Introduction
Acute lymphoblastic leukemia (ALL) is the most
common type of childhood cancer (1).
In all cases of childhood leukemia, ~80% are ALL, which represents
~25% of cancers in children diagnosed <15 years of age in the
United States in 2014 (2). Although
the etiology of ALL is largely unknown, it has been proposed that
multiple gene alterations, including inactivation of tumor
suppressor genes, activation of proto-oncogenes and chromosomal
rearrangements, in addition to gene-environmental interplay, are
involved in disease development (1,3,4). Chromosomal abnormalities, including
homozygous deletions and loss of heterozygosity, are among the most
common characteristics of human tumors. The fragile histidine triad
(FHIT) gene is located on the short arm of human
chromosome-3 (3p14.2) which is a major site of chromosomal
rearrangement (5).
FHIT protein has previously been proposed to act as
a tumor suppressor (6–8). However, the exact mechanisms by which
FHIT mediates its suppressive functions are not well understood.
Several researchers have indicated that restoration of FHIT
expression suppresses tumorigenicity, and transfection of
FHIT in FHIT-deficient human cancer cells appears to induce
apoptosis and inhibit cell growth (8–11). DNA
hypermethylation may occur in genes involved in the cell cycle, DNA
damage repair and signaling pathways. DNA methylation is a
well-studied epigenetic modification, which significantly
contributes to leukemia development (12). Several studies have demonstrated an
association between FHIT hypermethylation and an increased
risk of non-small-cell lung carcinoma (NSCLC) (13), breast cancer (14,15),
cervical cancer (16), hepatocellular
carcinoma (17) and thyroid carcinoma
(18). Furthermore, downregulation of
FHIT expression has been observed in acute lymphoblastic
leukemia (ALL) (19), gastric cancer
(20), colon adenocarcinoma (21), nasopharyngeal carcinoma (22) and colorectal cancer (23).
It has been proposed that the loss of normal FHIT
function may be involved in the pathogenesis of several human
leukemias, and the aberrant expression of FHIT is specific
and frequent in leukemia samples (24,25). The
frequent loss of FHIT expression in ALL suggests that
inactivating alterations at the FHIT locus may contribute to
the development of leukemias (26).
The present study aimed to assess promoter methylation status and
expression levels of the FHIT gene in childhood ALL.
Materials and methods
Patients
A case-control study was conducted, in which 100
children diagnosed with ALL (54 male, 46 female; age, 5.5±3.6
years) and 120 age and sex matched healthy controls (58 male, 62
female; age, 5.6±2.9 years) in Zahedan, Iran were enrolled. All
subjects were selected from the Ali-Ebne-Abitaleb Hospital, Zahedan
University of Medical Sciences (Zahedan, Iran) between February
2013 and May 2014. Details of the study design and enrolment
process have been previously described (27–29).
Analysis of DNA methylation in the FHIT promoter was
performed in 100 ALL cases and 120 healthy age and sex matched
cases. Analysis of FHIT mRNA expression levels was conducted
in 30 new cases of childhood ALL (20 males and 10 females; mean
age, 5.5±3.4 years; age range, 1–15 years) and 32 healthy age and
sex matched children (15 males and 17 females; mean age, 5.0±4.1
years; age range, 1–15 years). The subjects were selected from the
Ali-Ebne-Abitaleb Hospital, Zahedan University of Medical Sciences
(Zahedan, Iran) between February 2013 and May 2014. The local
ethics committee of Zahedan University of Medical Sciences
(Zahedan, Iran) approved the studies and written informed consent
was obtained from parents of cases and controls.
Promoter methylation
A methylation specific PCR (MSP) technique was used
for determination of promoter DNA methylation status. Whole blood
samples (2 ml) were collected in EDTA-containing tubes, and genomic
DNA was extracted using the salting out method as described
previously (30). DNA was modified by
sodium bisulfite using the CPGenome™ Direct Prep Bisulfite
Modification kit (Merck KGaA, Darmstadt, Germany). An MSP was
established for the evaluation of FHIT promoter methylation.
