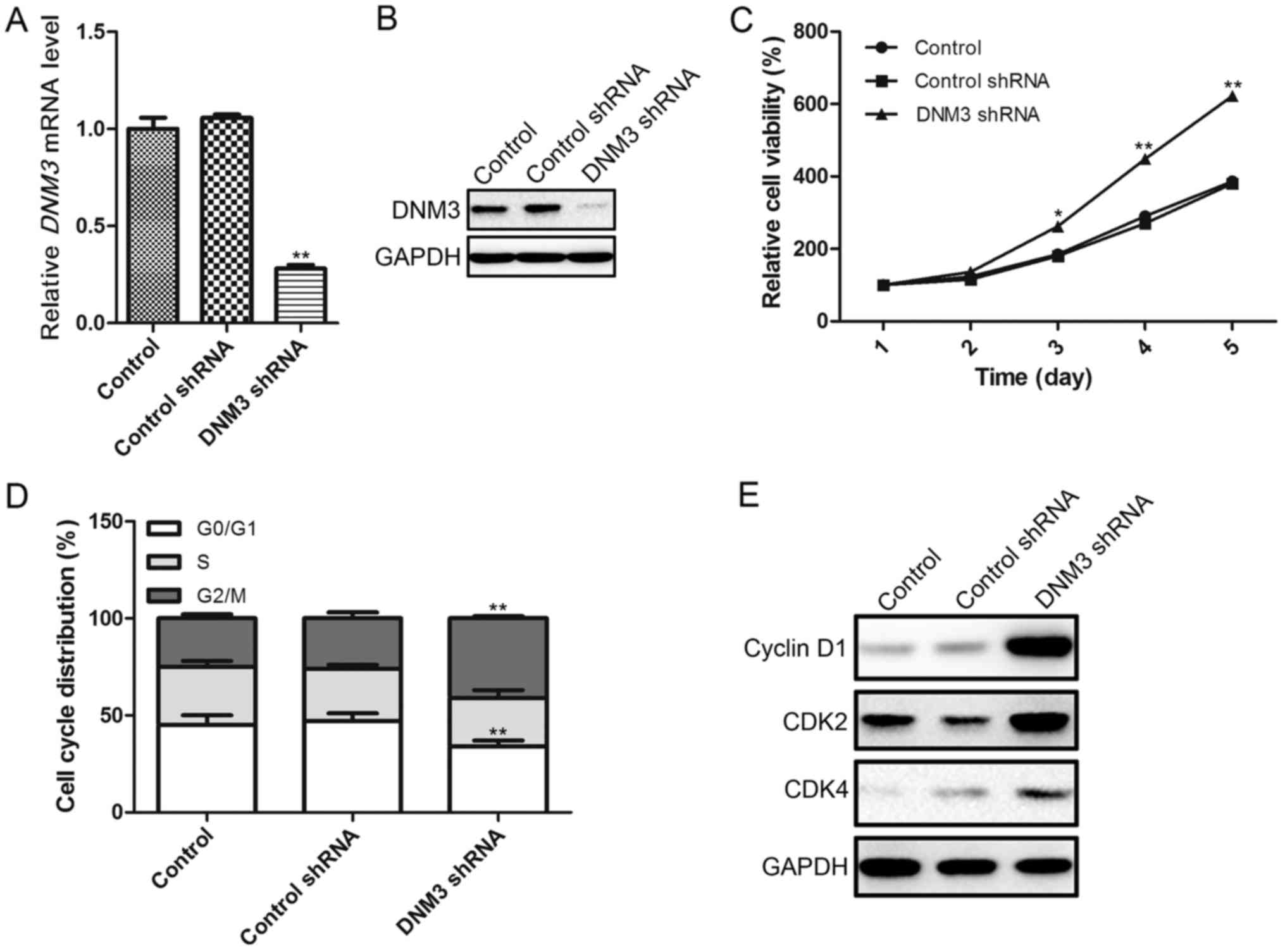
Inhibition of DNM3 in SMMC-7721 cells
promotes cell proliferation
In order to investigate the effects of DNM3
downregulation on HCC cells, the present study used DNM3
shRNA to interrupt DNM3 expression in SMMC-7721 cells, as
these cells exhibited a moderate DNM3 expression level. The
interference efficiency of DNM3 shRNA was confirmed by
RT-qPCR and western blotting, which demonstrated that DNM3
shRNA significantly downregulated DNM3 at the mRNA and protein
levels (Fig. 2A and B). As previous
studies had suggested that DNM3 may serve as a tumor suppressor of
HCC, we hypothesized that the downregulation of DNM3 may promote
HCC cell proliferation. To verify this hypothesis, a CCK-8 assay
was used to evaluate cell viability. It was revealed that the
downregulation of DNM3 increased cell proliferation capacity
(Fig. 2C). To investigate the
possible mechanism by which the downregulation of DNM3 promoted HCC
cell proliferation, the cell cycle distribution of SMMC-7721 cells
was analyzed by flow cytometry. The shRNA-mediated downregulation
of DNM3 significantly reduced the percentage of SMMC-7721
cells in the G0/G1 cell cycle phase, and
increased the percentage of cells in the G2/M phase
(Fig. 2D). Furthermore, the protein
expression levels of cell cycle-associated regulatory molecules
were determined, demonstrating that a decrease in DNM3 increased
the expression levels of cyclin D1, CDK2 and CDK4 compared with the
control groups (Fig. 2E).
