Introduction
Leukemia is a cancer of the white blood cells, and
can be classified as acute myeloid leukemia (AML), acute
lymphoblastic leukemia or chronic lymphoblastic leukemia (1). AML is the most common type of acute
leukemia, and accounts for ~80% of all cases of acute leukemia in
adults (2). AML is a clonal disorder
caused by rapid proliferation, accumulation and differentiation
arrest of hematopoietic progenitors in the bone marrow and blood
(3). It was predicted that in 2015,
there would be 20,830 new cases and 10,460 mortalities due to AML
in the United States (4).
AML can be divided into 3 risk-based categories
based on cytogenetic information: Favorable, intermediate and poor,
with a 5-year overall survival rate of 55, 24–42 and 11%,
respectively (5). Currently, the main
treatment for patients with AML is cytarabine/anthracycline-based
chemotherapy. However, the majority of patients cannot be cured by
this approach (6). Furthermore, the
majority of AML patients will relapse, with the major cause of
relapse and therapeutic failure in AML being resistance to
chemotherapy (7). Therefore, it is
important to further investigate the molecular mechanisms behind
chemoresistance and develop effective new therapeutic strategies
for the enhancement of efficacy in AML chemotherapy.
MicroRNAs (miRs) are highly conserved,
non-protein-coding, single-stranded small RNAs (~22 nucleotides in
length) that post-transcriptionally regulate gene expression in
cancerous and non-cancerous cells (8). miRs typically decrease the expression
level of target mRNA by interacting preferentially with the 3′
untranslated region (3′UTR) of target mRNA, resulting in
translation repression or degradation (9,10). miRs
act as either oncogenes or tumor suppressors in the development and
progression of human carcinogenesis (11). Certain miRs that are upregulated in
cancer directly target oncogenes, and function in a proliferative
and anti-apoptotic manner. Conversely, miRs that are downregulated
in certain types of cancer function as tumor suppressors and
inhibit cancer initiation and progression (12–14).
Studies have revealed that miRs control various key cellular
physiological and pathological processes, including the cell cycle,
cellular proliferation, apoptosis, differentiation and development,
and are involved in several human diseases, including cancer
(15,16). Additionally, miRs have been
demonstrated to perform important roles in the regulation of
chemoresistance (17,18). Therefore, miRs may be investigated as
targets for anticancer drug resistance in AML.
Abnormal expression of miR-217 has been demonstrated
in numerous human malignancies, however the mechanisms behind the
expression and function of miR-217 in AML have not yet been
recognized. The present study demonstrated that miR-217 was
downregulated in patients with AML compared with healthy controls.
In addition, upregulation of miR-217 suppressed cell proliferation
and enhanced the chemosensitivity of AML cells to doxorubicin (DOX)
through the cell apoptosis pathway. In addition, the Kirsten rat
sarcoma viral oncogene homolog (KRAS) was identified as a direct
target of miR-217. The present findings have therapeutic
implications and may be explored for implications for the treatment
of AML.
Materials and methods
Clinical specimens
In the present study, samples of bone marrow from 62
patients with AML were collected at Tongji Hospital (Wuhan, China).
A total of 25 healthy subjects were also used in this study as the
control group. The diagnosis of AML was made based on standard
diagnostic methods, including morphological assessment and
cytochemical studies of bone marrow smears. No patients received
anti-leukemic therapy during bone marrow aspiration. The present
study was approved by the Ethics Committee of Tongji Hospital and
informed written consent was also obtained from all patients in the
AML and control groups of this study.
Cell culture
Human leukemia HL-60 and K562 cell lines, were
obtained from the American Type Culture Collection (Manassas, VA,
USA). Cells were cultured in RPMI-1640 medium supplemented with 10%
fetal bovine serum, 100 U/ml penicillin, 100 mg/ml streptomycin and
2 mM L-glutamine (All from Gibco; Thermo Fisher Scientific, Inc.)
in a humidified air atmosphere of 5% CO2 at 37°C.
Cell transfection
Mature miR-217 mimics, negative control (NC), KRAS
small interfering (si) RNA, NC siRNA, and luciferase reporter
plasmids were obtained from Shanghai GenePharma Co., Ltd.
(Shanghai, China). To evaluate the functions of miR-217 and KRAS in
AML cells, cells were transfected with miR-217 mimics or NC, and
KRAS siRNA or NC siRNA using Lipofectamine® 2000
(Invitrogen; Thermo Fisher Scientific, Inc.), according to the
manufacturer's protocol. Subsequent to transfection at 37°C for 6
h, cell culture medium was replaced with RPMI-1640 medium
containing 10% FBS and 2 mM L-glutamine.
