Introduction
Human malignant brain tumors, glioblastomas as well
as brain metastasis and anaplastic meningiomas, are highly
vascularized tumors (1–4). Therefore, the tumor vasculature is a
reasonable therapeutic target. To date, the VEGF neutralizing
monoclonal antibody bevacizumab is the only FDA approved
antiangiogenic drug for the treatment of recurrent glioblastoma
(5,6).
However, bevacizumab failed to prolong overall survival (OS) in two
phase III trials for newly diagnosed glioblastoma and in
glioblastoma at first recurrence (7,8). Beyond
bevacizumab there has been a wealth of antiangiogenic compounds in
clinical trials interfering with different pathways of angiogenesis
(cilengitide, cediranib, enzastaurin, etc.) (5,9–12). ASA404 belongs to the class of vascular
disrupting agents (VDA) and showed encouraging preclinical activity
in a number of solid tumor models (13–20).
Treatment with VDAs usually results in a rapid shut down of blood
flow, resulting in extensive tumor necrosis without affecting
normal blood vessels (21). In
addition, ASA404 is able to induce an anti-tumor immunity in
vivo (18,22–25).
Several studies showed that stimulator of interferon gene (STING)
is the molecular target of ASA404, mediating the increase of
inflammatory cytokines (23,24,26–29).
Moreover, it has been shown that only murine STING but not human
STING has the ability to sense ASA404 (23,24,26,27,29–30).
This might explain why ASA404 has failed in a larger clinical trial
(31,32). Further, tumor associated macrophages
seem to be the cellular mediator of ASA404 activity (22,33).
Here we report on our findings on the treatment of
several subcutaneous and intracranial brain tumor models with
ASA404, including colon carcinoma, malignant gliomas and
meningioma.
Materials and methods
Cell lines
The malignant human glioma cell lines (U-87, LN-229,
U-251, LN-308) were kindly provided by Professor N. de Tribolet
(Lausanne, Switzerland). Tu-2449 is a glioma cell line derived from
a spontaneous tumor in GFAP-v-src-transgenic mice and was provided
by J. Weissenberger (Frankfurt, Germany) (34). HT-29 colon carcinoma cells were
purchased from the American Type Culture Collection (ATCC;
Manassas, VA, USA). The malignant meningioma cell line IOMM-Lee was
kindly provided by David H. Gutmann (Department of Neurology,
Washington University School of Medicine, St. Louis, MO, USA)
(35–39). Mouse brain endothelial cells bEnd.3
immortalized with the polyoma virus middle T oncogene were kindly
provided by Werner Risau (Max-Planck Institute for Physiological
and Clinical Research, Bad Nauheim, Germany) (40–42). All
cells were maintained in DMEM containing 10% fetal calf serum, 2 mM
glutamine and penicillin (100 IU/ml)/streptomycin (100 µg/ml)
(43).
Reagents
ASA404 (DMXAA, vadimezan) was provided as a dry
powder by Novartis (Nürnberg, Germany). ASA404 was dissolved in 50
mM Tris buffer at a concentration of 10 mg/ml and the pH was
adjusted to 7.8–8.2. For the used concentrations ASA404 stock
solution was diluted with PBS.
Cell viability and cell death
assays
For cytotoxicity assays, the cells were seeded at a
density of 104 cells/well in 96-well plates, adhered for
24 h and then exposed to ASA404 for 72 h in medium including 5%
fetal calf serum. Cell density was assessed by crystal violet
staining.
Animal models
The animal studies in this study have been approved
by our local authority (Regierungspräsidium, Darmstadt, Germany).
Experiments have been performed in accordance with European
guidelines on animal experiments and the ARRIVE guidelines.
For generation of subcutaneous tumors
3×106 tumor cells in 100 µl PBS were injected
subcutaneously in the lower back sides of 6 weeks old female
athymic nude mice (Foxn1nu, Harlan, Indianapolis, IN, USA). Tumor
size was evaluated twice per week using a manual caliper. Tumor
volume was estimated by multiplying the three dimensions of the
tumor (x*y*z) and dividing the result by 2. The tumors were allowed
to grow to a diameter of 10 mm before randomized treatment started.
