Introduction
Gliomas are serious primary brain tumors arising
from glial cells. According to the type of cells, they are
histologically classified into astrocytomas, oligodendrogliomas and
oligoastrocytomas. Gliomas are further categorized into grades I to
IV, according to the World Health Organization (WHO) grading system
(1). The most common form of gliomas
are WHO grade IV tumors (glioblastoma and its variants), which
account for 82% of cases of malignant glioma (2). Incidence rates of gliomas vary
significantly with histological type, age at diagnosis, gender,
ethnicity and nationality (3–7). High-grade gliomas typically recur after
an average period of only 6.9 months, resulting in a median patient
survival of just 12–15 months following diagnosis (8). Despite having an improved prognosis
compared with increased grade glial tumors, 50–75% of patients with
low-grading glioma also eventually succumb to the disease. Median
survival times have been reported to range between 5 and 10 years,
and estimates of 10-year survival rates range between 5 and 50%
(9).
The last decade has witnessed a significant
improvement in the understanding of the molecular mechanisms of
glioma progress. For example, integrin inhibition leads to
inactivation of the transforming growth factor (TGF)-β pathway,
which controls features of malignancy, including immunosuppression,
invasiveness and stemness in human glioblastoma (10). A previous study has reported that
Crk-like protein efficiently regulates cell proliferation,
migration and invasion induced by the TGF-β pathway in glioblastoma
(11). Additionally, zinc finger
E-box binding homeobox 1 expression is responsible for the invasion
and chemoresistance of glioblastoma cells by regulating the
downstream effectors roundabout, axon guidance receptor, homolog 1
(Drosophila), v-Myb avian myeloblastosis viral oncogene
homolog and O-6-methyl-guanine-DNA methyl transferase
(MGMT). Furthermore, previous studies have identified that
epigenetic modifications occur in gliomas (12), and hypermethylated genes in gliomas
include pithelial membrane protein 3, thrombospondin 1, excitatory
amino-acid transporter 2 and MGMT (13–15). Based
on the microarray dataset GSE31262, Sandberg et al have
identified 423 upregulated and 414 downregulated genes in
glioblastoma stem cells (GSCs) using the Rank Product algorithm,
comparing adult human neural stem cells (ahNSCs), and inhibition of
the Wnt-signaling pathway reduces proliferation and sphere forming
capacity in GSCs; additionally, they also identified a 30-gene
signature which is highly overexpressed in GSCs (16). Despite this, dysregulated gene
networks and pathways associated with glioma progression remain
undetected, and the molecular mechanisms of glioma remain
elusive.
To further identify the potential key gene networks
and pathways implicated in gliomas, based on the microarray data
GSE31262 deposited by Sandberg et al (16), the differentially expressed genes
(DEGs) were identified using the linear models for microarray data
(limma) package, which is a popular choice for DEGs identification
using microarray and high-throughput PCR data (17). Furthermore, co-expression interactions
of DEGs were analyzed and a co-expression network was constructed.
Functional and pathway enrichment analyses of DEGs in the
co-expression network were also performed. Additionally, drugs
closely associated with DEGs in the network were identified. The
present study may provide more novel information for the
investigation of the molecular mechanisms underlying gliomas, which
contributes to a better understanding of the pathogenesis of
gliomas.
Materials and methods
Obtaining and preprocessing of mRNA
expression profile data
The mRNA expression profile dataset GSE31262 was
downloaded from the GEO database (http://www.ncbi.nlm.nih.gov/geo/), which derived from
9 individual samples of GSCs and 5 individual samples of ahNSCs
taken from the adult human brain (16). The annotation platform used here was
GPL2986 ABI Human Genome Survey Microarray Version 2 (Applied
Biosystems; Thermo Fisher Scientific, Inc., Waltham, MA, USA). The
probe ID was first converted into a gene symbol. For probes being
mapped to the same gene, the average expression value was
calculated as the final expression value. Then, the data were
translated into log2 logarithms, and the median
normalization was performed using the robust multichip averaging
method (18).
DEG screening
The limma package (19) in R language (http://www.bioconductor.org/packages/release/bioc/html/limma.html)
was applied to identify DEGs between GSC samples and normal NSC
samples. The false discovery rate (FDR) for each gene was obtained
by adjusting the raw P-value with the Benjamini Hochberg method
(20). FDR ≤0.01 and |log2
fold change (FC)| ≥1 were set as the thresholds.
Hierarchical clustering analysis of
DEGs
To identify the sample-specificity of DEGs,
hierarchical clustering (21) was
conducted. Euclidean distance (22)
was chosen as a measure of distance for DEGs between GSC samples
and normal NSC samples. Pheatmap package (23) in R was used to visualize the result of
hierarchical clustering.
Co-expression network of predicted
target genes
An essential prerequisite for understanding cellular
functions at the molecular-level is to correctly uncover all
functional interactions among various proteins in the cells. The
Search Tool for the Retrieval of Interacting Genes (STRING)
database (http://string-db.org/) (24) was used to select co-expression
interactions of DEGs, with a co-expression coefficient >0.6 as
the cut-off. Based on the expression values of DEGs, the Pearson
Correlation Coefficient (PCC) (25)
between DEGs were calculated and only pairs with |PCC| >0.6 were
retained. Then, the common pairs obtained by the two aforementioned
methods were retained to construct the co-expression network, which
was visualized using the Cytoscape software (http://www.cytoscape.org/) (26). In the network, a node represents a
protein (gene), and lines represent the interactions of the
proteins. The degree of each node represents the number of nodes
that interact with this node.
Functional and pathway enrichment
analyses
To reveal the functions of DEGs in the co-expression
network, the online biological tool Database for Annotation,
Visualization and Integrated Discovery (27) was used to perform Gene Ontology (GO)
(28) functional enrichment analysis
in biological process (BP). Kyoto Encyclopedia of Genes and Genomes
(KEGG) Orthology-based Annotation System (KOBAS) was used to
identify the pathways that were significantly enriched by genes.
KOBAS provides the most comprehensive set of functionalities,
including input by both IDs and sequences, identifying both
frequent and statistically enriched pathways, offering the choices
of four statistical tests and the option of multiple testing
correction (29). P<0.05 was
selected as the cut-off for the enrichment analysis.
Analysis of DEGs associated with
glioblastoma prognosis
Preprocessed RNA-sequencing data and clinical data
of glioblastoma were downloaded from the The Cancer Genome Atlas
(TCGA) database (https://cancergenome.nih.gov/). Based on the clinical
data, sample files that provided survival time and status of
patients were chosen for subsequent analysis.
Cox proportional hazard model (30,31) was
utilized to predict the association of DEGs and patient prognosis.