RNA isolation, reverse transcription
and quantitative polymerase chain reaction (qPCR)
Bone marrow mononuclear cells from bone marrow
aspirates were isolated using Ficoll-Hypaque density gradient
centrifugation (400 × g for 30 min at 20°C followed by 100 × g for
10 min at 20°C) (Ficoll, Pharmacia LKB Biotechnology, Piscataway,
NY, USA). Total RNA was extracted from bone marrow mononuclear
cells using TRIzol reagent (Invitrogen; Thermo Fisher Scientific,
Inc.) according to the manufacturer's protocol. Single-stranded
cDNA for the miR analysis was synthesized by reverse-transcription
using PrimeScripts RT reagent kit (Takara Biotechnology Co., Ltd.,
Dalian, China) according to the manufacturer's protocol. qPCR was
performed using a SYBR premix Ex Taq kit (Takara Biotechnology Co.,
Ltd.) on an Applied Biosystems 7300 Real-time PCR system (Thermo
Fisher Scientific, Inc.) according to the manufacturer's protocol.
The thermocycling conditions for qPCR were as follows: 95°C for 30
sec; 40 cycles of 95°C for 5 sec; and 60°C for 30 sec. The relative
expression level was calculated using the 2−ΔΔCq method
(19). U6 small nuclear RNA and GAPDH
were used as an internal control. The primer sequences used were as
follows: miR-217 forward, 5′-TACTCAACTCACTACTGCATCAGGA-3′ and
reverse, 5′-TATGGTTGTTCTGCTCTCTGTGTC-3′; and U6 forward,
5′-GCTTCGGCAGCACATATACTAAA-3′ and reverse,
5′-GCTTCACGAATTTGCGTGTCAT-3′. KRAS forward,
5′-GACTCTGAAGATGTACCTATGGTCCTA-3′ and reverse,
5′-CATCATCAACACCCTGTCTTGTC-3′; and GAPDH forward,
5′-ATAGCACAGCCTGGATAGCAACGTAC-3′ and reverse,
5′-CACCTTCTACAATGAGCTGCGTGTG-3′. Each sample was analyzed in
triplicate.
MTT assay
Subsequent to transfection for 24 h, transfected
cells were harvested and seeded into 96-well culture plates at a
density of 3×104 cells. Following the incubation at 37°C
for various time periods (24–96 h), an MTT assay (5 mg/ml,
Sigma-Aldrich; Merck Millipore, St. Louis, MO, USA) was performed
according to the manufacturer's protocol. Briefly, 20 µl MTT assay
solution was added into each well. Subsequent to a 4 h incubation
at 37°C, the 96-well plate was centrifuged at 100 × g for 5 min at
room temperature, and the purple colored precipitate of formazan
was dissolved in 200 µl dimethyl sulfoxide. Subsequent to being
slowly spun at 37°C for 15 min, the absorbance at 490 nm wavelength
was detected using an automatic multi-well spectrophotometer
(Bio-Rad Laboratories, Inc., Hercules, CA, USA). All experiments
were performed in triplicate. Each experiment was repeated at least
3 times.
Chemosensitivity assay
Following transfection for 48 h, transfected cells
were harvested and seeded into 96-well culture plates at a density
of 5×104 cells. Cells were treated with DOX
(Sigma-Aldrich; Merck Millipore) at various concentrations
(32–1048576 ng/ml). Subsequent to incubation at 37°C for 48 h, a
chemosensitivity assay was performed using the MTT assay as
described previously. The dose-response curve was charted at
different concentrations. Each concentration was analyzed in
triplicate. Each experiment was repeated at least 3 times.
Cell apoptosis assay
Following transfection for 48 h, transfected cells
were harvested and seeded into 6-well plates at a density of
2×106 cells. Cells were then treated with DOX at 32
ng/ml. Subsequent to incubation for 48 h at 37°C, cells were
collected and washed with PBS (Gibco; Thermo Fisher Scientific).
Subsequently, cells were centrifuged at 100 × g for 5 min at room
temperature, all PBS was carefully removed and the cells were fixed
in 80% ice-cold ethanol in PBS. Subsequently, cells were
re-suspended in 1X binding buffer to a concentration of
1×104 cells/µl. Then cells were treated with 5 µl of
Annexin V-fluorescein isothiocyanate and 10 µl of propidium iodide
(PI) and incubated for 15 min at room temperature in dark. Cells
were then analyzed with flow cytometry (BD FACS Calibur; BD
Biosciences, Franklin Lakes, NJ, USA). Apoptotic cells were
recognized by a high Annexin V fluorescence signal combined with a
low PI signal and analyzed using CellQuest version 5.1 (BD
Biosciences).
Western blot analysis
Subsequent to transfection for 72 h, transfected
cells were lysed with RIPA lysis buffer (Beyotime Institute of
Biotechnology, Haimen, China). Protein concentration was measured
using a BCA assay kit (Pierce; Thermo Fisher Scientific, Inc.).
Equal amounts of protein (20 µg) were then separated by 10%
SDS-PAGE and transferred to polyvinylidene difluoride membranes
(Merck Millipore). Subsequent to blocking with 5% non-fat dry milk
in TBS saline containing 0.05% Tween-20 (Beyotime Institute of
Biotechnology), membranes were incubated at 4°C overnight with
mouse anti-human KRAS (cat. no. ab157255) or β-actin (cat. no.
ab8226) monoclonal antibody (both 1:1,000 dilution; Abcam,
Cambridge, MA, USA), followed by incubation with horseradish
peroxidase conjugated secondary antibody (1:3,000 dilution; cat.
no. ab6789; Abcam) at room temperature for 2 h. Bands were then
visualized with an enhanced chemiluminescence detection system
(Pierce; Thermo Fisher Scientific, Inc.). β-actin was used as an
internal control.