The primer sequences are listed in Table
I. To each 0.2 ml PCR reaction tube, 1 µl bisulfite-converted
DNA, 1 µl of forward and reverse primer (10 µM) and 17 µl dH2O were
added with HotStart PCR PreMix (AccuPower, Bionner Corp, Daejean,
South Korea). The PCR conditions were set as follows: 95°C for 5
min, 30 cycles of 95°C for 30 sec, 58°C for 30 sec, and 72°C for 30
sec, with a final extension step of 72°C for 5 min. The PCR product
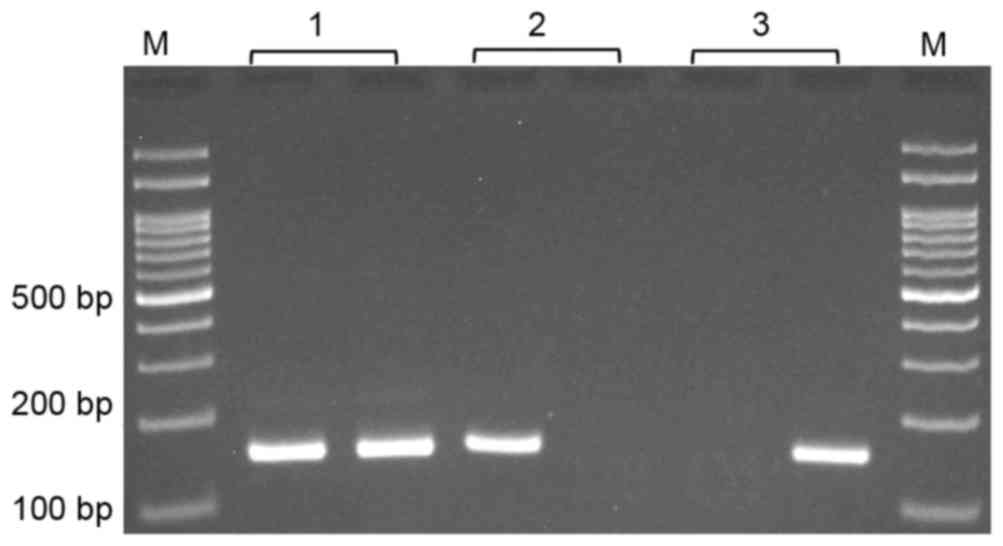
(623 bp) was used as a template for MSP. MSP was performed using
two pairs of primers; one pair specific for methylated and the
other for unmethylated template (Table
I). In each 0.2 ml PCR reaction tube, 0.5 µl nested PCR
product, 1 µl of each primer (10 µM) and 10 µl 2X Prime Taq Premix
(GeNet Bio, Daejean, South Korea) and 7 µl dH2O were added. MSP
conditions included an initial denaturation step at 95°C for 5 min,
followed by 30 cycles of 30 sec at 95°C, annealing at 58°C for 30
sec, and extension at 72°C for 30 sec, with a final extension step
at 72°C for 5 min. The methylated and unmethylated PCR products
were 157 and 159 bp, respectively (Fig.
1).
 | Table I.Primer sequences for MS-PCR and
RT-qPCR. |
Table I.
Primer sequences for MS-PCR and
RT-qPCR.
| Gene | Primers
(5′-3′) | Amplicon size
(bp) |
|---|
| Nested | F:
TTGATGGATTAAGTTAGGGATTGTAA | 623 |
|
| R:
CCCCTACCTTCCAAAATATTAACA |
|
| FHIT M | F:
TTTTCGTTTTTGTTTTTAGATAAGC | 157 |
|
| R:
AAAAATATACCCACTAAATAACCGC |
|
| FHIT U | F:
TGGTTTTTGTTTTTGTTTTTAGATAAGT | 159 |
|
| R:
AAAATATACCCACTAAATAACCACC |
|
| FHIT
RT-qPCR | F:
ACCTGCGTCCTGATGAAGTG | 144 |
|
| R:
CGTGAACGTGCTTCACAGTC |
|
| GAPDH
RT-qPCR | F:
GAAGGTGAAGGTCGGAGTC | 226 |
|
| R:
GAAGATGGTGATGGGATTTC |
|
Analysis of FHIT gene expression by
RT-qPCR in patients with ALL and healthy controls
Total mRNA extraction from fresh whole blood was
conducted using the commercially available kit, GeneJET RNA
Purification kit (Thermo Fisher Scientific, Inc., Waltham, MA, USA)
according to the manufacturer's protocol. cDNA synthesis was
performed using the AccuPower CycleScript RT PreMix kit (Bioneer
Corp., Daejeon, Korea) in a final volume of 20 µl according to the
manufacturer's protocol. The mRNA expression levels of FHIT
and an internal control gene, glyceraldehyde-3-phosphate
dehydrogenase (GAPDH), were detected by qPCR using SYBR
Green 2X RealQ Plus master mix (Amplicon; Bio, Korea; www.amplicon.com) on the ABI quantitative Real-Time
PCR system (Applied Biosystems, Thermo Fisher Scientific, Inc.).
