Introduction
Hepatocellular carcinoma (HCC) is one of the most
common types of cancer and the second leading cause of
malignant-neoplasm-associated mortality, worldwide (1). However, the molecular targeted therapy,
sorafenib, has exhibited promising results in the treatment of
patients with advanced HCC (2).
Sorafenib is a small molecule that functions as an oral multikinase
inhibitor and is also approved for the treatment of advanced renal
cell carcinoma (3). Sorafenib
exhibits direct antiproliferative effects on tumor cells due to the
blockade of numerous intracellular serine/threonine kinases (e.g.,
C-Raf and B-Raf), and indirect effects due to the blockade of
receptor tyrosine kinases, including vascular endothelial growth
factor receptors (VEGFRs) and platelet-derived growth factor
receptor-β (PDGFR-β) on endothelial cells followed by the
inhibition of angiogenesis (1,4,5). Des-γ-carboxyprothrombin (DCP), also
known as protein induced by vitamin K absence or antagonist-II
(PIVKA-II), is an abnormal prothrombin routinely used as a tumor
marker for HCC, and may be predictive of aggressive tumor behavior
and a poor prognosis (6–8). Serum DCP levels also increase in
patients with a vitamin K deficiency, including those taking
warfarin or who exhibit obstructive jaundice. The DCP molecules in
these conditions were demonstrated to contain more
γ-carboxyglutamic (Gla) residues and was termed NX-DCP. A number of
previous studies reported that serum NX-DCP was also useful for the
detection of HCC (6,9). We previously revealed that patients with
serum NX-DCP-positive HCC exhibited significantly larger tumors,
more frequent portal vein invasion and a poorer prognosis (10). The evaluation of α-fetoprotein (AFP)
level is already used for routine surveillance and noninvasive
diagnosis of HCC, and the prediction of prognosis and monitoring of
recurrence subsequent to treatment (11,12). As
the DCP level is not always correlated with the AFP level, the
recommendations in previous studies include combined testing of DCP
and Lens culinaris agglutinin-reactive fraction of AFP has
been established (13,14).
Sorafenib is widely used for patients with advanced
HCC. However, at present, there are no clinical parameters to
predict sorafenib efficacy. DCP levels were reported to be
increased in patients treated with sorafenib (15–17), and
the elevation of DCP may indicate a high therapeutic effect of
sorafenib (16,17). Although the precise mechanism remains
unknown, the ischemic and/or hypoxic conditions of HCC may be
associated with the elevation of DCP subsequent to sorafenib
treatment. In the present study, the mechanism of DCP level
elevation in hypoxic conditions and sorafenib treatment was
investigated using the HCC KYN-2 cell line, which produces DCP.
Materials and methods
Cell lines and cell culture
The present study used 11 HCC cell lines KIM-1,
KYN-1, KYN-2, KYN-3, HAK-1A, HAK-1B, HAK-2, HAK-3, HAK-4, HAK-5 and
HAK-6, and 2 combined hepatocellular-cholangiocarcinoma (CHC) cell
lines, KMCH-1 and KMCH-2. These cell lines were originally
established in the Department of Pathology (Kurume University
School of Medicine, Kurume, Japan), and each cell line retains the
morphological and functional features of the original tumor as
described elsewhere (18–26). The cells were grown in Dulbecco's
modified Eagle's medium (Nissui, Tokyo, Japan) supplemented with
2.5% heat-inactivated (56°C, 30 min) fetal bovine serum (FBS;
Bioserum, Victoria, Australia), 100 U/ml penicillin, 100 µg/ml
streptomycin (Gibco; Thermo Fisher Scientific, Inc., Waltham, MA,
USA) and 12 mmol/l sodium bicarbonate, in a humidified atmosphere
of 5% CO2 at 37°C.
Measurement of DCP, NX-DCP and
prothrombin levels
The cultured cells were seeded on 6-well plates
(Falcon, BD Biosciences Labware, Tokyo, Japan) at a density of
3–10×104 cells/well, and cells on the plates were
cultured in normoxic or hypoxic, 1% O2, conditions for 3
days. The prothrombin, DCP and NX-DCP levels in the supernatant
(centrifuged for 10 min, 12,000 × g, 4°C) and cell lysate were
determined using an ELISA kit (EIDIA Co., Ltd., Tokyo, Japan) for
prothrombin and an electro-chemiluminescence immunoassay (ECLIA)
kit (EIDIA Co., Ltd.) for DCP and NX-DCP, according to the protocol
of the manufacturer. The polyclonal antibody for prothrombin was
sourced from the ELISA kit, the monoclonal antibody for DCP (MU-3)
and monoclonal antibodies for NX-DCP (P-11 and P-16) were sourced
from the ECLIA kit.
Effects of vitamin K on the secretion
of DCP, NX-DCP and prothrombin
KYN-2 cells were seeded on 10 cm dishes (Falcon, BD
Biosciences Labware) at a density of 1.3×105 cells/well.
The media were replaced the subsequent day with medium alone or
medium containing 100 nM vitamin K (Sigma-Aldrich; Merck Millipore,
Darmstadt, Germany) and the cells were cultured in normoxic or
hypoxic condition for 72 h. The cell number was then determined and
the supernatant was collected (centrifuged for 10 min, 12,000 × g,
4°C) and used for DCP, NX-DCP and prothrombin measurements by ECLIA
and ELISA.
Effect of sorafenib on proliferation
and secretion of DCP, NX-DCP, prothrombin and VEGF
A total of 5×104 cells per well KYN-2
cells were seeded on 6-well plates. The medium was replaced the
next day with medium alone or medium containing 1.25 µM sorafenib
(Cell Signaling Technology, Danvers, MA) and the cells were
cultured in normoxic or hypoxic condition for 24, 48, or 72 h. Then
the cell number was determined and the supernatant was collected
and used for DCP, NX-DCP, prothrombin and vascular endothelial
growth factor (VEGF) measurements by ECLIA and ELISA. The
supernatant was also obtained from cells cultured with sorafenib
(0.313, 0.625 or 1.25 µM) for 72 h and used for the evaluation
performed by ECLIA and ELISA.