In the present study, each DEG was defined as a covariant, and
survival time of each chosen glioblastoma sample was defined as a
dependent variable. Multivariate Cox proportional hazard regression
analysis was performed for all DEGs. The obtained probability value
for each covariant <0.005 was set as the cut-off, indicating
that the DEG was associated with glioblastoma prognosis. The
analysis was conducted using SPSS 19.0 software (IBM Corp., Armonk,
NY, USA).
Identification of small-molecule drugs
based on DEGs
Web-based Gene Set Analysis Toolkit (WebGestalt)
incorporates information from different public resources and
provides an easy way for biologists to make sense out of gene lists
(32). The current version of
WebGestalt includes disease and drug-associated genes. In the
present study, drugs associated with genes in the co-expression
network were obtained from WebGestalt. The raw P-value for the
association between drugs and genes was calculated by the
hypergeometric test, and adjusted using the multiple test
adjustment. An adjusted P-value <0.05 was set as the
cut-off.
Results
Identification of DEGs and
hierarchical clustering analysis
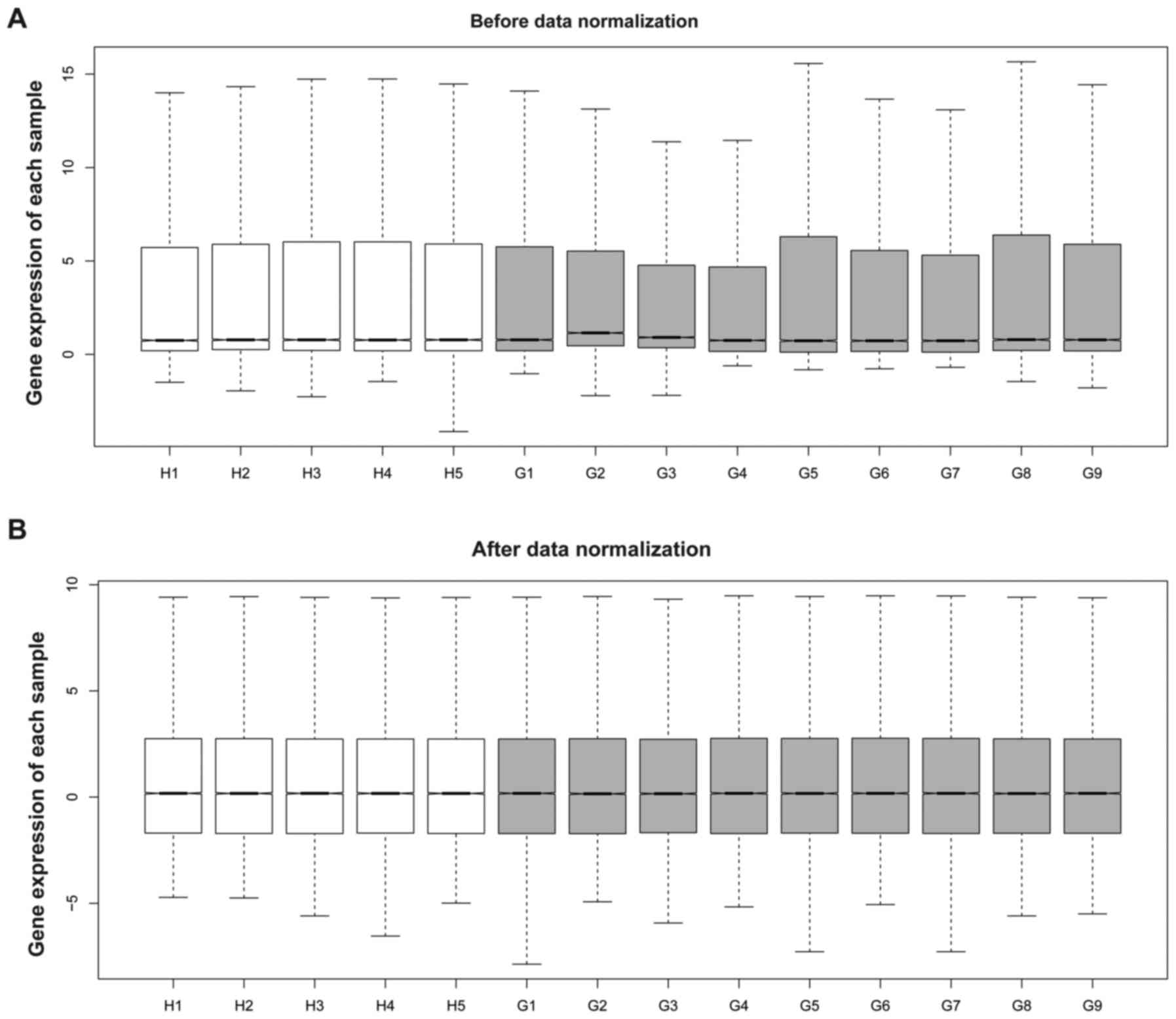
Subsequent to preprocessing, standard DEG expression
profile data was illustrated (Fig.
1). Based on the criteria of FDR <0.05 and |log2
FC| ≥1, a total of 431 DEGs were identified in the GSC samples,
including 98 upregulated DEGs and 333 downregulated DEGs.
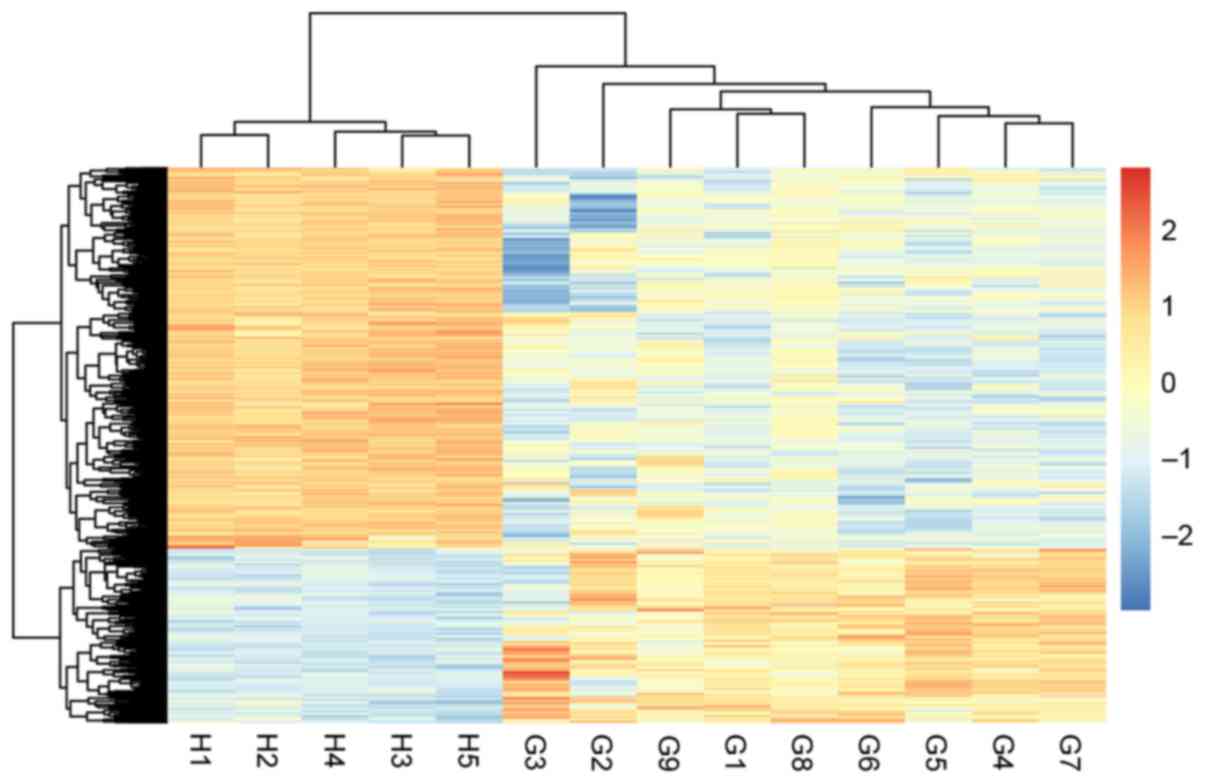
Hierarchical clustering analysis illustrated that the GSCs samples
and normal samples can be distinguished using the identified DEGs,
according to their differential expression status (Fig. 2).