All primer sequences are presented in Table I. To each 0.2 ml PCR reaction tube, 1
µl of cDNA, 1 µl of forward and reverse primers, 10 µl 2X RealQ
Plus Master Mix Green High Rox (Amplicon Bio, Korea) and 7 µl
dH2O was added. The PCR conditions were set as follows:
95°C for 6 min, 40 cycles of 95°C for 40 sec, 63°C for 40 sec, and
72°C for 35 sec, with a final extension step of 72°C for 10 min.
Results are presented as gene expression fold change of ALL samples
compared with controls, according to the 2−∆∆Cq method
(31).
Statistical analysis
Statistical analysis was performed using SPSS 20.0
software (IBM Corp., Armonk, NY, USA). Data were analyzed using an
independent sample t-test for continuous data and Fisher's exact
test or χ2 test for categorical data. The odds ratio
(OR) and 95% confidence intervals (CI) were calculated from
logistic regression analysis to identify the associations between
methylation status and ALL. P<0.05 was considered to indicate a
statistically significant difference.
Results
The case-control study was conducted on 100
childhood ALL cases (54 male, 46 female; age, 5.5±3.6 years) and
120 healthy children (58 male, 62 female; age, 5.6±2.9 years). No
significant differences were observed between groups regarding sex
and age (P=0.419 and 0.761, respectively). Table II presents the promoter DNA
methylation status of the FHIT gene in patients with ALL and
controls. The frequency distribution of non-methylation, partial
methylation and hypermethylation in cases and controls were 12.0,
65.0, and 23.0%, and 25.0, 62.5, and 12.5%, respectively.
Hypermethylation of the FHIT gene was significantly more
frequent in children with ALL than in healthy controls (OR=3.83,
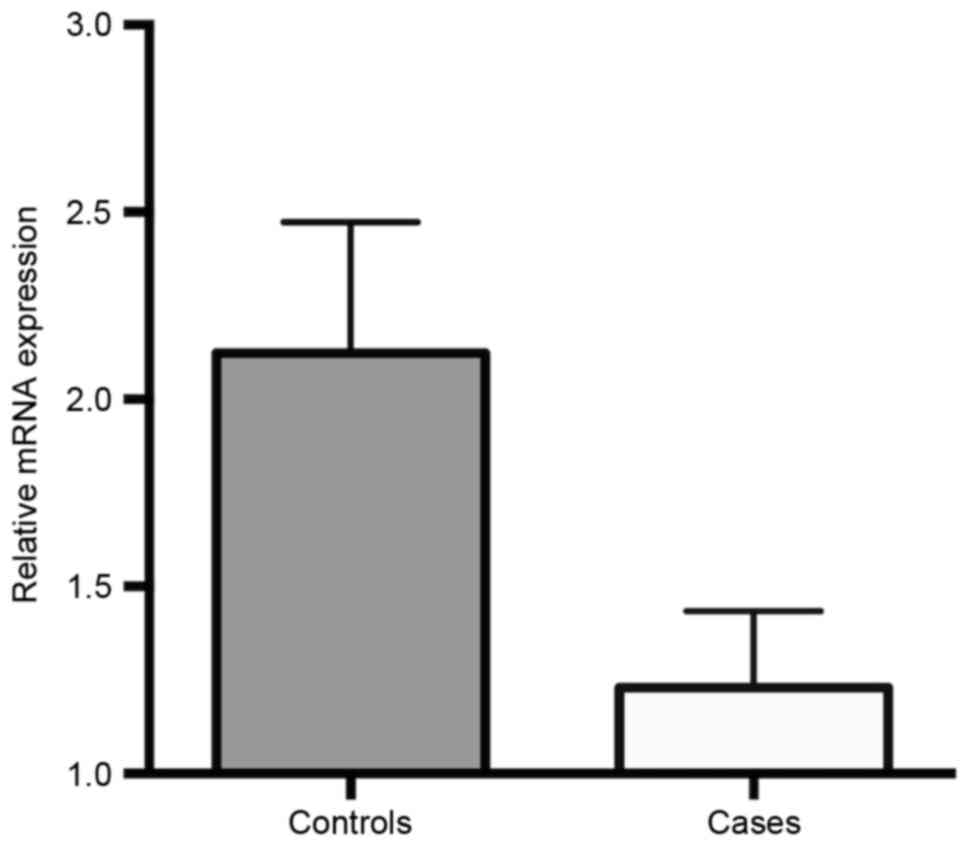
95% CI=1.51–9.75, P=0.007). FHIT mRNA expression levels in
ALL cases and controls are presented in Fig. 2. These results revealed that the gene
expression of FHIT was significantly downregulated in ALL
cases compared with that in healthy children (1.231±0.204 vs.