Dual-luciferase reporter assay
A dual-luciferase reporter assay was performed to
explore whether KRAS was a direct target of miR-217. Cells were
transfected with miR-217 mimics or NC, and co-transfected with
PGL3-KRAS-3′UTR wild type (Wt) or PGL3-KRAS-3′UTR mutant (Mut)
using Lipofectamine® 2000. Following incubation at 37°C
for 48 h, a Dual-Luciferase Reporter assay (Promega Corporation,
Madison, WI) was performed to detect Firefly and Renilla
luciferase activity according to the manufacturer's protocol.
Renilla luciferase activity was measured as an internal
control. Each experiment was repeated at least 3 times.
Statistical analysis
Data are presented as the mean ± standard deviation
and compared using the Student's t-test or one-way analysis of
variance with SPSS software (version 13.0; SPSS, Inc., Chicago, IL,
USA). Double-tailed P<0.05 was considered to indicate a
statistically significant difference.
Results
miR-217 was downregulated in AML
miR-217 expression was measured in patients with AML
and healthy controls using qPCR. As shown in Fig. 1, miR-217 was significantly lower in
patients with AML compared with healthy controls (P=0.001). The
results indicate that miR-217 may perform an important role in
AML.
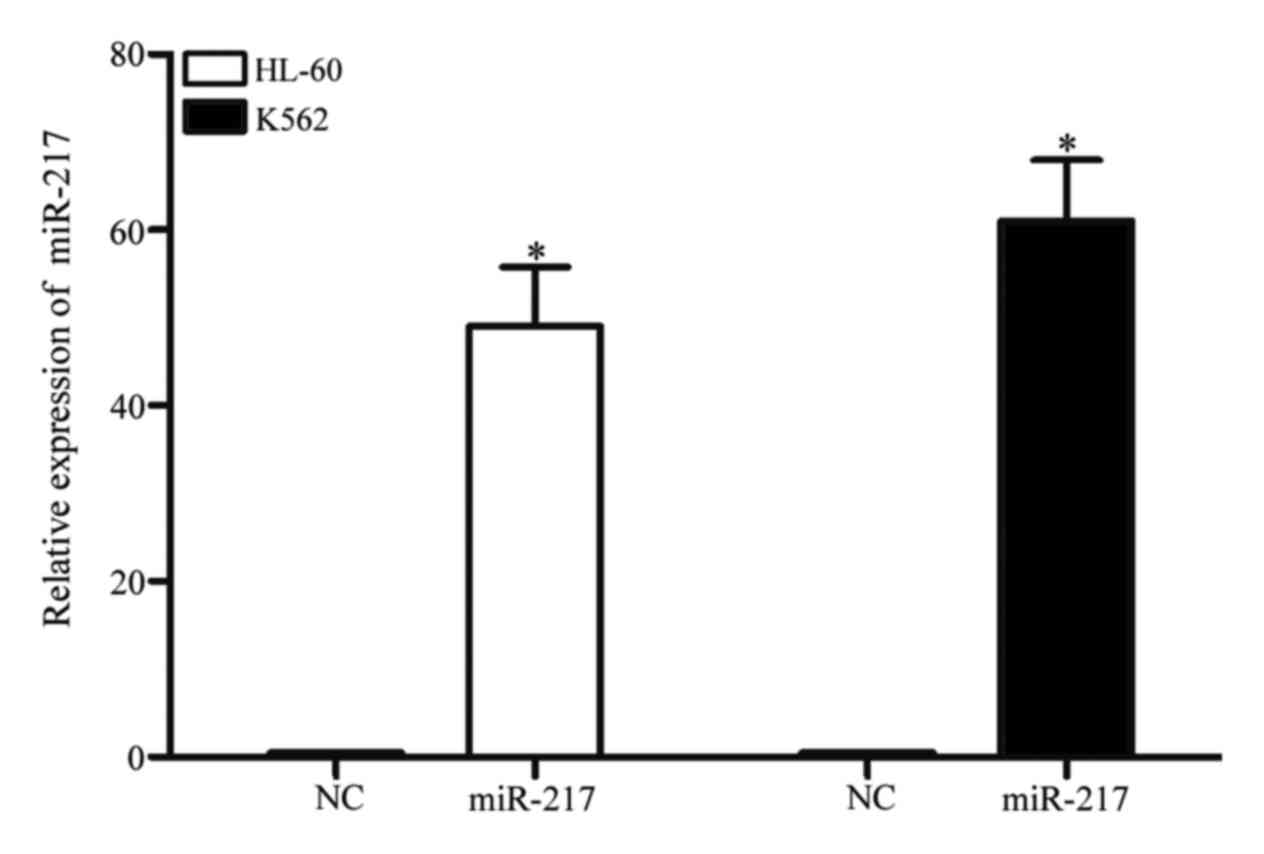
miR-217 was markedly upregulated in
HL-60 and K562 cells following transfection with miR-217
mimics
To investigate the functions of miR-217 on AML
cells, miR-217 mimics or NC was transfected into HL-60 and K562
cells. qPCR was performed to evaluate transfection efficiency. As
demonstrated in Fig. 2, miR-217 was
markedly upregulated in HL-60 and K562 (both P<0.001) cells
transfected with miR-217 mimics, in comparison with cells
transfected with NC.