Mice were treated with a single intraperitoneal dose of ASA404 with
25 mg/kg body weight or with PBS. 24 h after treatment mice were
sacrificed. The subcutaneous tumors were removed for further
histopathologic analyses. For longitudinal tumor volumetric
measurements, mice were treated twice per week.
For orthotopic tumors, 1×105 cells (U-87
and U-251) in 4 µl PBS were injected stereotactically into the
right striatum. Animals were observed daily for weight loss or
neurological deterioration. At first symptoms three mice were
treated with ASA404 (25 mg/kg) and three mice with PBS. After 24 h
mice were sacrificed and brains removed for further histologic
analyses. For evaluation of symptom-free survival mice were
continuously treated with ASA404 (25 mg/kg) or PBS twice a week
starting at day 7 after tumor implantation. When mice developed
neurologic symptoms or showed a weight loss of more than 10% they
were sacrificed.
As a syngeneic glioma model Tu-2449 cells were
injected into the right striatum of B6C3F1 hybrid mice (Taconic
Farms, Cologne, Germany). For a brain metastasis model we
stereotactically injected HT-29 cells to the right striatum and
treated the animals with ASA404 at first symptoms. For a malignant
meningioma model, orthotopic, subarachnoidal tumor cell inoculation
with IOMM-Lee cells was done as previously described (36).
Histology/Immunohistochemistry
Subcutaneous tumors and mouse brains were
formalin-fixed and paraffin-embedded. Thick sections (8 µm) were
cut and deparaffination procedures were performed according to
standard protocols. Hematoxylin and eosin (H&E) stainings were
analyzed by experienced neuropathologists (P.N.H, M.M. and C.M.).