2.124±0.349; P=0.032).
| Table II.Promoter DNA methylation of the
FHIT gene in patients with ALL and controls. |
Table II.
Promoter DNA methylation of the
FHIT gene in patients with ALL and controls.
| Methylation
status | Cases, n (%) | Controls, n
(%) | OR (95% CI) | P-value |
|---|
| Absent | 12 (12.0) | 30 (25.0) | 1.00 |
|
| Partial | 65 (65.0) | 75 (62.5) | 2.17
(1.03–4.58) | 0.050a |
| Present | 23 (23.0) | 15 (12.5) | 3.83
(1.51–9.75) | 0.007a |
| Partial +
present | 88 (88.0) | 90 (75.0) | 2.44
(1.18–5.08 | 0.016a |
Discussion
FHIT is a candidate tumor suppressor gene in
multiple types of cancer, although the mechanism of tumor
suppression by FHIT is not fully understood. Several
investigations have revealed that FHIT overexpression leads to the
activation of the two main apoptotic pathways, the extrinsic
caspase-8 pathway and the intrinsic mitochondrial pathway (32–34). The
inactivation of programmed cell death may lead to tumorigenesis
(35).
In the present study, hypermethylation of the
FHIT gene was demonstrated to be significantly more frequent
in children with ALL than in healthy controls. Furthermore, the
gene expression of FHIT was significantly lower in patients
with childhood ALL compared with healthy controls. The results of
the present study indicated that epigenetic modifications and
altered gene expression of FHIT in childhood ALL, may
contribute to the development of this disease.
In agreement with results of the present study,
Malak et al (19) demonstrated
that the expression of FHIT was significantly lower in
childhood ALL compared with healthy children. In addition, Chen
et al (36) reported that mRNA
expression of FHIT was significantly lower and methylation
frequency of FHIT was significantly higher in ALL samples
compared with controls.
Previous studies have provided evidence that DNA
methylation is the most commonly detected alteration in adult ALL
(37–40). Genome-wide DNA methylation profiles
are commonly altered in pediatric ALL (41). It has been demonstrated that
hypermethylation of multiple genes may be involved in the relapse
of childhood ALL (42). Suppression
of FHIT may be involved in the development of rearranged acute
lymphoblastic leukemia, an aggressive type of ALL (43).
A meta-analysis performed by Wu et al
(13) indicated that FHIT
hypermethylation, which induces the inactivation of the FHIT
gene, is associated with an increased risk and adverse clinical
outcome of NSCLC. It has been demonstrated that FHIT
promoter hypermethylation is associated with the development of
breast cancer and certain poor prognostic features of the disease
(14). FHIT may be a potential drug
target for the development of demethylation treatment for patients
with breast cancer (44).
Furthermore, hypermethylation of the FHIT promoter was
previously demonstrated to be involved in the development and
transformation of fetal mesenchymal stem cells into F6 tumor cells
(45). In conclusion, the results of
the present study suggested that hypermethlation and mRNA
downregulation of FHIT gene may be involved in the
development of pediatric ALL. Further studies are required to
reveal the exact role of FHIT on ALL risk.
Acknowledgements
The present study was supported by a dissertation
grant (no. 6858) from the Zahedan University of Medical
Sciences.