Collectively, these results indicate that the downregulation of
DNM3 may promote HCC cell proliferation by increasing cell
cycle-associated regulatory proteins.
Overexpression of DNM3 in SMMC-7721
cells induces apoptosis and suppresses tumor growth
To further evaluate the tumor-suppressive effect of
DNM3 on HCC cells, DNM3 was overexpressed by introducing the DNM3
cDNA vector into SMMC-7721 cells. At 96 h after transfection, the
overexpression efficiency was determined by RT-qPCR and western
blotting. The transfection of the DNM3 cDNA vector increased DNM3
mRNA and protein expression levels in SMMC-7721 cells compared with
the controls (Fig. 3A and B). As
expected, the CCK-8 assay revealed that overexpression of DNM3
inhibited SMMC-7721 cell viability (Fig.
3C).
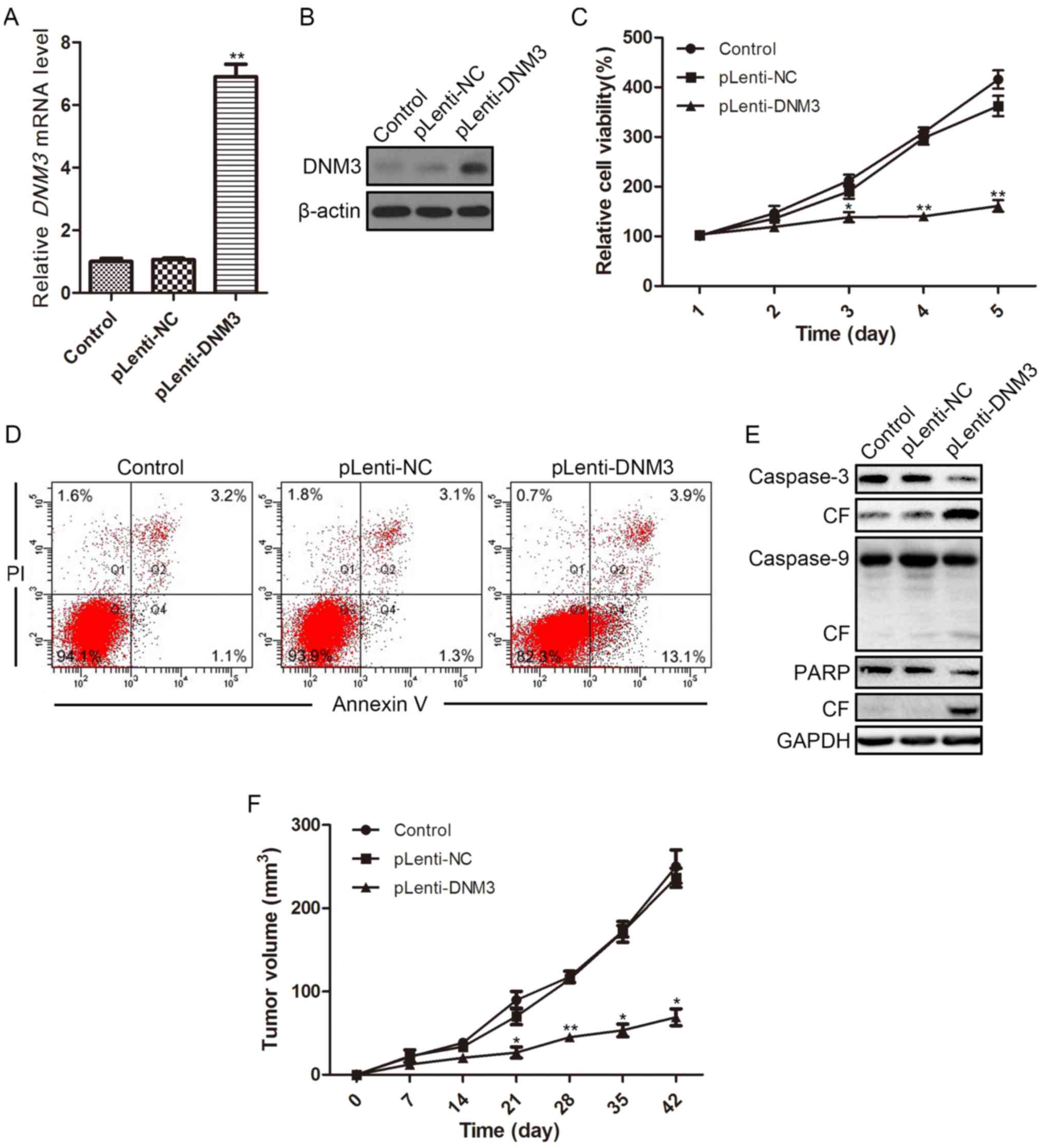 | Figure 3.Overexpression of DNM3 in SMMC-7721
cells reduces cell viability and induces apoptosis. Overexpression
of DNM3 in SMMC-7721 cells was accomplished by transfecting
SMMC-7721 cells with lentivirus containing DNM3. DNM3
overexpression efficiency was determined by (A) reverse
transcription-quantitative polymerase chain reaction and (B)
western blotting. (C) Cell viability of SMMC-7721 cells
overexpressing DNM3 was determined by Cell Counting Kit-8 assay.