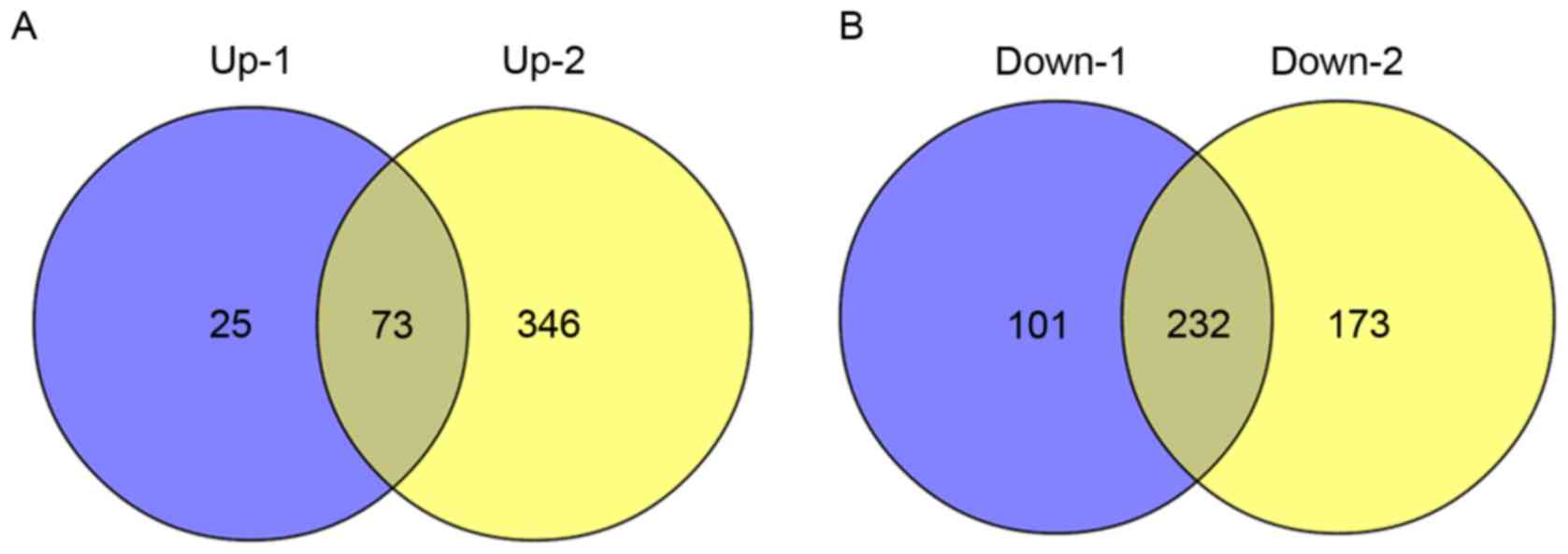
Furthermore, comparing with DEGs that identified by
Sandberg et al (16), 73
upregulated DEGs and 232 downregulated DEGs were common in the
present study and the study by Sandberg et al (16) (Fig.
3).
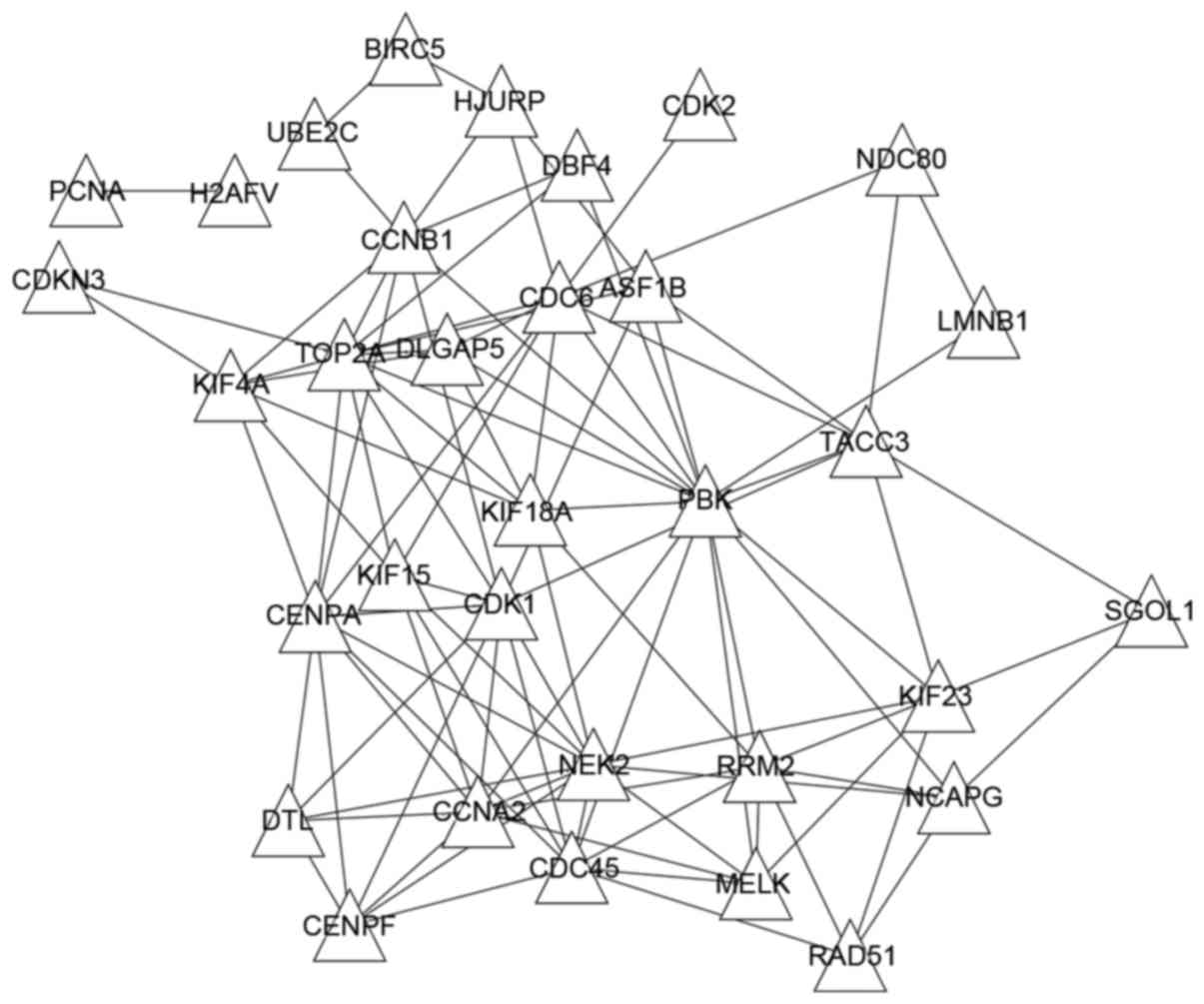
Co-expression network of the predicted
target genes
To analyze the interactions of DEGs, STRING database
and PCC were used to predict co-expressed interactions of DEGs. A
total of 678 pairs of co-expression genes with co-expression score
>0.6 were identified from the STRING database. In addition,
3,973 gene pairs with |PCC| >0.6 were extracted. Subsequent to
comparison, the 100 common gene pairs were selected. The Cytoscape
software was used to visualize the co-expression network (Fig. 4), which includes a total of 33 genes
and 100 interactions. Notably, all genes in this network were
upregulated, and the genes PDZ binding kinase (PBK),
topoisomerase (DNA) II α (TOP2A), cyclin dependent kinase
(CDK) 1, cell division cycle (CDC) 6 and NIMA related
kinase 2 (NEK2) had a higher degree that was >10.
PBK, TOP2A and CDK1 were co-expressed with
each other.
Functional and pathway enrichment
analyses of genes in the co-expression network
To further reveal the functions of DEGs in the
co-expression network, the GO functional and KEGG pathway
enrichment analyses were conducted. The majority of the GO BP terms
were associated with the cell cycle, including cell cycle
procession, nuclear division and chromosome segregation (Table I). Additionally, two significantly
enriched pathways (cell cycle and p53 signaling pathway) were
obtained (Table II). The upregulated
genes involved in the cell cycle pathway included cyclin B1
(CCNB1), CDK1, CDC6, CDC45,
DBF4, proliferating cell nuclear antigen, cyclin A2 and
CDK2. Genes enriched in the p53 signaling pathway were
CCNB1, CDK1, ribonucleotide reductase regulatory
subunit M2 and CDK2.
 | Table I.Biological processes of
differentially expressed genes in the co-expression network. |
Table I.
Biological processes of
differentially expressed genes in the co-expression network.
| Term | Process | Count | P-value | FDR |
|---|
| GO:0022403 | Cell cycle
phase | 21 |
2.02×10−23 |
2.79×10−20 |
| GO:0007049 | Cell cycle | 24 |
1.03×10−22 |
1.42×10−19 |
| GO:0000278 | Mitotic cell
cycle | 20 |
1.31×10−22 |
1.80×10−19 |
| GO:0022402 | Cell cycle
progression | 22 |