Immunohistochemistry was carried out using a monoclonal mouse
antibody against the human Ki67-antigen (clone MIB-1; Dako,
Glostrup, Denmark), against cleaved caspase-3 (Asp175, 5A1E, #9664;
Cell Signaling, Leiden, The Netherlands), a polyclonal antibody
against human and rodent STING (PA5-23381; Thermo Fisher, Dreieich,
Germany) and a rabbit antibody against Iba1 (Wako Chemicals, Neuss,
Germany) on the Ventana Discovery IHC system (Ventana, Strasbourg,
France). Iba1 staining was performed using the Leica BOND III
automated IHC system (Leica Biosystems, Nussloch, Germany). Only
nuclear Ki67/MIB-1 staining was counted as positive. Ten high-power
fields (HPF) were evaluated for the percentage of Ki67/MIB-1
positive cells.
Results
ASA404 shows low cytotoxicity in
vitro
As shown in Fig. 1
ASA404 results in a reduced cell density after 72 h of treatment.
This effect is limited to higher concentrations of ASA404 (≥0.1
mg/ml). No relevant differences between effects on the tumor cell
lines (U-87, LN-229, HT-29) and a brain endothelial cell line
(bEnd.3) were observed.
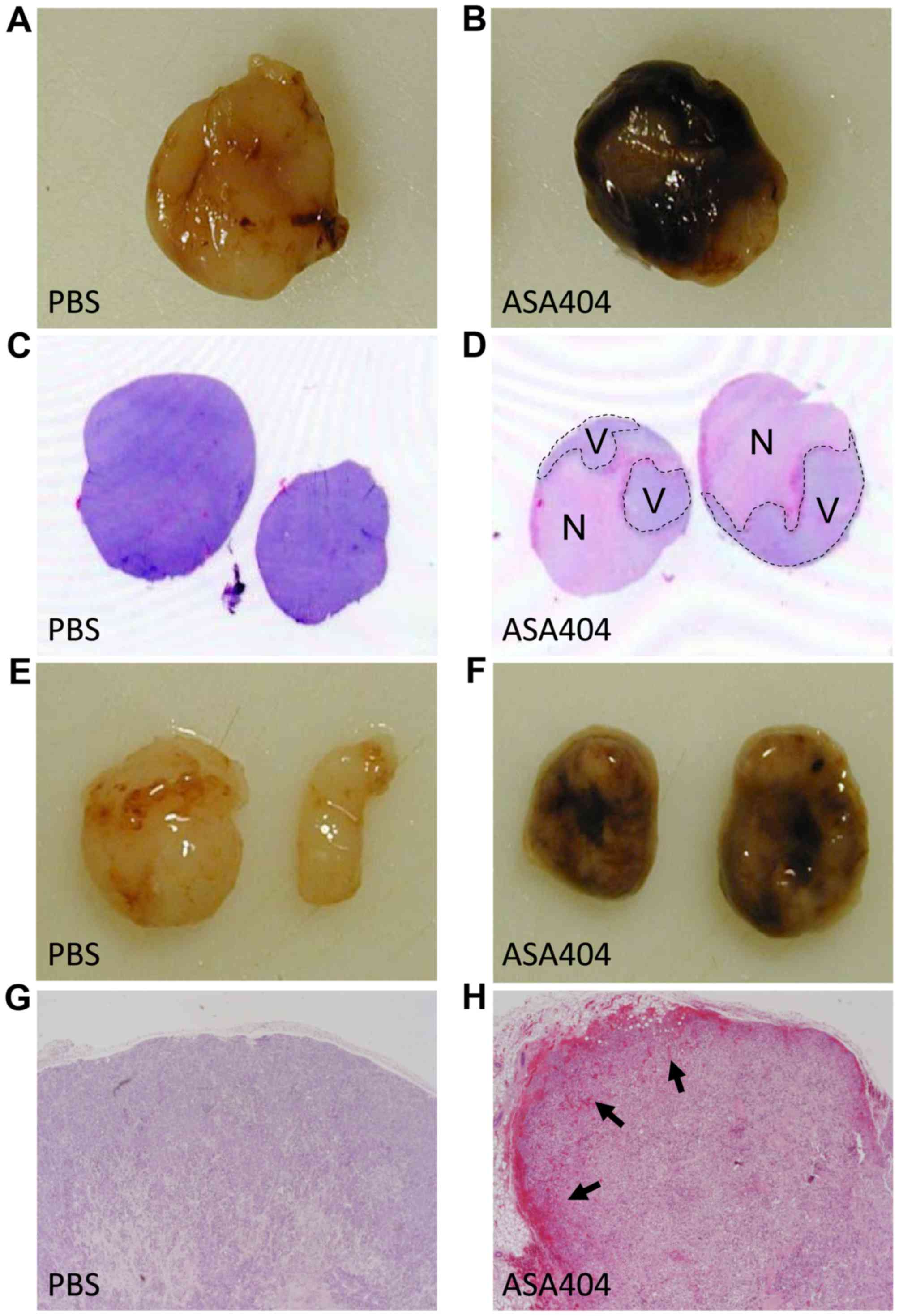
ASA404 induces large necrosis and
hemorrhage in subcutaneous tumors
Acute effects of ASA404 were evaluated in
subcutaneous tumor models. Tumors were excised 24 h after a single,
intraperitoneal treatment with ASA404 (25 mg/kg) or PBS as control.
All examined tumors showed large areas of necrotic tissue and
hemorrhages shortly after treatment. Fig.
2 shows macroscopic and microscopic effects in U-87 and HT-29
tumors. Similar results have been observed in LN-229, U-251 and
LN-308 tumors (data not shown).