(D) Flow cytometric analysis revealed increased apoptosis in
DNM3-overexpressing SMMC-7721 cells. (E) Western blotting results
for caspase-3, caspase-9 and PARP, as well as their respective CF,
in SMMC-7721 cells overexpressing DNM3. (F) For the in vivo
experiments, SMMC-7721 cells with DNM3 overexpression and control
cells were injected into the subcutaneous tissues of nude mice
(n=8). The tumor volume of each subcutaneous tumor was determined
every 7 days. Overexpression of DNM3 inhibited tumor growth
compared with the control groups. *P<0.05 and **P<0.01 vs.
pLenti-NC group. DNM3, dynamin 3; pLenti-DNM3, lentivirus
containing DNM3; pLenti-NC, negative control lentivirus; PI,
propidium iodide; CF, cleaved form; PARP, poly ADP-ribose
polymerase. |
Flow cytometric analysis revealed that the ratio of
apoptotic cells was increased in DNM3-overexpressing cells (13.1%)
as compared with NC-transfected cells (1.3%; Fig. 3D). Subsequently, the protein
expression levels of apoptotic proteins were evaluated by western
blotting. In SMMC-7721 cells with DNM3 overexpression, the cleaved
forms of caspase-3, caspase-9 and PARP increased compared with the
control and NC groups (Fig. 3E),
indicating that the overexpression of DNM3 activated
caspase-dependent apoptosis. The tumor-suppressive effect of DNM3
in HCC was further verified in animal models; DNM3 overexpression
significantly inhibited subcutaneous tumor growth relative to the
control groups in nude mice (Fig.
3F).
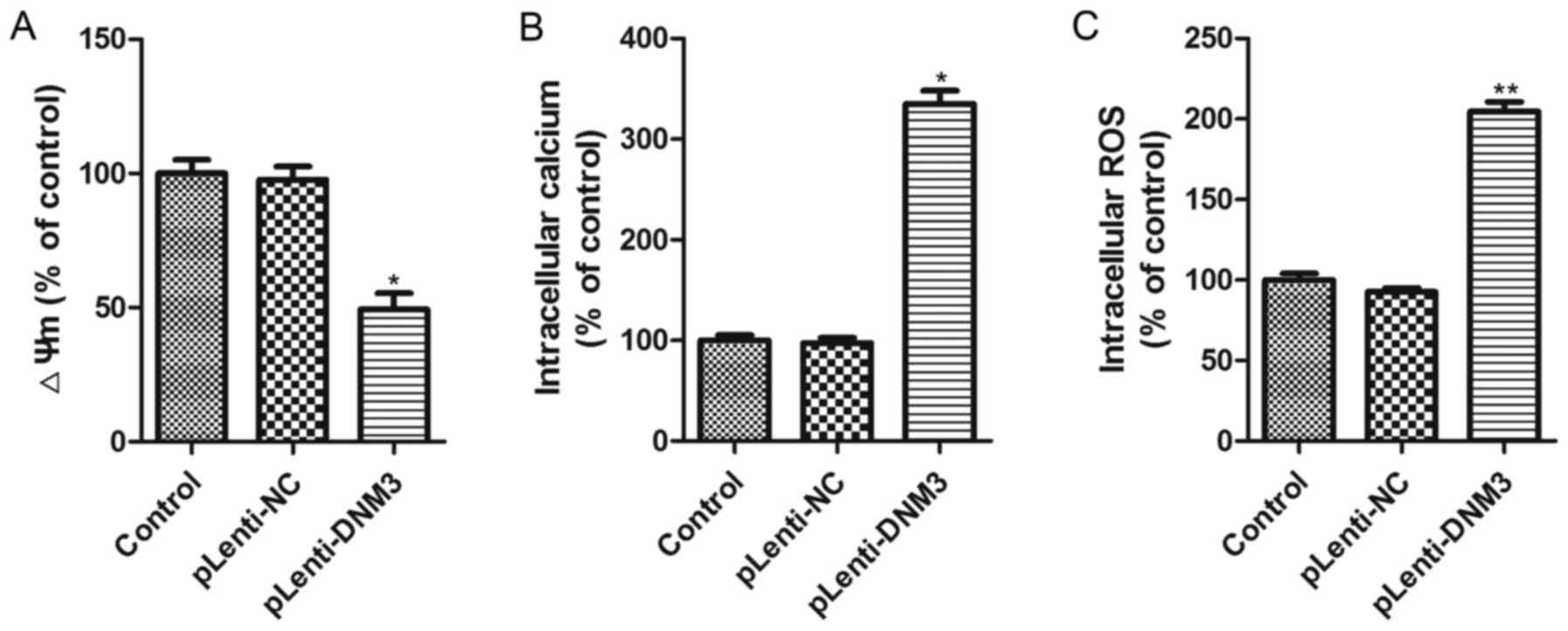
Overexpression of DNM3 induces
intracellular ROS accumulation
The aforementioned results revealed that the
downregulation of DNM3 in HCC cells promoted cell proliferation,
whereas the overexpression of DNM3 induced HCC cell apoptosis and
inhibited HCC growth. Subsequently, the present study aimed to
elucidate the potential mechanism by which DNM3 upregulation
induced HCC cell apoptosis. It has previously been reported that
mitochondrial damage is an early event in cellular death and that
intracellular ROS accumulation is a biochemical mediator of
apoptosis which could trigger cell death (18–20); thus,
the present study investigated the mitochondrial membrane