2.23×10−22 |
3.08×10−19 |
| GO:0000279 | M phase | 19 |
8.50×10−22 |
1.17×10−18 |
| GO:0007067 | Mitosis | 17 |
3.30×10−21 |
4.56×10−18 |
| GO:0000280 | Nuclear
division | 17 |
3.30×10−21 |
4.56×10−18 |
| GO:0000087 | M phase of mitotic
cell cycle | 17 |
4.43×10−21 |
6.12×10−18 |
| GO:0048285 | Organelle
fission | 17 |
6.35×10−21 |
8.77×10−18 |
| GO:0007059 | Chromosome
segregation | 10 |
1.13×10−13 |
1.57×10−10 |
| GO:0051301 | Cell division | 13 |
9.02×10−13 |
1.25×10−9 |
| GO:0051726 | Regulation of cell
cycle | 13 |
3.51×10−12 |
4.85×10−9 |
| GO:0006260 | DNA
replication | 10 |
2.72×10−10 |
3.75×10−7 |
| GO:0051276 | Chromosome
organization | 11 |
7.20×10−8 |
9.95×10−5 |
| GO:0006259 | DNA metabolism | 10 |
1.29×10−6 |
1.78×10−3 |
| Table II.Pathway enrichment analysis of the
differentially expressed genes enriched in the co-expression
network. |
Table II.
Pathway enrichment analysis of the
differentially expressed genes enriched in the co-expression
network.
| ID | Pathway | P-value | FDR | Genes |
|---|
| hsa04110 | Cell cycle |
6.96×10−9 |
1.39×10−7 | CCNB1, CDK1,
CDC6, CDC45, DBF4, PCNA, CCNA2, CDK2 |
| hsa04115 | p53 signaling
pathway |
5.94×10−4 |
5.93×10−3 | CCNB1, CDK1,
RRM2, CDK2 |
Screening of DEGs associated with
glioblastoma prognosis
To further analyze whether the identified DEGs were
associated with the prognosis of patients with glioblastoma, the
survival data from the TCGA database were used in the present
analysis. Among the sample files obtained from TCGA, a total of 107
files provided survival time and status of patients. Based on the
Cox proportional hazard regression analysis, 69 DEGs identified in
the present study were significantly associated with glioblastoma
prognosis, 4 of which were present in the co-expression network,
including CDK2, kinesin family member 15, kinesin family
member 18A and TOP2A.
Identification of small-molecule drugs
based on DEGs
To investigate potential drugs that may be used to
treat human glioblastoma, WebGestalt was used in the present study
to identify the drugs associated with the DEGs in the co-expression
network. A total of 9 drugs were obtained, including paclitaxel
(for genes including TOP2A, BIRC5 and CDK1)
and etoposide (for genes including TOP2A and CDC6)
(Table III).
| Table III.The significant drugs associated with
differentially expressed genes in co-expressed genes. |
Table III.