Effects of sorafenib on tumor
proliferation and serum DCP and NX-DCP levels in nude mice
A total of 1×107 cells/100 µl cultured
KYN-2 cells were subcutaneously injected into the backs of
4-week-old female BALB/c nude mice. Following two weeks, the mice
were divided into 3 groups of 12 in order to equalize the mean
diameter of tumors in each group. Each group was assigned to 1 of
the 3 treatment groups: Control; 300 µg/mouse/day sorafenib; 600
µg/mouse/day sorafenib. The 300 µg dose of sorafenib in proportion
to the average body weight of a mouse, 20 g, was 15 mg/kg, which is
comparable to a clinical dose of 800 mg total/day. The sorafenib
was diluted with 12.5% Cremophor EL (Sigma-Aldrich; Merck
Millipore)/12.5% ethanol/75% water for oral dosing in mice, and was
administered by tube feeding once a day. In the control group, 0.2
ml Cremophor EL/ethanol/water (12.5/12.5/75) alone was administered
by tube feeding once a day. Tumor size was measured in two
directions using calipers, and tumor volume (mm3) was
estimated by using the equation: Length × (width)2 ×
0.5. This measurement was performed every 2 or 3 days. On day 4, 9
or 14, subsequent to drawing blood for measuring the serum DCP and
NX-DCP level, 4 mice in each group were sacrificed by cervical
dislocation and the tumors were resected. The tumor weight was
measured.
Immunohistochemical analysis
The resected tumors were fixed in formalin and
prepared into paraffin sections, and underwent hematoxylin and
eosin staining and immunochemistry. Immunohistochemical staining
with monoclonal rat anti-mouse cluster of differentiation (CD)34
(cat. no. ab8185; dilution, 1:50; Abcam, Cambridge, UK) was
performed using the standard avidin-biotin-peroxidase complex
method and 3,3′-diaminobenzidine solution was used for color
development. The microvessel density (MVD) was evaluated within the
tumor. To quantify MVD, the slides stained with CD34 were observed
at low power field, magnification, ×10-100, using a light
microscope. A total of 5 areas with high MVD were selected at a
high-power field, magnification, ×200, and the MVD of these areas
in each specimen was measured using the WinROOF software package
(version 6.1; Mitani Corporation, Fukui, Japan).
Statistical analysis
Data are expressed as the mean ± standard deviation.
Comparisons between groups were performed using un-paired Student's
t-test and two-factor factorial analysis of variance. P<0.05 was
considered to indicate a statistically significant difference.
Ethical statement
The present study was approved by the Ethics
Committee of Kurume University (approval no. 10007). Animal
experiments for this study were approved by the Ethics Review
Committee for Animal Experimentation of Kurume University School of
Medicine (Kurume, Japan).
Results
Measurement of DCP, NX-DCP and
prothrombin
The levels of DCP secretion in the supernatant of
KYN-2 cells under normoxic conditions per µg protein was 11.2
mAU/ml/µg protein, and the expression in the cell lysate was 165.4
mAU/ml/µg protein. Under hypoxic conditions, the DCP expression
levels were 66.7 mAU/ml/µg protein in the supernatant, and 80.9
mAU/ml/µg protein in the cell lysate. In the KIM-1 cells, the level
of DCP secretion in the supernatant cultured under normoxic
conditions was 0.8 mAU/ml/µg protein, and the expression in the
cell lysate was 126.7 mAU/ml/µg protein. In hypoxic conditions, the
levels of DCP expression were 19.1 mAU/ml/µg protein in the
supernatant and 872.4 mAU/ml/µg protein in the cell lysate. The
levels of NX-DCP secretion in the supernatant of the KYN-2 cells
was 0.3 mAU/ml/µg protein under normoxic conditions, and 0.9
mAU/ml/µg protein under hypoxic condition. In the KIM-1 cells, the
levels of NX-DCP secretion in the supernatant were 0.2 mAU/ml/µg
protein under normoxic conditions, and 0.7 mAU/ml/µg protein under
hypoxic condition. The levels of prothrombin secretion under
normoxic conditions were 0.6 mAU/ml/µg protein in KYN-2 cells, and
0.6 mAU/ml/µg protein in the KIM-1 cells. Under hypoxic conditions,
the levels of prothrombin expression were 0.9 mAU/ml/µg protein and
0.9 mAU/ml/µg protein for KYN-2 and KIM-1, respectively. The
expression levels of DCP, NX-DCP and prothrombin in all of the
other 11 cell lines were less than 0.1 mAU/ml/µg protein (data not
shown).
Effects of vitamin K on secretion of
DCP, NX-DCP and prothrombin
The addition of vitamin K to the culture medium
suppressed cell proliferation in the KYN-2 cells under normoxic and
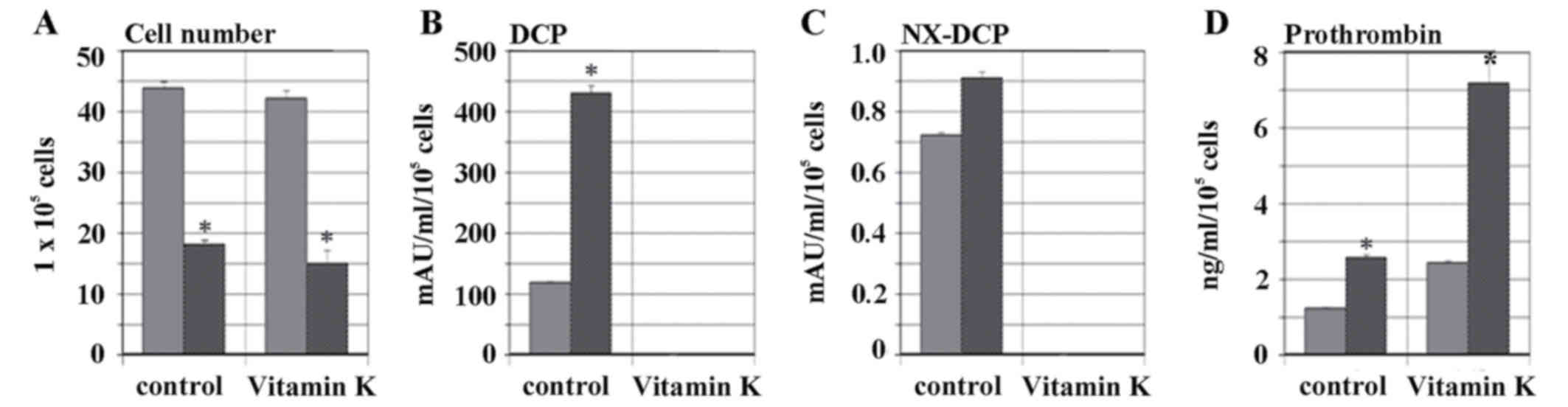
hypoxic conditions, as demonstrated in Fig. 1A. The level of secretion of DCP under
normoxic condition without the addition of vitamin K was 118.0±2.2
mAU/ml/105 cells, whereas levels of DCP increased
significantly to 428.2±14.2 mAU/ml/105 cells under
hypoxic condition (P<0.001). However, when vitamin K was added
to the cell cultures, the levels DCP secretion decreased below 0.5
mAU/ml/105 cells in normoxic and hypoxic condition, as
illustrated in Fig. 1B. NX-DCP
expression in normoxic and hypoxic conditions was <1.0
mAU/ml/105 cells. NX-DCP expression was not detected
with the addition of vitamin K, as demonstrated in Fig. 1C. The levels of prothrombin secretion
were 1.2±0.1 mAU/ml/105 cells under normoxic conditions
without vitamin K, and increased to 2.6±0.1 mAU/ml/105
cells under hypoxic conditions (P<0.001). Subsequent to the
addition of vitamin K, the prothrombin levels were 2.4±0.1
mAU/ml/105 cells under normoxic conditions, but
increased to 7.1±0.7 mAU/ml/105 cells in hypoxic
conditions (P<0.001), as illustrated in Fig. 1D.