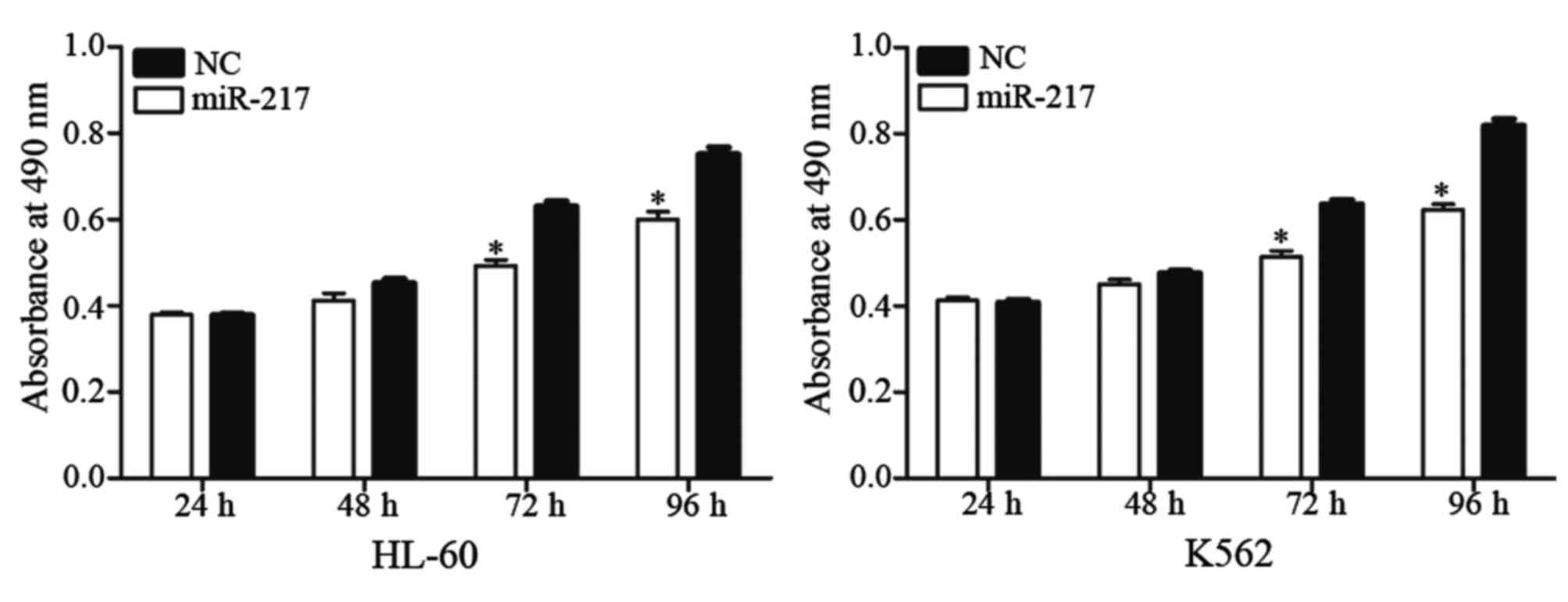
miR-217 decreased cell proliferation
in HL-60 and K562 cells
An MTT assay was performed to explore the effect of
miR-217 on cell proliferation. As shown in Fig. 3, enforced miR-217 expression resulted
in growth inhibition relative to NC in HL-60 (P=0.023) and K562
(P=0.015) cell lines. These results indicate that miR-217 was a
negative regulator of AML cell proliferation.
miR-217 enhanced cell chemosensitivity
to DOX in HL-60 and K562 cells
It has previously been demonstrated that miRs
perform important roles in the regulation of chemoresistance.
Therefore, the present study investigated the effect of miR-217 on
cell chemoresistance in AML using a chemosensitivity assay.
Following transfection with miR-217 mimics or NC, cells were
treated with DOX at various concentrations (32–1048576 ng/ml) for
48 h. As shown in Fig. 4, miR-217
enhanced cell chemosensitivity of HL-60 (P=0.012) and K562
(P=0.018) cells to DOX compared to cells transfected with NC.