References
|
1
|
Belson M, Kingsley B and Holmes A: Risk
factors for acute leukemia in children: A review. Environ Health
Perspect. 115:138–145. 2007. View
Article : Google Scholar : PubMed/NCBI
|
|
2
|
Ward E, DeSantis C, Robbins A, Kohler B
and Jemal A: Childhood and adolescent cancer statistics, 2014. CA
Cancer J Clin. 64:83–103. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Guo LM, Xi JS, Ma Y, Shao L, Nie CL and
Wang GJ: ARID5B gene rs10821936 polymorphism is associated with
childhood acute lymphoblastic leukemia: A meta-analysis based on
39,116 subjects. Tumour Biol. 35:709–713. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Ma Y, Sui Y, Wang L and Li H: Effect of
GSTM1 null genotype on risk of childhood acute leukemia: A
meta-analysis. Tumour Biol. 35:397–402. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Pekarsky Y, Zanesi N, Palamarchuk A,
Huebner K and Croce CM: FHIT: From gene discovery to cancer
treatment and prevention. Lancet Oncol. 3:748–754. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Ohta M, Inoue H, Cotticelli MG, Kastury K,
Baffa R, Palazzo J, Siprashvili Z, Mori M, McCue P, Druck T, et al:
The FHIT gene, spanning the chromosome 3p14.2 fragile site and
renal carcinoma-associated t (3;8) breakpoint, is abnormal in
digestive tract cancers. Cell. 84:587–597. 1996. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Brenner C, Bieganowski P, Pace HC and
Huebner K: The histidine triad superfamily of nucleotide-binding
proteins. J Cell Physiol. 181:179–187. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Siprashvili Z, Sozzi G, Barnes LD, McCue
P, Robinson AK, Eryomin V, Sard L, Tagliabue E, Greco A, Fusetti L,
et al: Replacement of Fhit in cancer cells suppresses
tumorigenicity. Proc Natl Acad Sci USA. 94:13771–13776. 1997.
View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Ji L, Fang B, Yen N, Fong K, Minna JD and
Roth JA: Induction of apoptosis and inhibition of tumorigenicity
and tumor growth by adenovirus vector-mediated fragile histidine
triad (FHIT) gene overexpression. Cancer Res. 59:3333–3339.
1999.PubMed/NCBI
|
|
10
|
Sard L, Accornero P, Tornielli S, Delia D,
Bunone G, Campiglio M, Colombo MP, Gramegna M, Croce CM, Pierotti
MA and Sozzi G: The tumor-suppressor gene FHIT is involved in the
regulation of apoptosis and in cell cycle control. Proc Natl Acad
Sci USA. 96:8489–8492. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Sevignani C, Calin GA, Cesari R, Sarti M,
Ishii H, Yendamuri S, Vecchione A, Trapasso F and Croce CM:
Restoration of fragile histidine triad (FHIT) expression induces
apoptosis and suppresses tumorigenicity in breast cancer cell
lines. Cancer Res. 63:1183–1187. 2003.PubMed/NCBI
|
|
12
|
Agrawal S, Unterberg M, Koschmieder S, zur
Stadt U, Brunnberg U, Verbeek W, Büchner T, Berdel WE, Serve H and
Müller-Tidow C: DNA methylation of tumor suppressor genes in
clinical remission predicts the relapse risk in acute myeloid
leukemia. Cancer Res. 67:1370–1377. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Wu X, Wu G, Yao X, Hou G and Jiang F: The
clinicopathological significance and ethnic difference of FHIT
hypermethylation in non-small-cell lung carcinoma: A meta-analysis
and literature review. Drug Des Devel Ther. 10:699–709.