The significant drugs associated with
differentially expressed genes in co-expressed genes.
| Drug | ID | Parameters | Genes |
|---|
| Paclitaxel | DB_ID:PA450761 | O=5;
rawP=9.99×10−9; adjP=1.90×10−7 | TOP2A, BIRC5,
CDK1, TACC3, CENPF |
| Etoposide | DB_ID:PA449552 | O=5;
rawP=4.20×10−8; adjP=3.99×10−7 | TOP2A, CDC6,
BIRC5, CDK1, DBF4 |
| Hydroxyurea | DB_ID:PA449942 | O=4;
rawP=1.86×10−7; adjP=8.83×10−7 | RRM2, PCNA,
CDK2, RAD51 |
|
Mechlorethamine | DB_ID:PA450336 | O=3;
rawP=1.85×10−7; adjP=8.83×10−7 | CCNB1, CDK2,
CDK1 |
| Podofilox | DB_ID:PA450993 | O=4;
rawP=7.57×10−7; adjP=2.88×10−6 | TOP2A, CDC6,
BIRC5, DBF4 |
| Progesterone | DB_ID:PA451123 | O=4;
rawP=3.61×10−6; adjP=9.80×10−6 | TOP2A, CCNA2,
CCNB1, RAD51 |
| Cisplatin | DB_ID:PA449014 | O=4;
rawP=3.50×10−6; adjP=9.80×10−6 | TOP2A, RRM2,
BIRC5, RAD51 |
| Docetaxel | DB_ID:PA449383 | O=3;
rawP=4.93×10−6; adjP=1.17×10−5 | TOP2A, RRM2,
BIRC5 |
| Adenosine | DB_ID:PA448049 | O=5;
rawP=2.98×10−5; adjP=5.66×10−5 | TOP2A, PCNA,
CDK2, RAD51, NCAPG |
Discussion
Malignant gliomas are the most frequently occurring
primary brain tumors, and they have a poor survival rate (8). The present study aimed to investigate
the potential crucial genes correlated with glioblastoma. Based on
the microarray data, a total of 98 upregulated DEGs and 333
downregulated DEGs were identified from the GSCs, of which, 33
upregulated genes had co-expression interactions. In the
co-expression network, genes including PBK, TOP2A,
CDK1, CDC6 and NEK2 had a higher degree. A set
of genes including CCNB1, CDK1, CDC6 and
CDK2 were significantly enriched in the cell cycle.
The PBK gene encodes PBK, and it has been
previously suggested that mitotic phosphorylation is required for
its catalytic activity (33,34). The present results illustrated that
PBK expression is upregulated in gliomas. Multiple previous
studies have shown the present results (35–37). A
previous study has demonstrated that gene knockdown of PBK
leads to decreased viability and sphere formation of glioma
initiating cells (GICs), and the PBK inhibitor HITOPK-032
abolishes growth and elicits a large increase in apoptosis of GICs,
indicating an important role of PBK in the cell growth of
glioma (38). As a result, the
present study speculated that PBK may play a pivotal role in
glioblastoma via the regulation of the cell cycle. The TOP2A
gene encodes TOP2A which controls and alters the topologic states
of DNA during transcription (39).
The present results showed an upregulated expression of
TOP2A protein in glioma, which has also been identified in
numerous cancers (40–42). In addition to its roles in DNA
synthesis and transcription, and chromosomal segregation during
mitosis, Tsavaris et al (43)
has reported that TOP2A is also a sensitive and specific
marker of actively proliferating cells. In the present study,
TOP2A was found to be significantly involved in the
prognosis of patients with glioblastoma, which was consistent with
numerous previous studies (44–46).
NEK2 encodes a serine/threonine-protein kinase, which is
involved in mitotic regulation (47).
Laakso and Hautaniemi (48) has
previously reported that NEK2 is upregulated in
glioblastoma, which is consistent with the present findings. The
overexpression of NEK2 has also been found in human breast
and liver cancer, and colorectal carcinoma (49). Cappello et al (50) identified that NEK2 knockdown
induced aneuploidy and cell cycle arrest that further led to cell
death in various human breast cancer cell lines. Therefore,
inhibiting the expression of the PBK, TOP2A and
NEK2 genes may be a novel approach for glioma therapy.
According to the pathway enrichment analysis, the
majority of significantly altered pathways were involved in the
cell cycle, which is consistent with the BP analysis, and genes
involved in this pathway included CDK1, CDC6,
DBF4, CCNB1 and CDK2. Cancer cells differ from
normal cells by numerous important characteristics, including loss
of differentiation, increased invasiveness and decreased drug
sensitivity. The conversion of normal cells into cancer cells is
facilitated by the loss of fidelity in cell cycle progression,
which under normal conditions is achieved by the coordinated
activity of CDKs, checkpoint controls and repair pathways in the
cell cycle (51–53). Furthermore, numerous studies have
shown that this fidelity can be abrogated by specific genetic
changes (54–56). Both CDK1 and CDK2 encode
CDKs, which are highly conserved proteins functioning as a
serine/threonine kinase (57,58). Overexpression of CDK1 and
CDK2 promotes oncogenesis and progression of human gliomas,
whereas downregulation of CDK1 and CDK2 expression
inhibits the proliferation activities of human malignant gliomas
(59,60). Furthermore, CDK2 was identified
as a prognostic gene in the present study, which was consistent
with the study by Zhang et al (61). Additionally, CDK2 is involved
in the expression of other prognostic markers, including p27 and
c-Met (62,63). CDC6 encodes CDC6, which is a
protein essential for the initiation of DNA replication (64). It has been demonstrated that
CDC6 expression is a marker of proliferative activity in
brain tumors, including gliomas (65). CCNB1 encodes CCNB1, which is a
regulatory protein of cdk1 in mitosis (66). An abnormally high expression level of
CCNB1 has been reported to be correlated with high grades
and advanced stages of gliomas (67,68).
Collectively, genes such as CDK1, CDK2, CDC6
and CCNB1 may exert critical roles in the progress of
glioblastoma through mediating cell cycle.
In the present study, CDK1, CDK2 and
CCNB1 were also markedly enriched in the p53 signaling
pathway. P53 signaling pathway is the most commonly mutated pathway
in tumorigenesis (69). As a
well-known tumor suppressor, p53 responds to DNA damage and various
genotoxic and cytotoxic stresses by inducing cell cycle arrest and
apoptosis (70). Overall, the p53
pathway is disrupted in ~80% of high-grade gliomas (WHO Grades III
and IV) (71). Therefore, in the
progress of glioblastoma, CDK1, CDK2 and CCNB1
may also regulate the p53 signaling pathway.