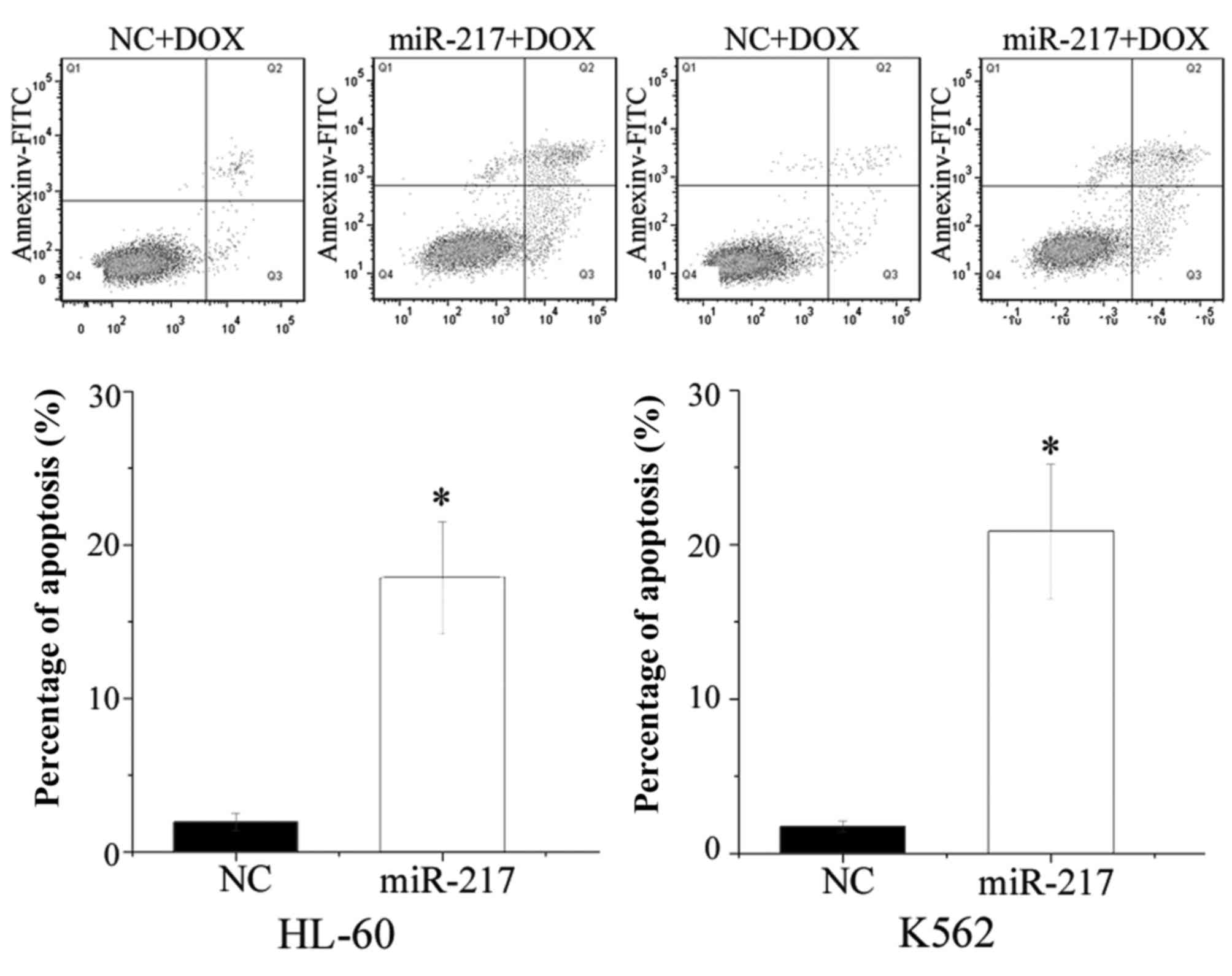
miR-217 enhanced cell apoptosis
induced by DOX in HL-60 and K562 cells
The present study performed flow cytometry to
evaluate the influence of miR-217 on cell apoptosis induced by DOX.
As shown in Fig. 5, ectopic
expression of miR-217 enhanced cell apoptosis induced by DOX in
HL-60 (P=0.010) and K562 (P=0.005) cells. The present findings are
consistent with the change of drug sensitivity, and demonstrate
that miR-217-increased cell chemosensitivity was possibly mediated
by the cell apoptosis pathway.
KRAS was a direct target gene of
miR-217 in vitro
To identify the target of miR-217, bioinformatic
algorithms (TargetScan, Whitehead Institute for Biomedical
Research, Cambridge, MA, USA) were performed. KRAS was predicted to
be a target of miR-217. As shown in Fig.
6A, 2 miR-217 putative binding sites were identified covering
the nucleotides 274–280 (site 1) and 4335–4341 (site 2) of KRAS
3′UTR.
In addition, a dual-luciferase reporter assay was
performed to explore whether KRAS was a direct target of miR-217.
As shown in Fig. 6B, miR-217 led to a
significant decrease of pGL3-KRAS-3′UTR site 1 Wt and
pGL3-KRAS-3′UTR site 2 Wt luciferase activity in HL-60 (site 1,
P=0.020; site 2, P=0.032) and K562 (site 1, P=0.035; site 2,
P=0.014) cells compared with NC cells. Mutation of the two miR-217
binding sites (site 1 Mut or site 2 Mut) restored normal luciferase
activity of KRAS-3′UTR in HL-60 and K562 cells. These results
indicate that KRAS was a direct target gene of miR-217 in
vitro.
miR-217 negatively regulated KRAS
protein expression at the post-transcriptional level
To determine the association between miR-217 and
KRAS at the mRNA and protein levels, miR-217 mimics or NC was
transfected into HL-60 and K562 cells. The expression of KRAS at
mRNA level was detected using qPCR. The expression of KRAS at
protein level was monitored using western blot analysis. As shown
in Fig. 7A, KRAS mRNA levels were not
significantly altered during these treatments (P>0.05). However,
western blot analysis revealed that compared to NC, the expression
of KRAS was significantly downregulated in HL-60 (P=0.022) and K562
(P=0.016) cells transfected with miR-217 mimics (Fig. 7B). These results indicated that
miR-217 did not affect KRAS mRNA stability, but decreased KRAS
expression at the post-transcriptional level.