potential, cytosolic free Ca2+ concentration and ROS
accumulation in SMMC-7721 cells with DNM3 overexpression. The
results demonstrated that mitochondrial membrane potential was
significantly decreased and free Ca2+ concentration
significantly increased following DNM3 overexpression (Fig. 4A and B). Significantly elevated
intracellular ROS accumulation in DNM3-overexpressing SMMC-7721
cells was also observed (Fig. 4C).
These results indicate that DNM3 overexpression can impair
mitochondrial function and promote ROS accumulation.
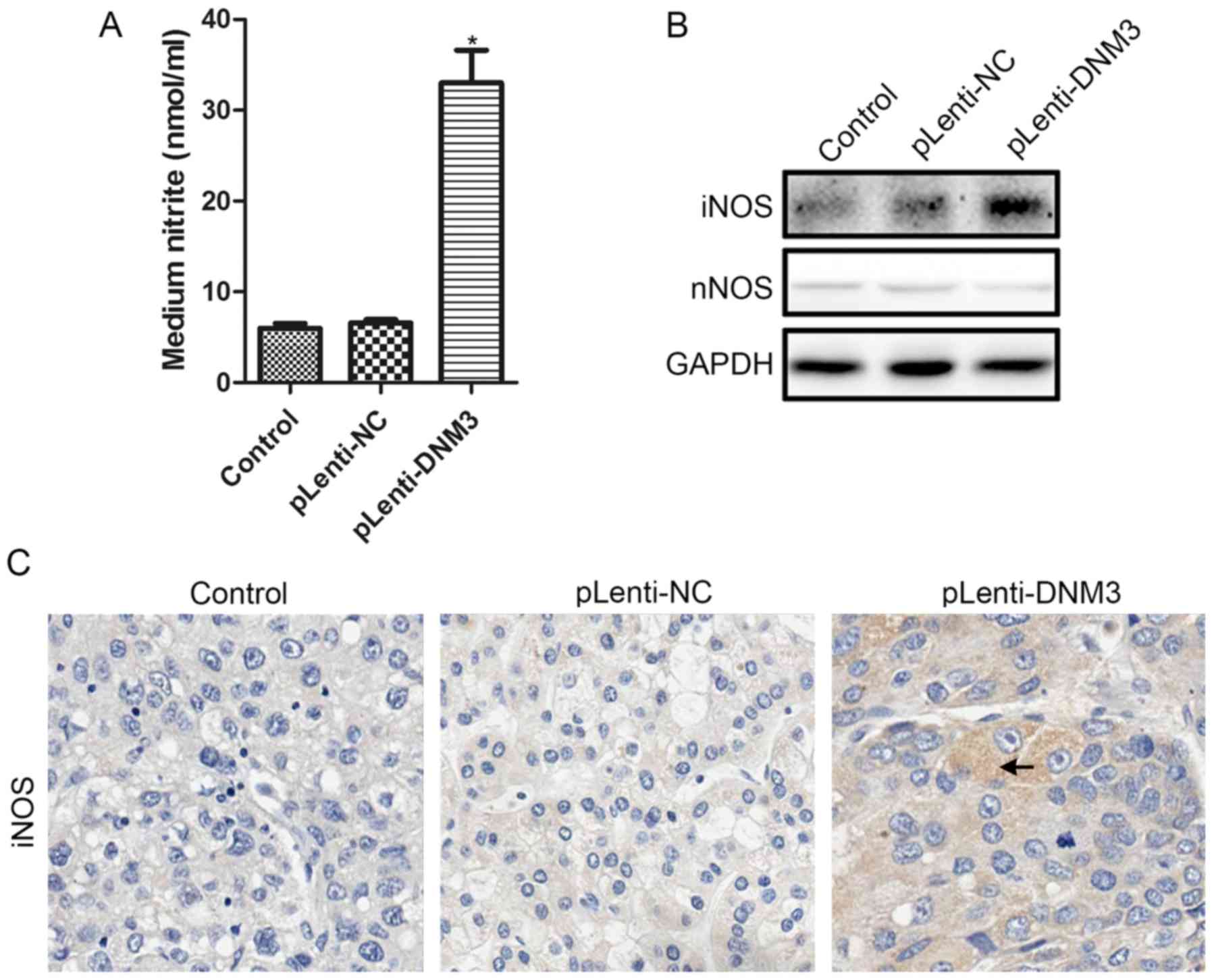
Overexpression of DNM3 induces iNOS
and NO production
It has been previously reported that the
accumulation of intracellular ROS increases with NO (21,22), and
that DNM3 can activate NO production (10). Therefore, we hypothesized that
increased ROS accumulation and apoptosis in DNM3-overexpressing
cells may be due to increased NO. Nitrite levels in the culture
supernatants of SMMC-7721 cells overexpressing DNM3 were detected.
Increased nitrite in the supernatants, indicating elevated NO
production, were detected following DNM3 upregulation in SMMC-7721
cells (Fig. 5A).
Subsequently, the protein expression levels of NOS
following DNM3 overexpression were investigated. According to a
study by Heymann and Hinshaw (7),
DNM3 may interact with nNOS directly and activate NO production.
However, in the present study, the protein expression level of nNOS
was the same following DNM3 overexpression. The protein expression
level of iNOS, by contrast, was markedly increased in
DNM3-overexpressing SMMC-7721 cells (Fig.
5B). Furthermore, the expression level of iNOS in subcutaneous
tumors generated from DNM3-overexpressing SMMC-7721 cells also
increased, as revealed by IHC staining (Fig. 5C). These results demonstrated that
increased NO production may induce ROS accumulation, and may result
from increased iNOS production.
Inhibition of iNOS attenuates
DNM3-induced apoptosis
To investigate whether the increased NO and ROS
expression levels were the results of iNOS activation, the specific
iNOS inhibitor L-canavanine was utilized for further experiments.
Incubation with L-canavanine significantly attenuated the