Additionally, the present study used the limma
package to identify DEGs, comparing the method (Rank Product
algorithm) used by Sandberg et al (16). Among the identified DEGs in the
present study, 73 upregulated genes and 232 downregulated DEGs were
common with those screened by Sandberg et al (16), indicating the potentially significant
roles of these common DEGs in glioblastoma. However, another 25
upregulated and 101 downregulated DEGs, which were not found by
Sandberg et al (16), were
also screened. The results suggest that it is necessary to
reanalyze the public microarray data using different methods. The
roles of these DEGs identified in the present study in glioblastoma
require additional investigation. Additionally, the expression
levels and functions of the genes associated with the cell cycle
and the p53 signaling pathway in glioblastoma require validation by
experiments, which will be conducted and reported later.
In conclusion, a total of 98 upregulated DEGs and
333 downregulated DEGs were identified from the GSCs compared with
the ahNSCs. The upregulated hub genes in the co-expression network
(including PBK, TOP2A, CDK1, CDC6 and
NEK2) and the DEGs (including CCNB1, CDK1,
CDC6, and CDK2) that correlated with cell cycle and
the p53 signaling pathway may have essential roles in the progress
of glioblastoma. These genes may be potentially therapeutic targets
of gliomas, and the findings may be helpful to identify the
molecular mechanisms of the pathogenesis of glioblastoma.
References
|
1
|
Lassman AB: Molecular biology of gliomas.
Curr Neurol Neurosci Rep. 4:228–233. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Dolecek TA, Propp JM, Stroup NE and
Kruchko C: CBTRUS statistical report: Primary brain and central
nervous system tumors diagnosed in the United States in 2005–2009.
Neuro Oncol. 14 Suppl 5:v1–v49. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Ostrom QT, Bauchet L, Davis FG, Deltour I,
Fisher JL, Langer CE, Pekmezci M, Schwartzbaum JA, Turner MC, Walsh
KM, et al: The epidemiology of glioma in adults: A ‘state of the
science’ review. Neuro Oncol. 16:896–913. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Ostrom QT, Gittleman H, Farah P, Ondracek
A, Chen Y, Wolinsky Y, Stroup NE, Kruchko C and Barnholtz-Sloan JS:
CBTRUS statistical report: Primary brain and central nervous system
tumors diagnosed in the United States in 2006–2010. Neuro Oncol. 15
Suppl 2:ii1–ii56. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Lee CH, Jung KW, Yoo H, Park S and Lee SH:
Epidemiology of primary brain and central nervous system tumors in
Korea. J Korean Neurosurg Soc. 48:145–152. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Gigineishvili D, Shengelia N, Shalashvili
G, Rohrmann S, Tsiskaridze A and Shakarishvili R: Primary brain
tumour epidemiology in Georgia: First-year results of a
population-based study. J Neurooncol. 112:241–246. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Dubrow R and Darefsky AS: Demographic
variation in incidence of adult glioma by subtype, United States,
1992–2007. BMC Cancer. 11:3252011. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Watkins S and Sontheimer H: Unique biology
of gliomas: Challenges and opportunities. Trends Neurosci.
35:546–556. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Sanai N and Berger MS: Recent surgical
management of gliomasGlioma. Yamanaka R: 746. Springer; New York:
pp. 12–25. 2012, View Article : Google Scholar
|
|
10
|
Roth P, Silginer M, Goodman SL, Hasenbach
K, Thies S, Maurer G, Schraml P, Tabatabai G, Moch H, Tritschler I
and Weller M: Integrin control of the transforming growth factor-β
pathway in glioblastoma. Brain. 136:564–576. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Lv S, Qin J, Yi R, Coreman M, Shi R, Kang
H and Yao C: CrkL efficiently mediates cell proliferation,
migration, and invasion induced by TGF-β pathway in glioblastoma. J
Mol Neurosci. 51:1046–1051. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Feinberg AP and Tycko B: The history of
cancer epigenetics. Nat Rev Cancer. 4:143–153. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Hegi ME, Diserens AC, Gorlia T, Hamou MF,
de Tribolet N, Weller M, Kros JM, Hainfellner JA, Mason W, Mariani
L, et al: MGMT gene silencing and benefit from temozolomide in
glioblastoma. N Engl J Med. 352:997–1003. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Kunitz A, Wolter M, Van den Boom J,
Felsberg J, Tews B, Hahn M, Benner A, Sabel M, Lichter P,
Reifenberger G, et al: DNA hypermethylation and aberrant expression
of the EMP3 gene at 19q13. 3 in human gliomas. Brain Pathol.
17:363–370. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Zschocke J, Allritz C, Engele J and Rein
T: DNA methylation dependent silencing of the human glutamate
transporter EAAT2 gene in glial cells. Glia. 55:663–674. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Sandberg CJ, Altschuler G, Jeong J,
Strømme KK, Stangeland B, Murrell W, Grasmo-Wendler UH, Myklebost
O, Helseth E, Vik-Mo EO, et al: Comparison of glioma stem cells to
neural stem cells from the adult human brain identifies
dysregulated Wnt-signaling and a fingerprint associated with
clinical outcome. Exp Cell Res. 319:2230–2243. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Ritchie ME, Phipson B, Wu D, Hu Y, Law CW,
Shi W and Smyth GK: Limma powers differential expression analyses
for RNA-sequencing and microarray studies. Nucleic Acids Res.
43:e472015. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Irizarry RA, Hobbs B, Collin F,
Beazer-Barclay YD, Antonellis KJ, Scherf U and Speed TP:
Exploration, normalization, and summaries of high density
oligonucleotide array probe level data. Biostatistics. 4:249–264.
2003. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Smyth G: Limma: Linear models for
microarray dataBioinformatics and computational biology solutions
using R and Bioconductor. Gentleman R, Carey V, Dudoit S, Irizarry
R and Huber W: Springer; New York: pp. 397–420. 2005, View Article : Google Scholar
|
|
20
|
Benjamini Y and Hochberg Y: Controlling
the false discovery rate: A practical and powerful approach to
multiple testing. J Royal Statistic Soci. Series B
(Methodological). 57:289–300. 1995.
|
|
21
|
Szekely GJ and Rizzo ML: Hierarchical
clustering via joint between-within distances: Extending Ward's
minimum variance method. Journal of Classification. 22:151–183.
2005. View Article : Google Scholar
|
|
22
|
Deza MM and Deza E: Encyclopedia of
distances. Springer; 2009, View Article : Google Scholar
|
|
23
|
Wang L, Cao C, Ma Q, Zeng Q, Wang H, Cheng
Z, Zhu G, Qi J, Ma H, Nian H and Wang Y: RNA-seq analyses of
multiple meristems of soybean: Novel and alternative transcripts,
evolutionary and functional implications. BMC Plant Biol.