KRAS was involved in miR-217-induced
effects in HL-60 and K562 cells
To determine whether KRAS serves as a critical
mediator of the effects of miR-217 in AML cells, KRAS siRNA and NC
siRNA were transfected into HL-60 and K562 cells. Following
transfection for 72 h, western blot analysis demonstrated that KRAS
was downregulated in HL-60 (P=0.010) and K562 (P=0.015) cells
transfected with KRAS siRNA (Fig.
8A).
In the MTT assay, the KRAS siRNA group significantly
inhibited cell growth (HL-60, P=0.008; K562, P=0.020) compared with
the NC siRNA group (Fig. 8B). In
addition, chemosensitivity assays revealed that the KRAS siRNA
group exhibited markedly enhanced cell chemosensitivity of HL-60
(P=0.024) and K562 (P=0.026) cells to DOX compared with cells
transfected with NC siRNA (Fig. 8C).
Additionally, the apoptosis assay verified that knockdown of KRAS
increased HL-60 (P=0.017) and K562 (P=0.030) cell apoptosis induced
via DOX (Fig. 8D). These data
indicate that effects of KRAS siRNA were similar to those induced
by miR-217 in AML cells, suggesting KRAS as a functional target of
miR-217 in AML.
Discussion
Abnormal expression of miRs has been verified to
contribute to carcinogenesis and the development of various types
of tumor (20). However, the role of
miRs in AML needs additional investigation. The present study
demonstrated that miR-217 was significantly downregulated in AML.
In addition, ectopic expression of miR-217 suppressed cellular
proliferation and enhanced the chemosensitivity of AML cells to DOX
through the cellular apoptosis pathway. To the best of our
knowledge, this is the first study to explore the expression and
functions of miR-217 in AML.
The abnormal expression of miR-217 has been
demonstrated in a number of human malignancies. For example, in
human osteosarcoma, the expression level of miR-217 was decreased
in tumor tissues and cell lines. Decreased miR-217 expression was
closely correlated with large tumor size, positive distant
metastasis, advanced clinical stage and shorter overall survival
(21,22). These findings suggest that in
osteosarcoma, miR-217 may be involved in the initiation and
progression of cancer, and could be investigated as a prognostic
biomarker in the future (23).
miR-217 was also found to be downregulated in colorectal cancer
tissues compared with corresponding noncancerous tissues. The low
expression level of miR-217 was notably correlated with tumor
differentiation and shorter overall survival for patients with
colorectal cancer (24).
Additionally, miR-217 was found to be downregulated in lung
(25), hepatocellular (26) and clear cell renal cell carcinoma
(27) and pancreatic ductal
adenocarcinoma (28). However, in
breast cancer, miR-217 was upregulated in tumor tissues in
comparison with normal breast tissues. High levels of miR-217 were
significantly associated with high histological grade, the triple
negative subtype and advanced tumor stage (29).
These conflicting studies indicate that miR
expression levels vary in different types of tumor, and that is has
tissue specificity. In addition, in gastric cancer, upregulation of
miR-217 significantly suppressed cell growth and invasion through
negative regulation of Glypican-5 (23). miR-217 has been verified as a tumor
suppressor in osteosarcoma via blockade of the WAS protein family
member 3 and Wnt5a (21,22,30). In
colorectal cancer, enforced miR-217 expression targeted
astrocyte-elevated gene-1 to decrease cell growth, colony
formation, migration and invasion by promoting apoptosis and
G0/G1 phase arrest (24). In hepatocellular carcinoma, endogenous
miR-217 expression inhibited cell invasion by directly targeting
E2F transcription factor 3 (26). In
clear cell renal cell carcinoma, ectopic expression of miR-217
suppressed cell proliferation, migration and invasion (27). These studies indicate that miR-217
mainly functions as a tumor suppressor in several types of cancer.
However, miR-217 also functions as an oncogene in human breast
cancer by targeting the Dachshund homolog 1 to enhance cell
proliferation (29). These ambivalent
results suggest that the functions of miR-217 in cancers are
tissue-type dependent. In addition, miR-217 enhanced the
sensitivity of lung cancer cells to cisplatin (25). The present study revealed that miR-217
decreased cell proliferation and enhanced chemosensitivity of AML
cells to DOX via the cell apoptosis pathway. The present results
therefore provide support for the use of DOX in combination with
miR-217 as a treatment therapy for patients with AML.
In addition, an important molecular link between
miR-217 and KRAS was demonstrated in the present study. miR-217
targeted KRAS to function as a tumor suppressor in AML. KRAS mainly
functions as a critical on-off switch in signaling networks that
transfer extracellular signals to the nucleus, and connect multiple
upstream signals to various downstream signaling pathways (31). These signaling pathways contribute to
cellular differentiation, proliferation, survival, cell cycle,
apoptosis, migration and invasion (32,33).