Effect of sorafenib on proliferation
and secretion of DCP, NX-DCP, prothrombin and VEGF
The cells cultured with 1.25 µM sorafenib decreased
in number to 25% of the control group on day 3 (P<0.001), as
demonstrated in Fig. 2A. Sorafenib
inhibited the levels of DCP secretion by the HCC cells: The levels
of DCP in the culture medium of the control cells at 24, 48 and 72
h were 0.21±0.04, 8.4±0.72 and 48.2±14.9 mAU/ml/104
cells, respectively, and in cells cultured with 1.25 µM sorafenib
the levels of DCP secretion at 24, 48 and 72 h were 0.16±0.03,
0.29±0.10 and 0.66±0.01 mAU/ml/104 cells, respectively
(P<0.001), as illustrated in Fig.
2B. Levels of NX-DCP expression demonstrated a similar trend to
DCP. The expression of NX-DCP was generally low, as the highest
level was 0.35±0.09 mAU/ml/104 cells at 72 h in the
non-sorafenib cell culture, as demonstrated in Fig. 2C. Prothrombin and VEGF secretion at
24, 48, and 72 h were all elevated in the sorafenib-treated cell
cultures. These expression levels peaked at 48 h and increased
>2-fold compared with the non-treated cells (P<0.001), as
illustrated in Fig. 2D and E.
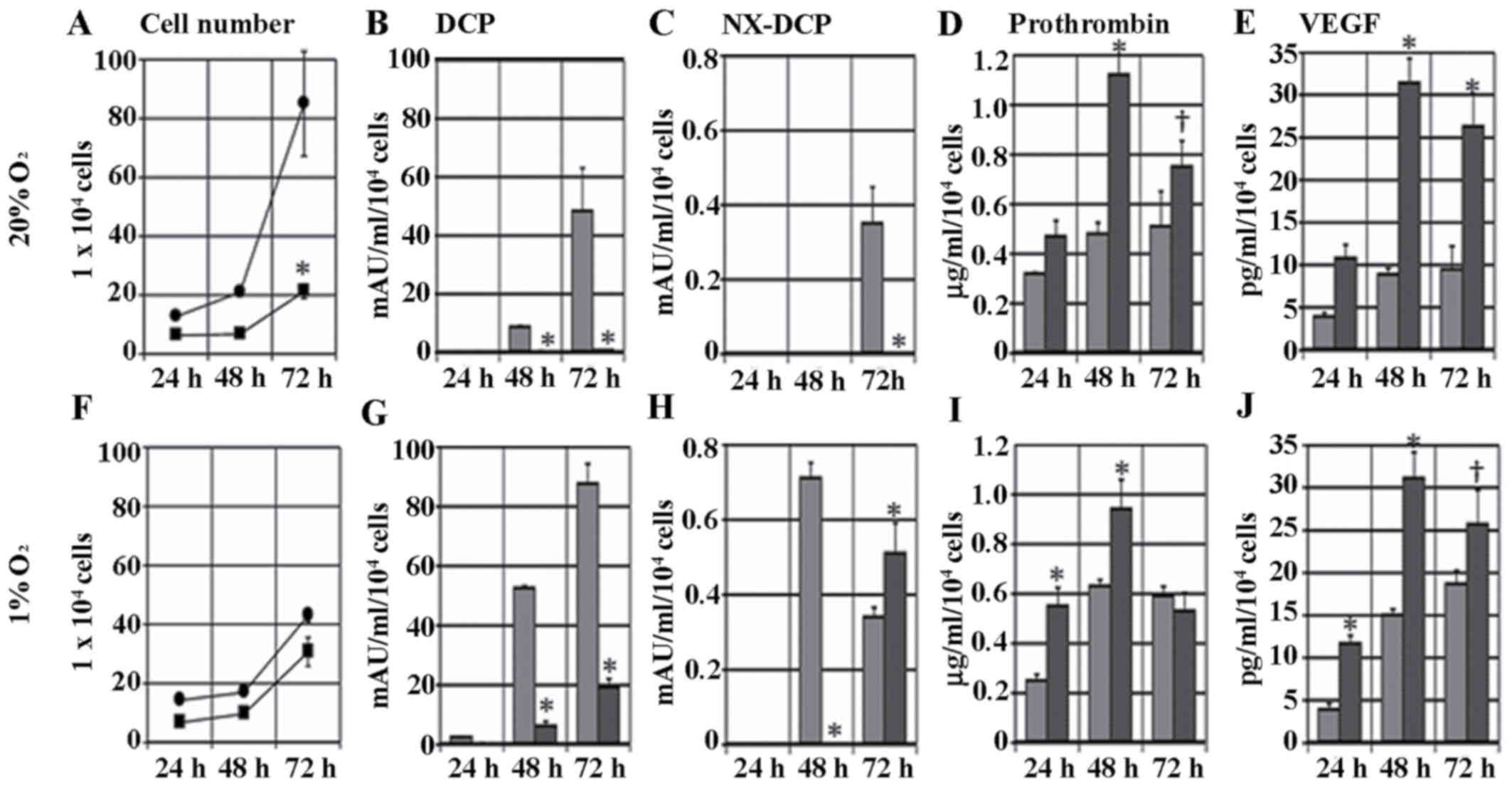 | Figure 2.Effect of sorafenib on the
proliferation and secretion of DCP, prothrombin, NX-DCP and VEGF.