14:1692014. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Szklarczyk D, Franceschini A, Kuhn M,
Simonovic M, Roth A, Minguez P, Doerks T, Stark M, Muller J, Bork
P, et al: The STRING database in 2011: Functional interaction
networks of proteins, globally integrated and scored. Nucleic Acids
Res. 39(Database Issue): D561–D568. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Obayashi T, Kinoshita K, Nakai K, Shibaoka
M, Hayashi S, Saeki M, Shibata D, Saito K and Ohta H: ATTED-II: A
database of co-expressed genes and cis elements for identifying
co-regulated gene groups in Arabidopsis. Nucleic Acids Res.
35(Database Issue): D863–D869. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Smoot ME, Ono K, Ruscheinski J, Wang PL
and Ideker T: Cytoscape 2.8: New features for data integration and
network visualization. Bioinformatics. 27:431–432. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Huang DW, Sherman BT and Lempicki RA:
Bioinformatics enrichment tools: Paths toward the comprehensive
functional analysis of large gene lists. Nucleic Acids Res.
37:1–13. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Ashburner M, Ball CA, Blake JA, Botstein
D, Butler H, Cherry JM, Davis AP, Dolinski K, Dwight SS, Eppig JT,
et al: Gene ontology: Tool for the unification of biology. The gene
ontology consortium. Nat Genet. 25:25–29. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Mao X, Cai T, Olyarchuk JG and Wei L:
Automated genome annotation and pathway identification using the
KEGG Orthology (KO) as a controlled vocabulary. Bioinformatics.
21:3787–3793. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Motakis E, Ivshina AV and Kuznetsov VA:
Data-driven approach to predict survival of cancer patients:
Estimation of microarray genes' prediction significance by Cox
proportional hazard regression model. IEEE Eng Med Biol Mag.
28:58–66. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Liu W, Hao XS, Fan Q, Li HX, Song LN, Wang
SJ, Wang PZ, Jin Y, Chen Y, Guan LY, et al: Cox proportional hazard
model analysis of prognosis in patients with carcinoma of esophagus
and gastric cardia after radical resection. Zhonghua Zhong Liu Za
Zhi. 30:921–925. 2008.(In Chinese). PubMed/NCBI
|
|
32
|
Zhang B, Kirov S and Snoddy J: WebGestalt:
An integrated system for exploring gene sets in various biological
contexts. Nucleic Acids Res. 33(Web Server Issue): W741–W748. 2005.
View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Park JH, Jeong YJ, Won HK, Choi SY, Park
JH and Oh SM: Activation of TOPK by lipopolysaccharide promotes
induction of inducible nitric oxide synthase through NF-κB activity
in leukemia cells. Cell Signal. 26:849–856. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Park JH, Lin ML, Nishidate T, Nakamura Y
and Katagiri T: PDZ-binding kinase/T-LAK cell-originated protein
kinase, a putative cancer/testis antigen with an oncogenic activity
in breast cancer. Cancer Res. 66:9186–9195. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Stangeland B, Mughal AA, Grieg Z, Sandberg
CJ, Joel M, Nygard S, Meling T, Murrell W, Vik Mo EO and Langmoen
IA: Combined expressional analysis, bioinformatics and targeted
proteomics identify new potential therapeutic targets in
glioblastoma stem cells. Oncotarget. 6:26192–26215. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Wei B, Wang L, Du C, Hu G, Wang L, Jin Y
and Kong D: Identification of differentially expressed genes
regulated by transcription factors in glioblastomas by
bioinformatics analysis. Mol Med Rep. 11:2548–2554. 2015.PubMed/NCBI
|
|
37
|
Freitas M, Malheiros S, Stávale JN, Biassi
TP, Zamunér FT, de Souza Begnami M, Soares FA and Vettore AL:
Expression of cancer/testis antigens is correlated with improved
survival in glioblastoma. Oncotarget. 4:6362013. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Joel M, Mughal AA, Grieg Z, Murrell W,
Palmero S, Mikkelsen B, Fjerdingstad HB, Sandberg CJ, Behnan J,
Glover JC, et al: Targeting PBK/TOPK decreases growth and survival
of glioma initiating cells in vitro and attenuates tumor growth in
vivo. Mol Cancer. 14:1212015. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Escargueil AE, Plisov SY, Filhol O, Cochet
C and Larsen AK: Mitotic phosphorylation of DNA topoisomerase II
alpha by protein kinase CK2 creates the MPM-2 phosphoepitope on
Ser-1469. J Biol Chem. 275:34710–34718. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Fasching PA, Weihbrecht S, Haeberle L,
Gasparyan A, Villalobos IE, Ma Y, Ekici AB, Wachter DL, Hartmann A,
Beckmann MW, et al: HER2 and TOP2A amplification in a
hospital-based cohort of breast cancer patients: Associations with
patient and tumor characteristics. Breast Cancer Res Treat.
145:193–203. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Jain M, Zhang L, He M, Zhang YQ, Nilubol
N, Shen M and Kebebew E: TOP2A is a therapeutic target for
adrenocortical carcinoma. Cancer Res. 73:35432013. View Article : Google Scholar
|
|
42
|
Kirk J, Schaarschuch K, Dalimov Z, Lasorsa
E, Ku S, Ramakrishnan S, Hu Q, Azabdaftari G, Wang J, Pili R and
Ellis L: Top2a identifies and provides epigenetic rationale for
novel combination therapeutic strategies for aggressive prostate
cancer. Oncotarget. 6:3136–3146. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Tsavaris N, Lazaris A, Kosmas C, Gouveris
P, Kavantzas N, Kopterides P, Papathomas T, Arapogiannis G, Zorzos
H, Kyriakou V and Patsouris E: Topoisomerase I and IIalpha protein
expression in primary colorectal cancer and recurrences following
5-fluorouracil-based adjuvant chemotherapy. Cancer Chemother
Pharmacol. 64:391–398. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Arivazhagan A, Kumar DM, Sagar V, Patric
IR, Sridevi S, Thota B, Srividya MR, Prasanna K, Thennarasu K,
Mondal N, et al: Higher topoisomerase 2 alpha gene transcript
levels predict better prognosis in GBM patients receiving
temozolomide chemotherapy: Identification of temozolomide as a
TOP2A inhibitor. J Neurooncol. 107:289–297. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Bie L, Zhao G, Cheng P, Rondeau G,
Porwollik S, Ju Y, Xia XQ and McClelland M: The accuracy of
survival time prediction for patients with glioma is improved by
measuring mitotic spindle checkpoint gene expression. PLoS One.
6:e256312011. View Article : Google Scholar : PubMed/NCBI
|
|
46
|
Zhang X, Yang H, Gong B, Jiang C and Yang
L: Combined gene expression and protein interaction analysis of
dynamic modularity in glioma prognosis. J Neurooncol. 107:281–288.