Therefore, it is important to pay close attention to KRAS as a
potential targeted therapy for inhibition in cancer.
DOX, a cytotoxic antiproliferative drug, is widely
used as cytotoxic agent for chemotherapy in a wide range of
cancers, including AML (34,35). It operates by intercalating into the
DNA and preventing DNA from being resealed; this stops replication
and eventually damages the DNA structure, resulting in the arrest
of the cell growth cycle and the cellular apoptosis pathway
(36). However, the use of DOX in
clinical settings is limited due to the risk of cardiotoxicity and
the ability of the cancer cells to develop resistance to the drug
(35). Additionally, drug resistance
is the major reason for treatment failure. This indicates that
intrinsic chemoresistance may have an important role in cancer. The
present study verified for the first time that upregulation of
miR-217 induced chemosensitivity to DOX through the cellular
apoptosis pathway in vitro. Above all, the present results
have the potential to lead to the development of novel strategies
in treating AML.
In summary, the present study was the first to show
that miR-217 was downregulated in AML and contributed to the
cellular proliferation. In addition, it was demonstrated that
miR-217 enhanced cell chemosensitivity of AML cells to DOX through
the cell apoptosis pathway. In the present study, KRAS was
identified as a direct target of miR-217. The identification of
candidate target genes of miR-217 may aid understanding of the
potential mechanisms involved in AML.
Additional studies are required to address whether
the potential of miR-217 may be fully realized in AML treatment. If
so, miR-217 may be beneficial for treatment of AML.
Acknowledgements
The present study was supported by the Natural
Science Foundation of Hubei (grant no. 2012FFB02435) and the
central university special funding (Huazhong University of Science
and Technology; grant no. 2013QN191).
References
|
1
|
Das RP, Konkimalla VB, Rath SN, Hansa J
and Jagdeb M: Elucidation of the molecular interaction between
miRNAs and the HOXA9 gene, involved in acute myeloid leukemia, by
the assistance of argonaute protein through a computational
approach. Genomics Inform. 13:45–52. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Estey EH: Acute myeloid leukemia: 2013
update on risk-stratification and management. Am J Hematol.
88:318–327. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Ferrara F and Schiffer CA: Acute myeloid
leukaemia in adults. Lancet. 381:484–495. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Siegel RL, Miller KD and Jemal A: Cancer
statistics, 2015. CA Cancer J Clin. 65:5–29. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Gregory TK, Wald D, Chen Y, Vermaat JM,
Xiong Y and Tse W: Molecular prognostic markers for adult acute
myeloid leukemia with normal cytogenetics. J Hematol Oncol.
2:232009. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Burnett A, Wetzler M and Löwenberg B:
Therapeutic advances in acute myeloid leukemia. J Clin Oncol.
29:487–494. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Stanisic S and Kalaycio M: Treatment of
refractory and relapsed acute myelogenous leukemia. Expert Rev
Anticancer Ther. 2:287–295. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
He L and Hannon GJ: MicroRNAs: Small RNAs
with a big role in gene regulation. Nat Rev Genet. 5:522–531. 2004.
View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Ameres SL and Zamore PD: Diversifying
microRNA sequence and function. Nat Rev Mol Cell Biol. 14:475–488.
2013. View
Article : Google Scholar : PubMed/NCBI
|
|
10
|
Berindan-Neagoe I, Pdel C Monroig,
Pasculli B and Calin GA: MicroRNAome genome: A treasure for cancer
diagnosis and therapy. CA Cancer J Clin. 64:311–336. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Ueda T, Volinia S, Okumura H, Shimizu M,
Taccioli C, Rossi S, Alder H, Liu CG, Oue N, Yasui W, et al:
Relation between microRNA expression and progression and prognosis
of gastric cancer: A microRNA expression analysis. Lancet Oncol.
11:136–146. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Braun J and Hüttelmaier S: Pathogenic
mechanisms of deregulated microRNA expression in thyroid carcinomas
of follicular origin. Thyroid Res. 4 Suppl 1:S12011. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Zhang B, Pan X, Cobb GP and Anderson TA:
microRNAs as oncogenes and tumor suppressors. Dev Biol. 302:1–12.
2007. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Esquela-Kerscher A and Slack FJ:
Oncomirs-microRNAs with a role in cancer. Nat Rev Cancer.
6:259–269. 2006. View
Article : Google Scholar : PubMed/NCBI
|
|
15
|
Bhardwaj A, Singh S and Singh AP:
MicroRNA-based cancer therapeutics: Big hope from small RNAs. Mol
Cell Pharmacol. 2:213–219. 2010.PubMed/NCBI
|
|
16
|
Ryan BM, Robles AI and Harris CC: Genetic
variation in microRNA networks: The implications for cancer
research. Nat Rev Cancer. 10:389–402. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Lu F, Zhang J, Ji M, Li P, Du Y, Wang H,
Zang S, Ma D, Sun X and Ji C: miR-181b increases drug sensitivity
in acute myeloid leukemia via targeting HMGB1 and Mcl-1. Int J
Oncol. 45:383–392. 2014.PubMed/NCBI
|
|
18
|
Nagano H, Tomimaru Y, Eguchi H, Hama N,
Wada H, Kawamoto K, Kobayashi S, Mori M and Doki Y: MicroRNA-29a
induces resistance to gemcitabine through the Wnt/β-catenin
signaling pathway in pancreatic cancer cells. Int J Oncol.