Upper row (A-E), normoxic conditions; lower row (F-J), hypoxic
conditions. (A and F) The number of KYN-2 cells cultured with or
without 1.25 µM sorafenib for 24, 48, or 72 h. The levels of (B and
G) DCP; (C and H) NX-DCP; (D and I) prothrombin; and (E and J) VEGF
secreted by KYN-2 cells cultured with medium alone or medium with
1.25 µM sorafenib for 24, 48, or 72 h. Data are present as the mean
± standard deviation from three independent experiments.
†P<0.01 and *P<0.001 medium alone vs. medium with
sorafenib. DCP, des-γ-carboxyprothrombin; NX-DCP,
des-γ-carboxyprothrombin with more γ-carboxyglutamic residues;
VEGF, vascular endothelial growth factor; ■/black bar, cells
cultured with 1.25 µM sorafenib; •/gray bar, cells cultured without
1.25 µM sorafenib. |
The same experiments were performed under hypoxic
conditions. The levels of cell proliferation decreased subsequent
to sorafenib treatment, but the difference was less compared with
normoxic conditions, as demonstrated in Fig. 2F. DCP expression was higher in the
sorafenib-treated and non-treated cells compared with the cells
under normoxic conditions. The level of DCP in the
sorafenib-treated cells was significantly lower compared with the
control group and the cells under normoxic conditions (P<0.001),
as illustrated in Fig. 2G. NX-DCP
expression in sorafenib-treated cells was higher compared with the
control at 72 h, as demonstrated in Fig.
2H. The levels of secretion of prothrombin and VEGF under
hypoxic conditions were almost the same as under normoxic
conditions, as illustrated in Fig. 2I and
J. VEGF secretion increased 1.5- to 2-fold compared with
non-treated cells under normoxic conditions.
Subsequent to culturing the cells for 72 h with the
addition of sorafenib at a range of concentrations, or without
sorafenib, cell proliferation was more significantly suppressed
under hypoxic conditions compared with under normoxic conditions.
Under these two conditions, sorafenib treatment significantly
suppressed cell proliferation (P<0.001), as demonstrated in
Fig. 3A. The level of DCP secretion
under normoxic conditions decreased subsequent to sorafenib
treatment in a dose-dependent manner, but this effect was enhanced
in hypoxic conditions (P<0.001), as illustrated in Fig. 3B. NX-DCP expression was low compared
with DCP, and tended to decrease subsequent to sorafenib treatment
under normoxic conditions. However, NX-DCP levels exhibited an