2012. View Article : Google Scholar : PubMed/NCBI
|
|
47
|
Schultz SJ, Fry AM, Sütterlin C, Ried T
and Nigg EA: Cell cycle-dependent expression of Nek2, a novel human
protein kinase related to the NIMA mitotic regulator of Aspergillus
nidulans. Cell Growth Differ. 5:625–635. 1994.PubMed/NCBI
|
|
48
|
Laakso M and Hautaniemi S: DEGs in
Glioblastoma. 2011, http://csbi.ltdk.helsinki.fi/camda/candidateReport-document.pdfFebruary
2–2015
|
|
49
|
Chen YJ, Lin HS and Jiang JK:
Overexpression of nek2 in colorectal carcinoma. Cancer Research.
68:55092008.
|
|
50
|
Cappello P, Blaser H, Gorrini C, Lin D,
Elia A, Wakeham A, Haider S, Boutros P, Mason J, Miller NA, et al:
Role of Nek2 on centrosome duplication and aneuploidy in breast
cancer cells. Oncogene. 33:2375–2384. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
51
|
Evan GI and Vousden KH: Proliferation,
cell cycle and apoptosis in cancer. Nature. 411:342–348. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
52
|
Kastan MB and Bartek J: Cell-cycle
checkpoints and cancer. Nature. 432:316–323. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
53
|
Vermeulen K, Van Bockstaele DR and
Berneman ZN: The cell cycle: A review of regulation, deregulation
and therapeutic targets in cancer. Cell Prolif. 36:131–149. 2003.
View Article : Google Scholar : PubMed/NCBI
|
|
54
|
Tang S, Bonaroti J, Unlu S, Liang X, Tang
D, Zeh HJ and Lotze MT: Sweating the small stuff: microRNAs and
genetic changes define pancreatic cancer. Pancreas. 42:740–759.
2013. View Article : Google Scholar : PubMed/NCBI
|
|
55
|
Gollin SM, Parikh RA and Huang X: Genetic
changes in ATM and ATR/CHEK1 as prognostic indicators in cancer US
Patent 20130338212 A1. June 10–2013, issued December 19. 2013
|
|
56
|
Cairns RA, Harris IS and Mak TW:
Regulation of cancer cell metabolism. Nat Rev Cancer. 11:85–95.
2011. View Article : Google Scholar : PubMed/NCBI
|
|
57
|
Fourest-Lieuvin A, Peris L, Gache V,
Garcia-Saez I, Juillan-Binard C, Lantez V and Job D: Microtubule
regulation in mitosis: Tubulin phosphorylation by the
cyclin-dependent kinase Cdk1. Mol Biol Cell. 17:1041–1050. 2006.
View Article : Google Scholar : PubMed/NCBI
|
|
58
|
Gu Y, Rosenblatt J and Morgan DO: Cell
cycle regulation of CDK2 activity by phosphorylation of Thr160 and
Tyr15. Embo J. 11:3995–4005. 1992.PubMed/NCBI
|
|
59
|
Chen H, Huang Q, Zhai DZ, Dong J, Wang AD
and Lan Q: CDK1 expression and effects of CDK1 silencing on the
malignant phenotype of glioma cells. Zhonghua Zhong Liu Za Zhi.
29:484–488. 2007.PubMed/NCBI
|
|
60
|
Zhang R, Banik NL and Ray SK: Combination
of all-trans retinoic acid and interferon-gamma upregulated
p27(kip1) and down regulated CDK2 to cause cell cycle arrest
leading to differentiation and apoptosis in human glioblastoma LN18
(PTEN-proficient) and U87MG (PTEN-deficient) cells. Cancer
Chemother Pharmacol. 62:407–416. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
61
|
Zhang J, Liu B, Jiang X, Zhao H, Fan M,
Fan Z, Lee JJ, Jiang T, Jiang T and Song SW: A systems
biology-based gene expression classifier of glioblastoma predicts
survival with solid tumors. PLoS One. 4:e62742009. View Article : Google Scholar : PubMed/NCBI
|
|
62
|
Kong DS, Song SY, Kim DH, Joo KM, Yoo JS,
Koh JS, Dong SM, Suh YL, Lee JI, Park K, et al: Prognostic
significance of c-Met expression in glioblastomas. Cancer.
115:140–148. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
63
|
Kirla RM, Haapasalo HK, Kalimo H and
Salminen EK: Low expression of p27 indicates a poor prognosis in
patients with high-grade astrocytomas. Cancer. 97:644–648. 2003.
View Article : Google Scholar : PubMed/NCBI
|
|
64
|
Saha P, Chen J, Thome KC, Lawlis SJ, Hou
ZH, Hendricks M, Parvin JD and Dutta A: Human CDC6/Cdc18 associates
with Orc1 and cyclin-cdk and is selectively eliminated from the
nucleus at the onset of S phase. Mol Cell Biol. 18:2758–2767. 1998.
View Article : Google Scholar : PubMed/NCBI
|
|
65
|
Ohta S, Koide M, Tokuyama T, Yokota N,
Nishizawa S and Namba H: Cdc6 expression as a marker of
proliferative activity in brain tumors. Oncol Rep. 8:1063–1066.
2001.PubMed/NCBI
|
|
66
|
Petri ET, Errico A, Escobedo L, Hunt T and
Basavappa R: The crystal structure of human cyclin B. Cell Cycle.
6:1342–1349. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
67
|
Zhang J, Zhao P, Fu Z, Chen X, Liu N, Lu
A, Li R, Shi L, Pu P, Kang C and You Y: Glioma cells enhance
endothelial progenitor cell angiogenesis via VEGFR-2, not VEGFR-1.
Oncol Rep. 20:1457–1463. 2008.PubMed/NCBI
|
|
68
|
Ducray F, Idbaih A, de Reyniès A, Bièche
I, Thillet J, Mokhtari K, Lair S, Marie Y, Paris S, Vidaud M, et
al: Anaplastic oligodendrogliomas with 1p19q codeletion have a
proneural gene expression profile. Mol Cancer. 7:412008. View Article : Google Scholar : PubMed/NCBI
|
|
69
|
Bode AM and Dong Z: Post-translational
modification of p53 in tumorigenesis. Nat Rev Cancer. 4:793–805.
2004. View Article : Google Scholar : PubMed/NCBI
|
|
70
|
Tanaka H, Arakawa H, Yamaguchi T,
Shiraishi K, Fukuda S, Matsui K, Takei Y and Nakamura Y: A
ribonucleotide reductase gene involved in a p53-dependent
cell-cycle checkpoint for DNA damage. Nature. 404:42–49. 2000.
View Article : Google Scholar : PubMed/NCBI
|
|
71
|
Mao H, LeBrun DG, Yang J, Zhu VF and Li M:
Deregulated signaling pathways in glioblastoma multiforme:
Molecular mechanisms and therapeutic targets. Cancer Invest.
30:48–56. 2012. View Article : Google Scholar : PubMed/NCBI
|