43:1066–1072. 2013.PubMed/NCBI
|
|
19
|
Livak KJ and Schmittgen TD: Analysis of
relative gene expression data using real-time quantitative PCR and
the 2(−Delta Delta C(T)) method. Methods. 25:402–408. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Wu D, Niu X, Pan H, Zhang Z, Zhou Y, Qu P
and Zhou J: MicroRNA-497 targets hepatoma-derived growth factor and
suppresses human prostate cancer cell motility. Mol Med Rep.
13:2287–2292. 2016.PubMed/NCBI
|
|
21
|
Sun B, Yang M, Li M and Wang F: The
microRNA-217 functions as a tumor suppressor and is frequently
downregulated in human osteosarcoma. Biomed Pharmacother. 71:58–63.
2015. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Wei R, Deng Z and Su J: miR-217 targeting
Wnt5a in osteosarcoma functions as a potential tumor suppressor.
Biomed Pharmacother. 72:158–164. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Wang H, Dong X, Gu X, Qin R, Jia H and Gao
J: The MicroRNA-217 functions as a potential tumor suppressor in
gastric cancer by targeting GPC5. PLoS One. 10:e01254742015.
View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Wang B, Shen ZL, Jiang KW, Zhao G, Wang
CY, Yan YC, Yang Y, Zhang JZ, Shen C, Gao ZD, et al: MicroRNA-217
functions as a prognosis predictor and inhibits colorectal cancer
cell proliferation and invasion via an AEG-1 dependent mechanism.
BMC Cancer. 15:4372015. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Guo J, Feng Z, Huang Z, Wang H and Lu W:
MicroRNA-217 functions as a tumour suppressor gene and correlates
with cell resistance to cisplatin in lung cancer. Mol Cells.
37:664–671. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Su J, Wang Q, Liu Y and Zhong M: miR-217
inhibits invasion of hepatocellular carcinoma cells through direct
suppression of E2F3. Mol Cell Biochem. 392:289–296. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Li H, Zhao J, Zhang JW, Huang QY, Huang
JZ, Chi LS, Tang HJ, Liu GQ, Zhu DJ and Ma WM: MicroRNA-217,
down-regulated in clear cell renal cell carcinoma and associated
with lower survival, suppresses cell proliferation and migration.
Neoplasma. 60:511–515. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Vychytilova-Faltejskova P, Kiss I, Klusova
S, Hlavsa J, Prochazka V, Kala Z, Mazanec J, Hausnerova J, Kren L,
Hermanova M, et al: MiR-21, miR-34a, miR-198 and miR-217 as
diagnostic and prognostic biomarkers for chronic pancreatitis and
pancreatic ductal adenocarcinoma. Diagn Pathol. 10:382015.
View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Zhang Q, Yuan Y, Cui J, Xiao T and Jiang
D: MiR-217 promotes tumor proliferation in breast cancer via
targeting DACH1. J Cancer. 6:184–191. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Shen L, Wang P, Yang J and Li X:
MicroRNA-217 regulates WASF3 expression and suppresses tumor growth
and metastasis in osteosarcoma. PLoS One. 9:e1091382014. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Zuber J, Tchernitsa OI, Hinzmann B,
Schmitz AC, Grips M, Hellriegel M, Sers C, Rosenthal A and Schäfer
R: A genome-wide survey of RAS transformation targets. Nat Genet.
24:144–152. 2000. View
Article : Google Scholar : PubMed/NCBI
|
|
32
|
Crespo P and León J: Ras proteins in the
control of the cell cycle and cell differentiation. Cell Mol Life
Sci. 57:1613–1636. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Wu Y, Zhuang Y, Han M, Xu T and Deng K:
Ras promotes cell survival by antagonizing both JNK and Hid signals
in the Drosophila eye. BMC Dev Biol. 9:532009. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Lagadinou ED, Ziros PG, Tsopra OA, Dimas
K, Kokkinou D, Thanopoulou E, Karakantza M, Pantazis P,
Spyridonidis A and Zoumbos NC: c-Jun N-terminal kinase activation
failure is a new mechanism of anthracycline resistance in acute
myeloid leukemia. Leukemia. 22:1899–1908. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Kweon SH, Song JH and Kim TS:
Resveratrol-mediated reversal of doxorubicin resistance in acute
myeloid leukemia cells via downregulation of MRP1 expression.
Biochem Biophys Res Commun. 395:104–110. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Tao J, Lu Q, Wu D, Li P, Xu B, Qing W,
Wang M, Zhang Z and Zhang W: ΜicroRNA-21 modulates cell
proliferation and sensitivity to doxorubicin in bladder cancer
cells. Oncol Rep. 25:1721–1729. 2011.PubMed/NCBI
|