increase under hypoxic conditions (P<0.001), as demonstrated in
Fig. 3C. The secretion of prothrombin
and VEGF was also significantly higher under hypoxic conditions in
cells treated with high concentrations of sorafenib compared with
normoxic conditions (P<0.001), as illustrated in Fig. 3D and E.
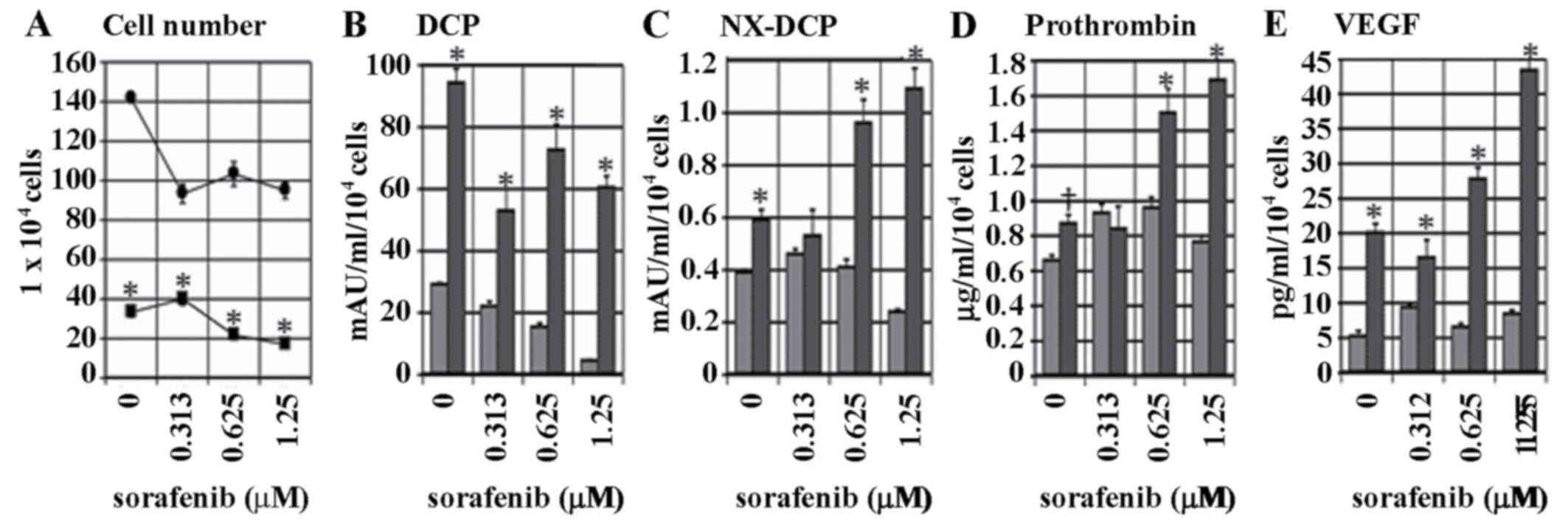 | Figure 3.Dose-dependent effect of 0.313, 0.625
or 1.25 µM sorafenib on the levels of proliferation and secretion
of DCP, prothrombin, NX-DCP and VEGF. (A) KYN-2 cells were cultured
with medium alone or medium with 0.313, 0.625, or 1.25 µM sorafenib
in normoxic or hypoxic conditions for 72 h. The (A) cell number and
levels of (B) DCP, (C) NX-DCP, (D) prothrombin and (E) VEGF
secreted by KYN-2 cells cultured with or without sorafenib in
normoxic or hypoxia conditions for 72 h. †P<0.01 and
*P<0.001 vs. normoxic condition. •/gray bar, normoxic
conditions; ■/black bar, hypoxic conditions; DCP,
des-γ-carboxyprothrombin; NX-DCP, des-γ-carboxyprothrombin with
more γ-carboxyglutamic residues; VEGF, vascular endothelial growth
factor. |
Effects of sorafenib on tumor
proliferation and serum DCP and NX-DCP levels in nude mice
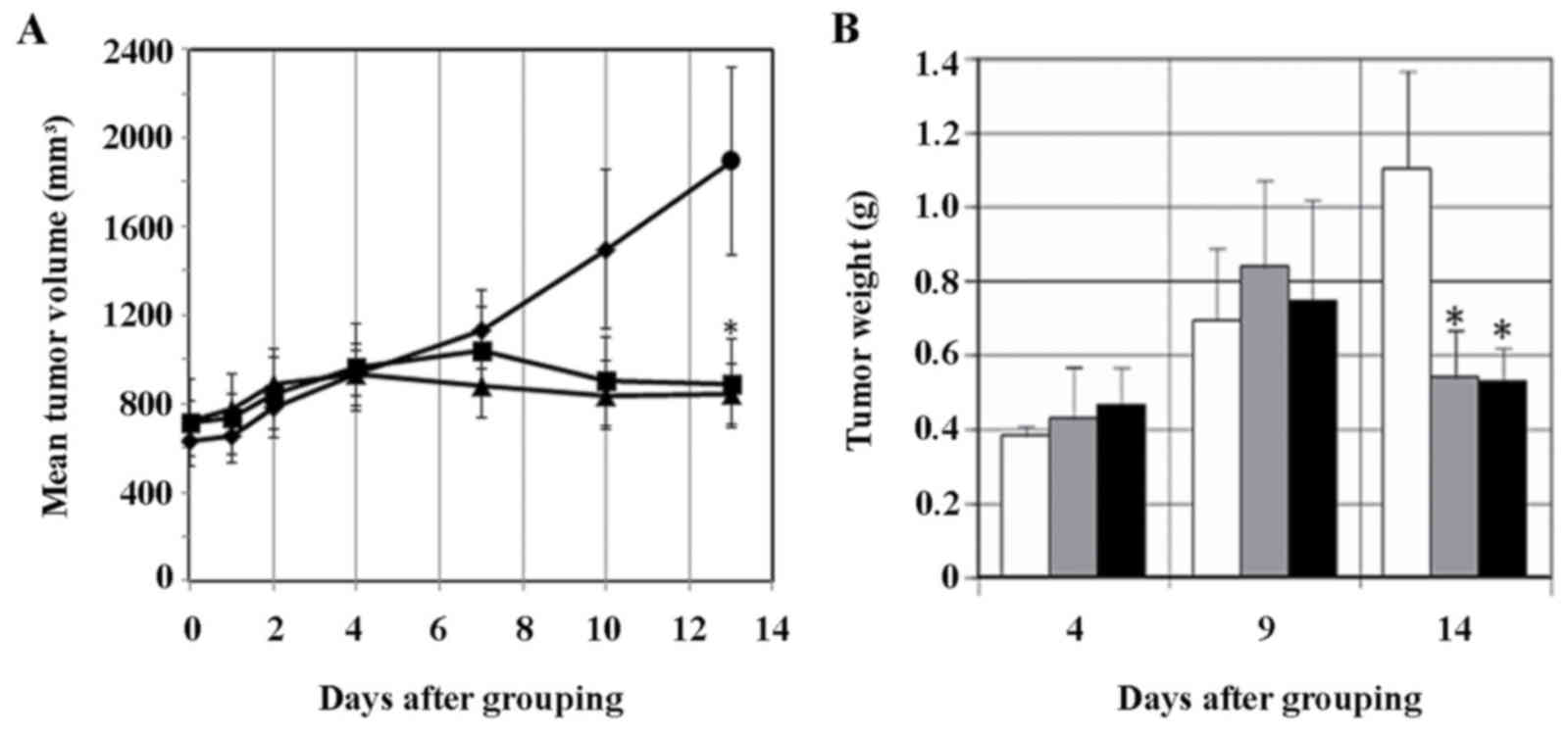
The growth of the tumor (tumor volume) was
suppressed in sorafenib-treated mice from day 7 compared with the
control, and this difference was significant at day 13 (P<0.05),
as demonstrated in Fig. 4A. Tumor
weight also decreased significantly at day 14 in sorafenib-treated
mice compared with the control (P<0.05), as illustrated in
Fig. 4B. The tumor volume and weight
decreased ~50% of the control level subsequent to sorafenib
treatment, however no difference was observed between the two dose
levels, 300 µg/mouse/day or 600 µg/mouse/day, used in the present
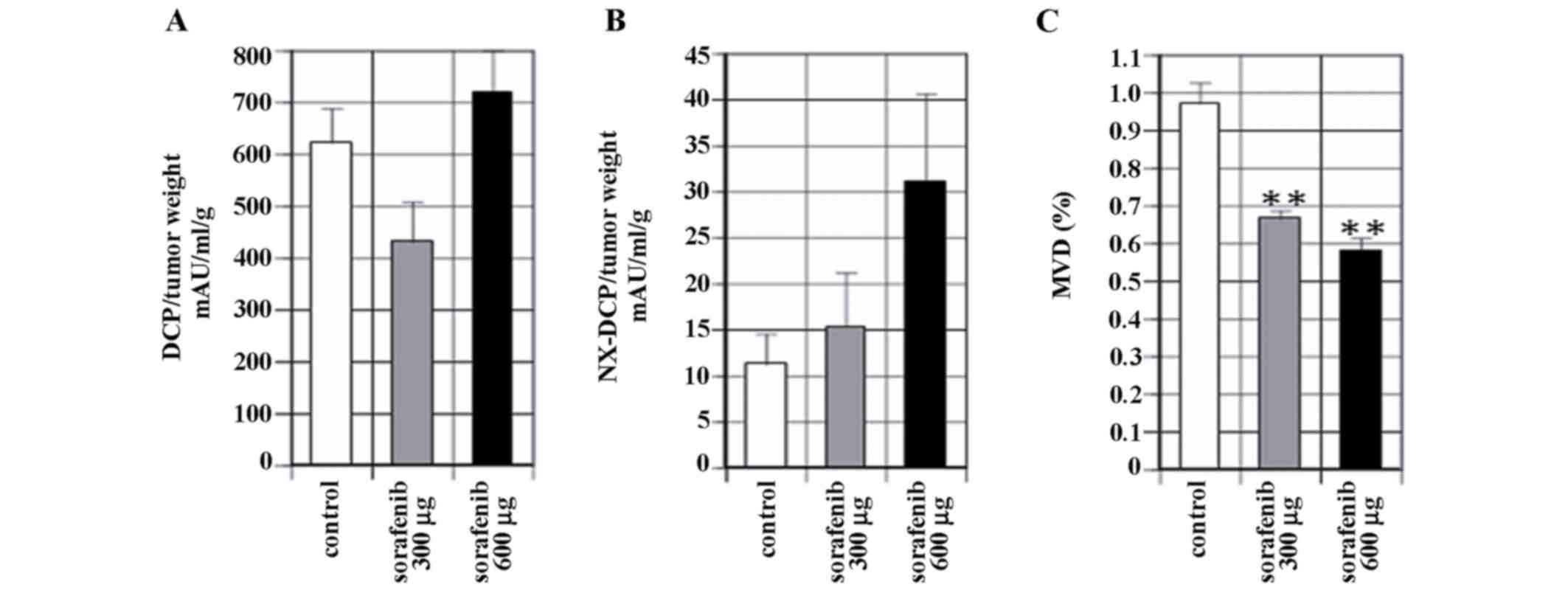
study. The levels of DCP expression by tumor weight was 622.2±66.4
mAU/ml/g in the control, 430.7±77.7 mAU/ml/g in the sorafenib 300
µg/mouse/day and 763.1±78.4 mAU/ml/g in the sorafenib 600
µg/mouse/day group, as illustrated in Fig. 5A. The levels of NX-DCP expression per
unit tumor weight was 11.2±3.3 mAU/ml/g in the control, 15.3±5.8
mAU/ml/g in the sorafenib 300 µg/mouse/day group and 31.1±9.5
mAU/ml/gin the sorafenib 600 µg/mouse/day group, as demonstrated in
Fig. 5B. There were no significant
differences observed between the levels of expression of DCP or
NX-DCP between the sorafenib and control groups.