2016.PubMed/NCBI
|
|
14
|
Zaki SM, Abdel-Azeez HA, El Nagar MR,
Metwally KAS and Ahmed MM: Analysis of FHIT gene methylation in
egyptian breast cancer women: Association with clinicopathological
features. Asian Pac J Cancer Prev. 16:1235–1239. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Liu L, Sun L, Li C, Li X, Zhang Y, Yu Y
and Xia W: Quantitative detection of methylation of FHIT and BRCA1
promoters in the serum of ductal breast cancer patients. Biomed
Mater Eng. 26 Suppl 1:S2217–S2222. 2015.PubMed/NCBI
|
|
16
|
Banzai C, Nishino K, Quan J, Yoshihara K,
Sekine M, Yahata T and Tanaka K: Gynecological Cancer Registry of
Niigata: Promoter methylation of DAPK1, FHIT, MGMT, and CDKN2A
genes in cervical carcinoma. Int J Clin Oncol. 19:127–132. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Zhang X, Li HM, Liu Z, Zhou G, Zhang Q,
Zhang T, Zhang J and Zhang C: Loss of heterozygosity and
methylation of multiple tumor suppressor genes on chromosome 3 in
hepatocellular carcinoma. J Gastroenterol. 48:132–143. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Yin DT, Wang L, Sun J, Yin F, Yan Q, Shen
R, He G and Gao JX: Association of the promoter methylation and
protein expression of Fragile Histidine Triad (FHIT) gene with the
progression of differentiated thyroid carcinoma. Int J Clin Exp
Pathol. 3:482–491. 2010.PubMed/NCBI
|
|
19
|
Malak CA, Elghanam DM and Elbossaty WF:
FHIT gene expression in acute lymphoblastic leukemia and its
clinical significance. Asian Pac J Cancer Prev. 16:8197–8201. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Liao J, Wen S, Cao L, Zhou Y and Feng Z:
Effect of eradication of Helicobacter pylori on expression levels
of FHIT, IL-8 and P73 in gastric mucosa of first-degree relatives
of gastric cancer patients. PLoS One. 10:e01245762015. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Kapitanović S, Čačev T, Lončar B, Ivković
Catela T, Križanac Š and Pavelić K: Reduced FHIT expression is
associated with tumor progression in sporadic colon adenocarcinoma.
Exp Mol Pathol. 96:92–97. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Chen X, Li P, Yang Z and Mo WN: Expression
of fragile histidine triad (FHIT) and WW-domain oxidoreductase gene
(WWOX) in nasopharyngeal carcinoma. Asian Pac J Cancer Prev.
14:165–171. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Al-Temaimi RA, Jacob S, Al-Ali W, Thomas
DA and Al-Mulla F: Reduced FHIT expression is associated with
mismatch repair deficient and high CpG island methylator phenotype
colorectal cancer. J Histochem Cytochem. 61:627–638. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Sugimoto K, Yamada K, Miyagawa K, Hirai H
and Oshimi K: Decreased or altered expression of the FHIT gene in
human leukemias. Stem Cells. 15:223–228. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Iwai T, Yokota S, Nakao M, Nakazawa N,
Taniwaki M, Kimura T, Sonoda Y, Kaneko H, Okuda T, Azuma H, et al:
Frequent aberration of FHIT gene expression in acute leukemias.
Cancer Res. 58:5182–5187. 1998.PubMed/NCBI
|
|
26
|
Hallas C, Albitar M, Letofsky J, Keating
MJ, Huebner K and Croce CM: Loss of FHIT expression in acute
lymphoblastic leukemia. Clin Cancer Res. 5:2409–2414.
1999.PubMed/NCBI
|
|
27
|
Hashemi M, Sheybani-Nasab M, Naderi M,
Roodbari F and Taheri M: Association of functional polymorphism at
the miR-502-binding site in the 3′ untranslated region of the SETD8
gene with risk of childhood acute lymphoblastic leukemia, a
preliminary report. Tumour Biol. 35:10375–10379. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Hasani SS, Hashemi M, Eskandari-Nasab E,
Naderi M, Omrani M and Sheybani-Nasab M: A functional polymorphism
in the miR-146a gene is associated with the risk of childhood acute
lymphoblastic leukemia: A preliminary report. Tumour Biol.
35:219–225. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Hashemi M, Ebrahimi M, Amininia S, Naderi
M, Eskanadri-Nasab E and Taheri M: Evaluation of rs3102735 and
rs2073617 osteoprotegerin gene polymorphisms and the risk of
childhood acute lymphocytic leukemia in Zahedan Southeast Iran. Int
J Hematol Oncol Stem Cell Res. 8:39–44. 2014.PubMed/NCBI
|
|
30
|
Hashemi M, Moazeni-Roodi AK, Fazaeli A,
Sandoughi M, Bardestani GR, Kordi-Tamandani DM and Ghavami S: Lack
of association between paraoxonase-1 Q192R polymorphism and
rheumatoid arthritis in southeast Iran. Genet Mol Res. 9:333–339.