DNM3-induced mitochondrial membrane potential decrease and the
intracellular ROS accumulation (Fig. 6A
and B). The present study also investigated whether the
inhibition of iNOS could attenuate the apoptosis induced by DNM3
upregulation. It was revealed that inhibition of iNOS inhibited the
activation of PARP cleavage, indicating attenuation of apoptosis
(Fig. 6C). Thus, by inhibiting iNOS,
L-canavanine reversed DNM3-induced ROS accumulation and, as a
result, HCC cell apoptosis. This suggests that the activation of
iNOS underlies the apoptosis of SMMC-7721 cells resulting from DNM3
upregulation.
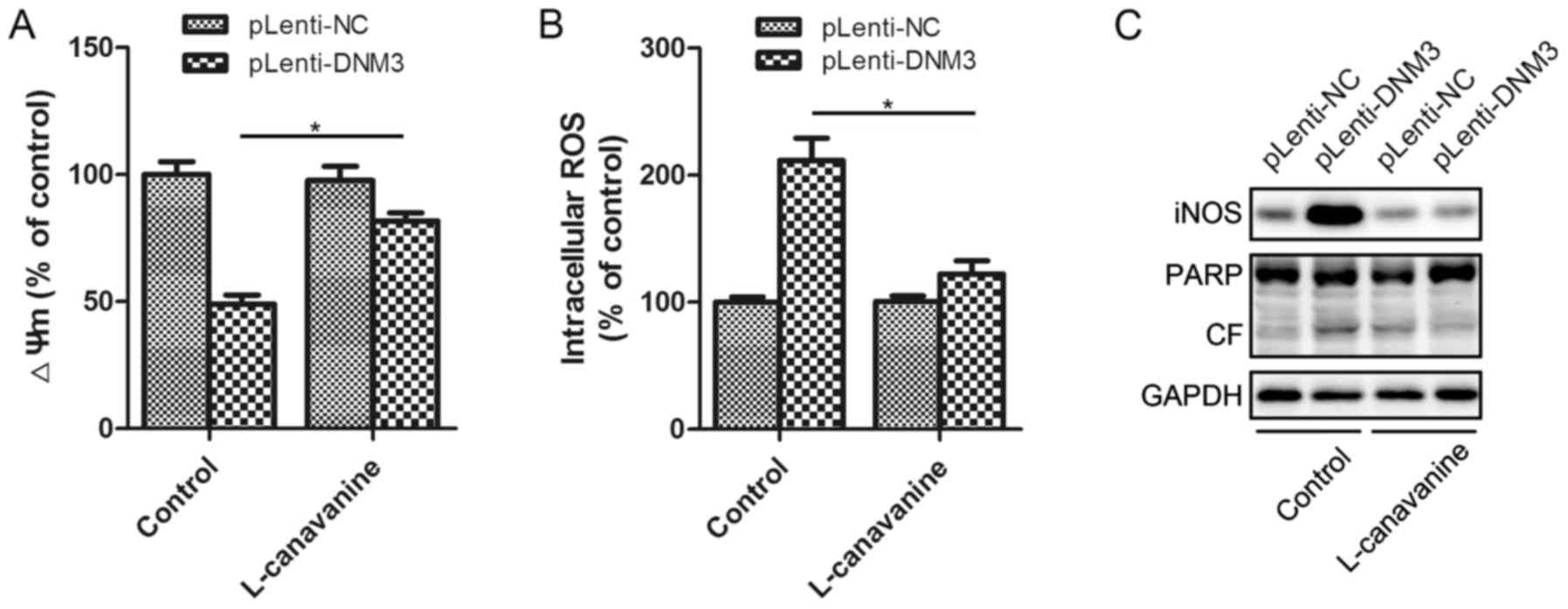 | Figure 6.Inhibition of iNOS attenuated
DNM3-overexpression-induced apoptosis. (A) The iNOS inhibitor,
L-canavanine, attenuated the DNM3-induced decrease in ΔΨm. (B)
L-canavanine inhibited the DNM3-induced increase in intracellular
ROS levels. (C) L-canavanine inhibited a DNM3-induced increase in
cleaved PARP in SMMC-7721 cells. *P<0.05. DNM3, dynamin 3; iNOS,
inducible nitric oxide synthase; ΔΨm, mitochondrial membrane
potential; ROS, reactive oxygen species; pLenti-DNM3, lentivirus
containing DNM3; pLenti-NC, negative control lentivirus; PARP, poly
ADP-ribose polymerase; CF, cleaved form. |
Discussion
The present study demonstrated that DNM3 was
downregulated in HCC tissues and cells. The downregulation of DNM3
promoted cell proliferation by increasing certain cell
cycle-associated proteins. By contrast, overexpression of DNM3
inhibited HCC cell viability and induced apoptosis. The in
vivo experiments also demonstrated that overexpression of DNM3
suppressed the growth of human HCC xenograft tumors in nude mice.
Furthermore, the mechanisms underlying the DNM3-mediated inhibition
of HCC growth was investigated. This revealed that increased iNOS
expression levels following DNM3 overexpression were associated
with decreased mitochondrial membrane potentials and increased
accumulation of intracellular ROS. These effects could be
attenuated by the iNOS inhibitor L-canavanine. Thus, DNM3 can
inhibit HCC by activating iNOS and subsequent NO production,
inducing ROS accumulation and HCC cell apoptosis.