Immunohistochemical analysis
MVD was significantly reduced to 68.4% of the
control level in the sorafenib 300 µg/mouse/day group, and 59.4% of
the control level in the sorafenib 600 µg/mouse/day group
(P<0.001). No significant difference in other parameters, such
as tumor volume and tumor weight, was observed between the two
concentrations of sorafenib used, as illustrated in Fig. 5C.
Discussion
The 10 glutamic acid (Glu) residues of human
prothrombin at the N-terminus are typically converted by
carboxylase to a Gla domain. DCP is an abnormal prothrombin as all
or part of the Gla domain remains as Glu residues. NX-DCP, which is
induced in conditions of vitamin K deficiency, is a protein with a
smaller number of Glu residues.
In the present study, the levels of DCP, NX-DCP and
prothrombin expression in 13 different cell lines were examined. It
was revealed that the expression levels were low in all cell lines
except KIM-1 and KYN-2. Comparing the expression levels of DCP,
NX-DCP and prothrombin under normoxic or hypoxic conditions, the
levels increased under hypoxic conditions in all categories except
DCP in the KYN-2 cell lysate. NX-DCP and prothrombin were expressed
under hypoxic conditions, but the levels were low compared with
levels of DCP. The production of DCP in HCC is hypothesized to be
associated with a reduction in the activity of vitamin K dependent
γ-carboxylase (27,28), excessive production of prothrombin
precursors (29,30) and to effects associated with vitamin K
concentration. In the present study, cell proliferation and DCP
expression were suppressed under normoxic and hypoxic conditions in
KYN-2 cells treated with vitamin K. This suggests that the addition
of vitamin K restored the metabolic pathways to near normal status
in the KYN-2 cells where the increased expression of prothrombin
lowered the uptake or reduced the activity of vitamin K. In studies
using cell lines, vitamin K treatment was demonstrated to suppress
DCP expression, whilst increasing the expression of prothrombin and
carboxylase mRNA (31,32). These results suggest that DCP
production in HCC may be caused by a reduction in vitamin K
concentration in the microenvironment of the cancer cells.
Conversely, vitamin K derivatives are not reduced in patients with
HCC, and one study reported that vitamin K administration reduced
DCP levels (33). However, whilst
vitamin K administration reduced DCP to normal levels in patients
exhibiting a high serum level of vitamin K derivatives, DCP
remained abnormal in patients exhibiting low serum levels of
vitamin K derivatives. These data indicate that high levels of DCP
are not caused by lowered levels of vitamin K, but by a reduced
utilization efficiency caused by a defect in vitamin K
metabolism.
A transitory increase in DCP has been reported in
certain patients with HCC subsequent to sorafenib treatment
(34). Additionally, progression-free
survival in those patients who exhibited an increase in DCP
expression levels was longer compared with the patients who
exhibited no increase in DCP (17,35). The
elevation of DCP subsequent to sorafenib treatment may be a
prognostic marker, and this elevation may be caused by tumor cell
ischemia. Sorafenib at concentrations of 0.3125–20 µM suppressed
cell proliferation in a dose-dependent manner in all 13 cell lines
used in the present study (data not shown). Similar to the results
revealed by Llobet et al (36)
and Fernando et al (37),
apoptosis was induced in 8 of the cell lines in between 5 to 50% of
the cells, however the induction of autocrine cell proliferation by
DCP or activation of hepatocyte growth factor receptor was not
observed such as the report of Suzuki et al (38) and Gao and Vande Woude (39).
In the in vivo experiment of the present
study, tumor volume and weight, and blood vessel density were
suppressed in mice receiving 600 µg/mouse/day sorafenib, compared
with the control group. The present study hypothesizes that the
inhibitory effect of sorafenib on neovascularization is responsible
for the suppression of tumor proliferation. Also, NX-DCP secretion
per unit weight increased in the sorafenib-treated mice, which may
be an additional mechanism of creating a hypoxic environment
through the inhibition of neovascularization.
The levels of DCP and NX-DCP secretion were
significantly decreased in sorafenib treated cell cultures compared
with the non-treated controls, whereas levels of prothrombin and
VEGF expression were increased. However, under normoxic conditions
levels of NX-DCP expression increased at 72 h subsequent to
sorafenib treatment. The DCP values increased under hypoxic
conditions compared with normoxic conditions, but decreased
significantly in the cells cultured with sorafenib. With regard to
the increase in levels of DCP expression under hypoxic conditions,
as reported by Murata et al (40), hypoxic conditions induce epithelial to
fibroblastoid conversion and epithelial mesenchymal transition,
which may inhibit vitamin K uptake and stimulate DCP production.