2010. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Livak KJ and Schmittgen TD: Analysis of
relative gene expression data using real-time quantitative PCR and
the 2(−Delta Delta C(T)) method. Methods. 25:402–408. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Ishii H, Dumon KR, Vecchione A, Trapasso
F, Mimori K, Alder H, Mori M, Sozzi G, Baffa R, Huebner K and Croce
CM: Effect of adenoviral transduction of the fragile histidine
triad gene into esophageal cancer cells. Cancer Res. 61:1578–1584.
2001.PubMed/NCBI
|
|
33
|
Roz L, Gramegna M, Ishii H, Croce CM and
Sozzi G: Restoration of fragile histidine triad (FHIT) expression
induces apoptosis and suppresses tumorigenicity in lung and
cervical cancer cell lines. Proc Natl Acad Sci USA. 99:3615–3620.
2002. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Dumon KR, Ishii H, Vecchione A, Trapasso
F, Baldassarre G, Chakrani F, Druck T, Rosato EF, Williams NN,
Baffa R, et al: Fragile histidine triad expression delays tumor
development and induces apoptosis in human pancreatic cancer.
Cancer Res. 61:4827–4836. 2001.PubMed/NCBI
|
|
35
|
Ghavami S, Hashemi M, Ande SR, Yeganeh B,
Xiao W, Eshraghi M, Bus CJ, Kadkhoda K, Wiechec E, Halayko AJ and
Los M: Apoptosis and cancer: Mutations within caspase genes. J Med
Genet. 46:497–510. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Chen X, Zhang H, Li P, Yang Z, Qin L and
Mo W: Gene expression of WWOX, FHIT and p73 in acute lymphoblastic
leukemia. Oncol Lett. 6:963–969. 2013.PubMed/NCBI
|
|
37
|
Garcia-Manero G, Daniel J, Smith TL,
Kornblau SM, Lee MS, Kantarjian HM and Issa JP: DNA methylation of
multiple promoter-associated CpG islands in adult acute lymphocytic
leukemia. Clin Cancer Res. 8:2217–2224. 2002.PubMed/NCBI
|
|
38
|
Yang Y, Takeuchi S, Hofmann WK, Ikezoe T,
van Dongen JJ, Szczepański T, Bartram CR, Yoshino N, Taguchi H and
Koeffler HP: Aberrant methylation in promoter-associated CpG
islands of multiple genes in acute lymphoblastic leukemia. Leuk
Res. 30:98–102. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Garcia-Manero G, Yang H, Kuang SQ, O'Brien
S, Thomas D and Kantarjian H: Epigenetics of acute lymphocytic
leukemia. Semin Hematol. 46:24–32. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Paulsson K, An Q, Moorman AV, Parker H,
Molloy G, Davies T, Griffiths M, Ross FM, Irving J, Harrison CJ, et
al: Methylation of tumour suppressor gene promoters in the presence
and absence of transcriptional silencing in high hyperdiploid acute
lymphoblastic leukaemia. Br J Haematol. 144:838–847. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Chatterton Z, Morenos L, Mechinaud F,
Ashley DM, Craig JM, Sexton-Oates A, Halemba MS, Parkinson-Bates M,
Ng J, Morrison D, et al: Epigenetic deregulation in pediatric acute
lymphoblastic leukemia. Epigenetics. 9:459–467. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Matsushita C, Yang Y, Takeuchi S,
Matsushita M, Van Dongen JJ, Szczepanski T, Bartram CR, Seo H,
Koeffler HP and Taguchi H: Aberrant methylation in
promoter-associated CpG islands of multiple genes in relapsed
childhood acute lymphoblastic leukemia. Oncol Rep. 12:97–99.
2004.PubMed/NCBI
|
|
43
|
Stam RW, den Boer ML, Passier MM,
Janka-Schaub GE, Sallan SE, Armstrong SA and Pieters R: Silencing
of the tumor suppressor gene FHIT is highly characteristic for MLL
gene rearranged infant acute lymphoblastic leukemia. Leukemia.
20:264–271. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Su Y, Wang X, Li J, Xu J and Xu L: The
clinicopathological significance and drug target potential of FHIT
in breast cancer, a meta-analysis and literature review. Drug Des
Devel Ther. 9:5439–5445. 2015.PubMed/NCBI
|
|
45
|
Xu XJ, Gao S, Wang M, Qian H, Gu GY, Zhang
K and Xu WR: Methylation status of the gene in the transformed
human mesenchymal F6 stem cell line. Oncol Lett. 9:2661–2666.
2015.PubMed/NCBI
|