NO is one of the smallest signaling molecules and is
involved in various physiological functions (23). As previously reported, NO serves a
dual role in tumor biological behavior: Prolonged and excessive NO
levels may lead to inflammation and tumor development, whereas
higher expression levels of NO could also trigger cell apoptosis
(24,25). NO may be generated in NOS-dependent
and NOS-independent manners (23,26). The
NOS-independent signaling pathway involves the non-enzymatic
generation of NO; NO produced from dietary sources of nitrate and
nitrite in the bloodstream and tissues complements the
NOS-dependent signaling pathway (26). NOSs are enzymes that catalyze the
formation of NO from nitrogen in guanidine and L-arginine (27,28). There
are three forms of NOS: nNOS, iNOS and eNOS (23). The present study demonstrated that
DNM3 increased the level of iNOS but not nNOS, in contrast to a
previous study by Hyndman et al which suggested that DNM3
activated nNOS (10). Previous
studies have revealed that nNOS and eNOS are constitutive NOS
isoforms (cNOS), and may be stimulated by a variety of signaling
pathways that are dependent or independent of Ca2+
(29). However, iNOS may be induced
by pathological stimuli which are Ca2+-independent in
numerous types of cells, including hepatocytes, Kupffer cells,
lymphocytes and macrophages (30).
One of the major differences between iNOS and cNOS is that iNOS is
capable of releasing a large amount of NO for relatively long
periods of time in a sustained manner, whereas cNOS produces a
small amount of NOS over a short period of time and is short-acting
(31–33). The present study demonstrated that
increased expression levels of iNOS after DNM3 upregulation induced
apoptosis, whereas the inhibition of iNOS via L-canavanine
attenuated the ROS accumulation and apoptosis. Janakiram and Rao
(23) reported that high levels of
iNOS expression may exert cytostatic or cytotoxic effects on tumor
cells; thus, the increased iNOS expression levels following DNM3
overexpression were likely a crucial step in the induction of
SMMC-7721 cell apoptosis. However, the present study did not
elucidate the pathway underlying DNM3-activated iNOS production.