Conversely, levels of NX-DCP secretion exhibited a different trend
compared with DCP in the sorafenib-treated cells under hypoxic
conditions. At 48 h, sorafenib suppressed NX-DCP secretion, but at
72 h an increase in NX-DCP was observed. In our previous study of
resected HCC tissues, NX-DCP originated from non-tumorous cells in
the background, whilst expression within the cancerous area was
limited (10). With regard to the
production of NX-DCP in HCC, it is possible that the direct effects
of sorafenib and blood vessel ischemia may be responsible for the
increase in levels of serum NX-DCP. However, NX-DCP from
non-cancerous hepatocytes may also be present in the sera to
varying degrees, so an accurate assessment of this effect may prove
difficult.
As a mechanism for the increase in DCP observed in
patients with HCC subsequent to sorafenib treatment, the present
study suggests that the suppression of neovascularization by
sorafenib promotes blood vessel ischemia, producing hypoxic
conditions whereby vitamin K uptake and utilization efficiency is
reduced. In these circumstances it is likely that the degree of the
increase in serum DCP will be determined according to a balance
between the direct suppression of DCP by sorafenib and the increase
in DCP secretion due to ischemia.
Acknowledgements
The authors would like to thank Ms. Akemi Fujiyoshi
for her assistance in the present study.
References
|
1
|
Liu L, Cao Y, Chen C, Zhang X, McNabola A,
Wilkie D, Wilhelm S, Lynch M and Carter C: Sorafenib blocks the
RAF/MEK/ERK pathway, inhibits tumor angiogenesis, and induces tumor
cell apoptosis in hepatocellular carcinoma model PLC/PRF/5. Cancer
Res. 66:11851–11858. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Llovet JM, Ricci S, Mazzaferro V, Hilgard
P, Gane E, Blanc JF, de Oliveira AC, Santoro A, Raoul JL, Forner A,
et al: Sorafenib in advanced hepatocellular carcinoma. N Engl J
Med. 359:378–390. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Escudier B, Eisen T, Stanler WM, Szczylik
C, Oudard S, Staehler M, Negrier S, Chevreau C, Desai AA, Rolland
F, et al: Sorafenib for treatment of renal cell carcinoma: Final
efficacy and safety results of the phase III treatment approaches
in renal cancer global evaluation trial. J Clin Oncol.
27:3312–3318. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Carlomagno F, Anaganti S, Guida T,
Salvatore G, Troncone G, Wilhelm SM and Santoro M: BAY 43-9006
inhibition of oncogenic RET mutants. J Natl Cancer Inst.
98:326–334. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Wilhelm SM, Carter C, Tang L, Wilkie D,
McNabola A, Rong H, Chen C, Zhang X, Vincent P, McHugh M, et al:
BAY 43-9006 exhibits broad spectrum oral antitumor activity and
targets the RAF/MEK/ERK pathway and receptor tyrosine kinases
involved in tumor progression and angiogenesis. Cancer Res.
64:7099–7109. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Liebman HA, Furie BC, Tong MJ, Blanchard
RA, Lo KJ, Lee SD, Coleman MS and Furie B: Des-gamma-carboxy
(abnormal) prothrombin as a serum marker of primary hepatocellular
carcinoma. N Engl J Med. 310:1427–1431. 1984. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Sakon M, Monden M, Gotoh M, Kanai T,
Umeshita K, Nakano Y, Mori T, Sakurai M and Wakasa K: Relationship
between pathologic prognostic factors and abnormal levels of
des-gamma-carboxy prothrombin and alpha-fetoprotein in
hepatocellular carcinoma. Am J Surg. 163:251–256. 1992. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Suehiro T, Sugimachi K, Matsumata T,
Itasaka H, Taketomi A and Maeda T: Protein induced by vitamin K
absence or antagonist II as a prognostic marker in hepatocellular
carcinoma. Comparison with alpha-fetoprotein. Cancer. 73:2464–2471.
1994. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Sakamoto N: NX-PVKA assay, a conventional
but refined prognostic biomarker for hepatocellular carcinoma. J
Gastroenterol Hepatol. 28:755–756. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Sumi A, Akiba J, Ogasawara S, Nakayama M,
Nomura Y, Yasumoto M, Sanada S, Nakashima O, Abe T and Yano H:
Des-γ-carboxyprothrombin (DCP) and NX-DCP expressions and their
relationship with clinicopathological features in hepatocellular
carcinoma. PLoS One. 10:e01184522015. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Kim BK, Ahn SH, Seong JS, Park JY, Kim DY,
Kim JK, Lee DY, Lee KH and Han KH: Early α-fetoprotein response as
a predictor for clinical outcome after localized concurrent
chemoradiotherapy for advanced hepatocellular carcinoma. Liver Int.
31:369–376. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Johnson PJ: The role of serum
alpha-fetoprotein estimation in the diagnosis and management of
hepatocellular carcinoma. Clin Liver Dis. 5:145–159. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Zhou L, Liu J and Luo F: Serum tumor
markers for detection of hepatocellular carcinoma. World J
Gastroenterol. 12:1175–1181. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Malaguarnera G, Giordano M, Paladina I,
Berretta M, Cappellani A and Malaguarnera M: Serum markers of
hepatocellular carcinoma. Dig Dis Sci. 55:2744–2755. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Kuzuya T, Asahina Y, Tsuchiya K, Tanaka K,
Suzuki Y, Hoshioka T, Tamaki S, Kato T, Yasui Y, Hosokawa T, et al:
Early decrease in α-fetoprotein, but not des-γ-carboxy prothrombin,
predicts sorafenib efficacy in patients with advanced
hepatocellular carcinoma. Oncology. 81:251–258. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Nakazawa T, Hidaka H, Shibuya A and
Koizumi W: Rapid regression of advanced hepatocellular carcinoma
associated with elevation of des-gamma-carboxy prothrombin after
short-term treatment with sorafenib-a report of two cases. Case Rep
Oncol. 3:298–303. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Ueshima K, Kudo M, Takita M, Nagai T,
Tatsumi C, Ueda T, Kita S, Ishikawa E, Yada N, Inoue T, et al:
Des-γ-carboxyprothrombin may be a promising biomarker to determine
the therapeutic efficacy of sorafenib for hepatocellular carcinoma.
Dig Dis. 29:321–325. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Murakami T: Establishment and
characterization of human hepatoma cell line (KIM-1). Acta Hepatol
Jpn. 25:532–539. 1984. View Article : Google Scholar
|
|
19
|
Yano H, Kojiro M and Nakashima T: A new
human hepatocellular carcinoma cell line (KYN-1) with a
transformation to adenocarcinoma. In Vitro Cell Dev Biol.