This may be due to a direct interaction between DNM3 and iNOS
(11).
We hypothesized that impaired mitochondrial function
and intracellular ROS accumulation may serve a vital role in
inducing apoptotic cell death following DNM3 upregulation.
Mitochondrial damage is an early event in cellular death (18). The decrease in mitochondrial membrane
potential occurs earlier than the morphological alterations of
mitochondria and DNA fragmentation. Increased membrane permeability
decreases mitochondrial membrane potential, and this is closely
associated with the opening of the mitochondrial permeability
transition pore (MPTP). Irreversible opening of the MPTP has
previously been revealed to be involved in cell apoptosis (34). Mitochondria are regarded as the main
source of ROS in tumor cells (35,36). It
was previously demonstrated that an increased expression level of
intracellular ROS was a biochemical mediator of apoptosis and was
sufficient to trigger cell death (37). Excessive ROS accumulation has also
been revealed to alter cell functions and disrupt the intracellular
homeostasis of the redox system via oxidative damage (38). Cohen and Vanhoutte (28) reported that ROS mediated the early and
late steps of apoptosis associated with mitochondrial dysfunction.
In the present study, an increased DNM3 expression level was
revealed to decrease mitochondrial membrane potential and increase
intracellular ROS accumulation.
In conclusion, the present study demonstrated that
DNM3 can inhibit the proliferation of SMMC-7721 cells and induce
apoptosis by activating iNOS production and, subsequently, an
increase in NO expression level and ROS accumulation. Further
studies are required in order to reveal the interactions between
DNM3 and iNOS.
Acknowledgements
The present study was supported by the Health and
Family Commission of Shanghai Jinshan District (grant no.
JSKJ-KTMS-2014-06).
References
|
1
|
Torre LA, Bray F, Siegel RL, Ferlay J,
Lortet-Tieulent J and Jemal A: Global cancer statistics, 2012. CA
Cancer J Clin. 65:87–108. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Earl TM and Chapman WC: Hepatocellular
carcinoma: Resection versus transplantation. Semin Liver Dis.
33:282–292. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Cusnir M and Patt YZ: Novel systemic
therapy options for hepatocellular carcinoma. Cancer J. 10:97–103.
2004. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Yu MC and Yuan JM: Environmental factors
and risk for hepatocellular carcinoma. Gastroenterology. 127 5
Suppl 1:S72–S78. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Shiraha H, Yamamoto K and Namba M: Human
hepatocyte carcinogenesis (Review). Int J Oncol. 42:1133–1138.
2013.PubMed/NCBI
|
|
6
|
El-Serag HB and Rudolph KL: Hepatocellular
carcinoma: Epidemiology and molecular carcinogenesis.
Gastroenterology. 132:2557–2576. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Heymann JA and Hinshaw JE: Dynamins at a
glance. J Cell Sci. 122:3427–3431. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Hinshaw JE: Dynamin and its role in
membrane fission. Annu Rev Cell Dev Biol. 16:483–519. 2000.
View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Schmid SL and Frolov VA: Dynamin:
Functional design of a membrane fission catalyst. Annu Rev Cell Dev
Biol. 27:79–105. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Hyndman KA, Musall JB, Xue J and Pollock
JS: Dynamin activates NO production in rat renal inner medullary
collecting ducts via protein-protein interaction with NOS1. Am J
Physiol Renal Physiol. 301:F118–F124. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Cao S, Yao J and Shah V: The proline-rich
domain of dynamin-2 is responsible for dynamin-dependent in vitro
potentiation of endothelial nitric-oxide synthase activity via
selective effects on reductase domain function. J Biol Chem.
278:5894–5901. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Kang-Decker N, Cao S, Chatterjee S, Yao J,
Egan LJ, Semela D, Mukhopadhyay D and Shah V: Nitric oxide promotes
endothelial cell survival signaling through S-nitrosylation and
activation of dynamin-2. J Cell Sci. 120:492–501. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Shen J, Wang S, Zhang YJ, Kappil M, Wu HC,
Kibriya MG, Wang Q, Jasmine F, Ahsan H, Lee PH, et al: Genome-wide
DNA methylation profiles in hepatocellular carcinoma. Hepatology.
55:1799–1808. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Inokawa Y, Nomoto S, Hishida M, Hayashi M,
Kanda M, Nishikawa Y, Takeda S, Fujiwara M, Koike M, Sugimoto H, et
al: Dynamin 3: A new candidate tumor suppressor gene in
hepatocellular carcinoma detected by triple combination array
analysis. Onco Targets Ther. 6:1417–1424. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Livak KJ and Schmittgen TD: Analysis of
relative gene expression data using real-time quantitative PCR and
the 2(−Delta Delta C(T)) method. Methods. 25:402–408. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Nie X, Song S, Zhang L, Qiu Z, Shi S, Liu
Y, Yao L and Zhu D: 15-Hydroxyeicosatetraenoic acid (15-HETE)
protects pulmonary artery smooth muscle cells from apoptosis via
inducible nitric oxide synthase (iNOS) pathway. Prostaglandins
Other Lipid Mediat. 97:50–59. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Yin XY, Jiang JM, Liu JY and Zhu JR:
Effects of endogenous nitric oxide induced by 5-fluorouracil and
L-Arg on liver carcinoma in nude mice. World J Gastroenterol.
13:6249–6253. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Lemasters JJ: V. Necrapoptosis and the
mitochondrial permeability transition: Shared pathways to necrosis
and apoptosis. Am J Physiol. 276:G1–G6. 1999.PubMed/NCBI
|
|
19
|
Pastorino JG and Hoek JB: Ethanol
potentiates tumor necrosis factor-alpha cytotoxicity in hepatoma
cells and primary rat hepatocytes by promoting induction of the
mitochondrial permeability transition. Hepatology. 31:1141–1152.
2000. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Circu ML and Aw TY: Reactive oxygen
species, cellular redox systems, and apoptosis. Free Radic Biol
Med. 48:749–762. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Lala PK and Chakraborty C: Role of nitric
oxide in carcinogenesis and tumour progression. Lancet Oncol.
2:149–156. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Soliman MK, Mazzio E and Soliman KF:
Levodopa modulating effects of inducible nitric oxide synthase and
reactive oxygen species in glioma cells. Life Sci. 72:185–198.
2002. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Vannini F, Kashfi K and Nath N: The dual
role of iNOS in cancer. Redox Biol. 6:334–343. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Bonavida B and Baritaki S: Dual role of NO
donors in the reversal of tumor cell resistance and EMT:
Downregulation of the NF-κB/Snail/YY1/RKIP circuitry. Nitric Oxide.
24:1–7. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Kim SF, Huri DA and Snyder SH: Inducible
nitric oxide synthase binds, S-nitrosylates, and activates
cyclooxygenase-2. Science. 310:1966–1970. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Lepore DA: Nitric oxide
synthase-independent generation of nitric oxide in muscle
ischemia-reperfusion injury. Nitric Oxide. 4:541–545. 2000.
View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Moncada S, Palmer RM and Higgs EA: Nitric
oxide: Physiology, pathophysiology, and pharmacology. Pharmacol
Rev. 43:109–142. 1991.PubMed/NCBI
|
|
28
|
Cohen RA and Vanhoutte PM:
Endothelium-dependent hyperpolarization. Beyond nitric oxide and
cyclic GMP. Circulation. 92:3337–3349. 1995. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Panda SP, Gao YT, Roman LJ, Martásek P,
Salerno JC and Masters BS: The role of a conserved serine residue
within hydrogen bonding distance of FAD in redox properties and the
modulation of catalysis by Ca2+/calmodulin of constitutive
nitric-oxide synthases. J Biol Chem. 281:34246–34257. 2006.
View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Bredt DS, Hwang PM, Glatt CE, Lowenstein
C, Reed RR and Snyder SH: Cloned and expressed nitric oxide
synthase structurally resembles cytochrome P-450 reductase. Nature.
351:714–718. 1991. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Dominiczak AF and Bohr DF: Nitric oxide
and its putative role in hypertension. Hypertension. 25:1202–1211.
1995. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Crane BR, Arvai AS, Gachhui R, Wu C, Ghosh
DK, Getzoff ED, Stuehr DJ and Tainer JA: The structure of nitric
oxide synthase oxygenase domain and inhibitor complexes. Science.
278:425–431. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Schulz R and Triggle CR: Role of NO in
vascular smooth muscle and cardiac muscle function. Trends
Pharmacol Sci. 15:255–259. 1994. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Crompton M: The mitochondrial permeability
transition pore and its role in cell death. Biochem J. 341:233–249.
1999. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Higuchi Y: Chromosomal DNA fragmentation
in apoptosis and necrosis induced by oxidative stress. Biochem
Pharmacol. 66:1527–1535. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Bartosz G: Reactive oxygen species:
Destroyers or messengers? Biochem Pharmacol. 77:1303–1315. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Buttke TM and Sandstrom PA: Oxidative
stress as a mediator of apoptosis. Immunol Today. 15:7–10. 1994.
View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Sabharwal SS and Schumacker PT:
Mitochondrial ROS in cancer: Initiators, amplifiers or an Achilles'
heel? Nat Rev Cancer. 14:709–721. 2014. View Article : Google Scholar : PubMed/NCBI
|