22:637–646. 1986. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Yano H, Maruiwa M, Murakami T, Fukuda K,
Ito Y, Sugihara S and Kojiro M: A new human pleomorphic
hepatocellular carcinoma cell line, KYN-2. Acta Pathol Jpn.
38:953–966. 1988.PubMed/NCBI
|
|
21
|
Murakami T, Maruiwa M, Fukuda K, Kojiro M,
Tanaka M and Tanikawa K: Establishment and characterization of a
new human hepatoma cell line (KYN-3) derived from the ascites of
the hepatoma paitient. Proceedings of the Japanese Cancer
Association. Jpn J Cancer Res. 292:1988;
|
|
22
|
Yano H, Iemura A, Fukuda K, Mizoguchi A,
Haramaki M and Kojiro M: Establishment of two distinct human
hepatocellular carcinoma cell lines from a single nodule showing
clonal dedifferentiation of cancer cells. Hepatology. 18:320–327.
1993. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Haramaki M, Yano H, Iemura A, Momosaki S,
Ogasawara S, Inoue M, Yamaguchi R, Kusaba A, Utsunomiya I and
Kojiro M: A new human hepatocellular carcinoma cell line (HAK-2)
forms various structures in collagen gel matrices. Hum Cell.
10:183–192. 1997.PubMed/NCBI
|
|
24
|
Utsunomiya I, Iemura A, Yano H, Akiba J
and Kojiro M: Establishment and characterization of a new human
hepatocellular carcinoma cell line, HAK-3, and its response to
growth factors. Int J Oncol. 15:669–675. 1999.PubMed/NCBI
|
|
25
|
Murakami T, Yano H, Maruiwa M, Sugihara S
and Kojiro M: Establishment and characterization of a human
combined hepatocholangiocarcinoma cell line and its heterologous
transplantation in nude mice. Hepatology. 7:551–556. 1987.
View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Yano H, Iemura A, Haramaki M, Momosaki S,
Ogasawara S, Higaki K and Kojiro M: A human combined hepatocellular
and cholangiocarcinoma cell line (KMCH-2) that shows the features
of hepatocellular carcinoma or cholangiocarcinoma under different
growth conditions. J Hepatol. 24:413–422. 1996. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Shah DV, Engelke JA and Suttie JW:
Abnormal prothrombin in the plasma of rats carrying hepatic tumors.
Blood. 69:850–854. 1987.PubMed/NCBI
|
|
28
|
Shah DV, Zhang P, Engelke JA, Bach AU and
Suttie JW: Vitamin K-dependent carboxylase activity, prothrombin
mRNA, and prothrombin production in two cultured rat hepatoma cell
lines. Thromb Res. 70:365–373. 1993. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Ono M, Ohta H, Ohhira M, Sekiya C and
Namiki M: Measurement of immunoreactive prothrombin,
des-gamma-carboxy prothrombin, and vitamin K in human liver
tissues: Overproduction of immunoreactive prothrombin in
hepatocellular carcinoma. Am J Gastroenterol. 85:1149–1154.
1990.PubMed/NCBI
|
|
30
|
Yamagata H, Nakanishi T, Furukawa M, Okuda
H and Obata H: Levels of vitamin K, immunoreactive prothrombin,
des-gamma-carboxy prothrombin and gamma-glutamyl carboxylase
activity in hepatocellular carcinoma tissue. J Gastroenterol
Hepatol. 10:8–13. 1995. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Wang Z, Wang M, Finn F and Carr BI: The
growth inhibitory effects of vitamins K and their actions on gene
expression. Hepatology. 22:876–882. 1995. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Okuda H, Obata H, Nakanishi T, Furukawa R
and Hashimoto E: Production of abnormal prothrombin
(des-gamma-carboxy prothrombin) by hepatocellular carcinoma. A
clinical and experimental study. J Hepatol. 4:357–363. 1987.
View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Sakon M, Monden M, Gotoh M, Kobayashi K,
Kanai T, Umeshita K, Endoh W and Mori T: The effects of vitamin K
on the generation of des-gamma-carboxy prothrombin (PIVKA-II) in
patients with hepatocellular carcinoma. Am J Gastroenterol.
86:339–345. 1991.PubMed/NCBI
|
|
34
|
Kuzuya T, Tsuchiya K, Tanaka K, Suzuki Y,
Hoshioka T, Tamaki S, Kato T, Yasui Y, Tanaka T, Hosokawa T, et al:
Significance of PIVKA-II in sorafenib therapy for advanced
hepatocellular carcinoma. Kanzo. 51:403–404. 2010. View Article : Google Scholar
|
|
35
|
Ueshima K and Kudo M: PIVKA-II is a
predictive marker in the treatment response of sorafenib to
hepatocellular carcinoma. Kanzo. 51:681–683. 2010. View Article : Google Scholar
|
|
36
|
Llobet D, Eritja N, Yeramian A, Pallares
J, Sorolla A, Domingo M, Santacana M, Gonzalez-Tallada FJ,
Matias-Guiu X and Dolcet X: The multikinase inhibitor Sorafenib
induces apoptosis and sensitises endometrial cancer cells to TRAIL
by different mechanisms. Eur J Cancer. 46:836–850. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Fernando J, Sancho P, Fernández-Rodriguez
CM, Lledó JL, Caja L, Campbell JS, Fausto N and Fabregat I:
Sorafenib sensitizes hepatocellular carcinoma cells to
physiological apoptotic stimuli. J Cell Physio. 227:1319–1325.
2012. View Article : Google Scholar
|
|
38
|
Suzuki M, Shiraha H, Fujikawa T, Takaoka
N, Ueda N, Nakanishi Y, Koike K, Takaki A and Shiratori Y:
Des-gamma-carboxy prothrombin is a potential autologous growth
factor for hepatocellular carcinoma. J Biol Chem. 280:6409–6415.
2005. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Gao CF and Woude GF Vande: HGF/SF-Met
signaling in tumor progression. Cell Res. 15:49–51. 2005.
View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Murata K, Suzuki H, Okano H, Oyamada T,
Yasuda Y and Sakamoto A: Hypoxia-induced des-gamma-carboxy
prothrombin production in hepatocellular carcinoma. Int J Oncol.
36:161–170. 2010.PubMed/NCBI
|