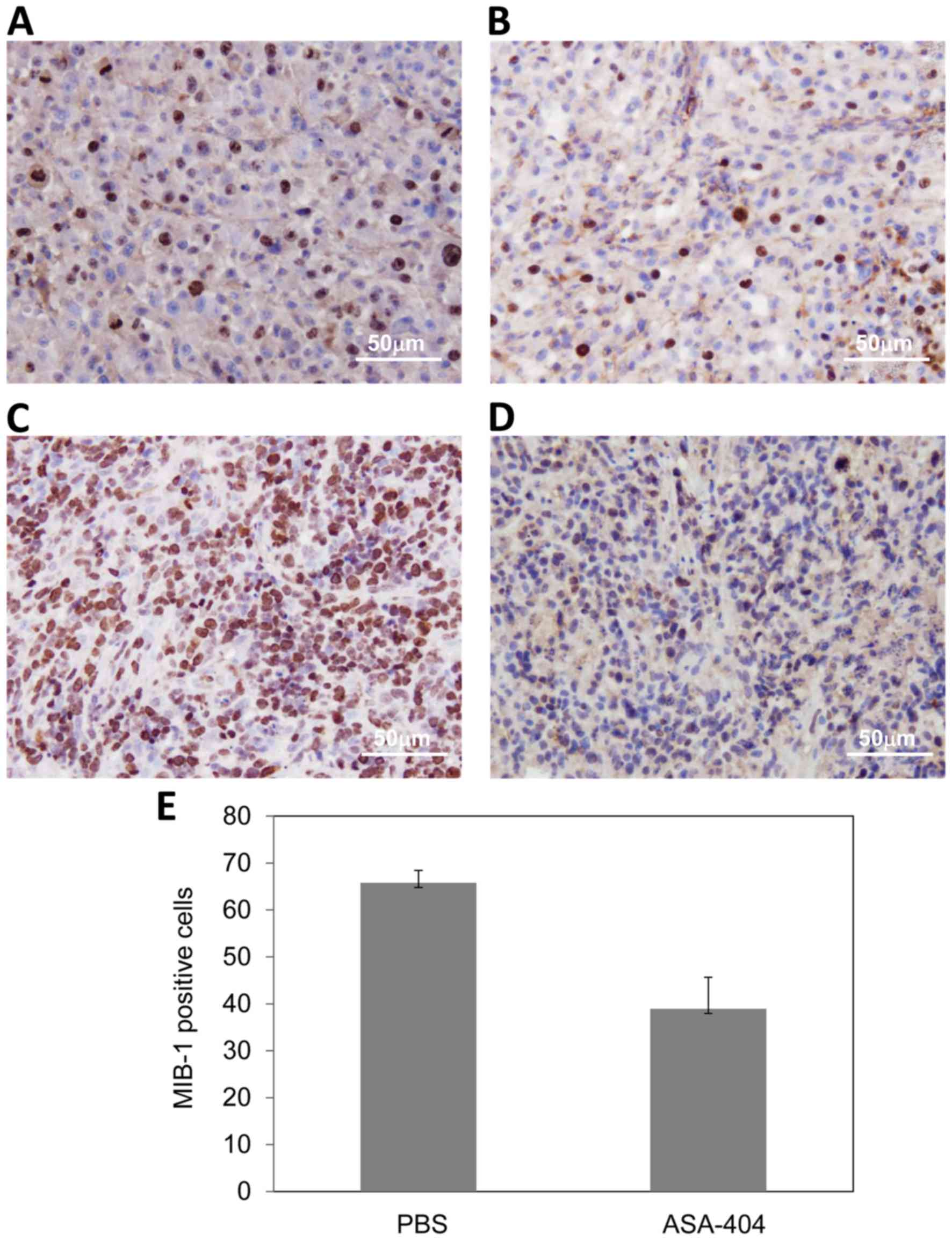
Decreased proliferation after
treatment with ASA404
We further analyzed the expression of the
proliferation marker Ki67/MIB-1 in subcutaneous tumors treated with
ASA404. As expected, Ki67/MIB-1 expression was not evaluable in
necrotic tumor areas. Interestingly, Ki67/MIB-1 expression was
markedly reduced in vital areas of the ASA404 treated tumors
compared to tumors treated with PBS. As shown in Fig. 3, immunohistochemical staining for
Ki67/MIB-1 in U-87 tumors treated with PBS (Fig. 3A) and treated with ASA404 (Fig. 3B). Fig. 3C
and D show LN-308 tumors treated with PBS (Fig. 3C) and ASA404 (Fig. 3D). In LN-308 tumors we quantified the
Ki67/MIB-1 expression by assessing the percentage of tumor cells
that show nuclear expression of Ki67/MIB-1.
ASA404 induces cleavage of caspase-3
in subcutaneous tumors
As we found apoptotic figures in the necrotic areas
of all subcutaneous tumors we analyzed the tumors for expression of
cleaved caspase-3, an indicator for apoptosis. As shown in Fig. 4, immunohistochemistry for cleaved
caspase-3 in subcutaneous U-87 tumors. The large necrotic areas on
the H&E slides (Fig. 4A and C)
show strong expression of cleaved caspase-3 (Fig. 4B and D). In vital tumor areas of
ASA404 treated tumors and in tumors treated with PBS cleaved
caspase-3 is absent.
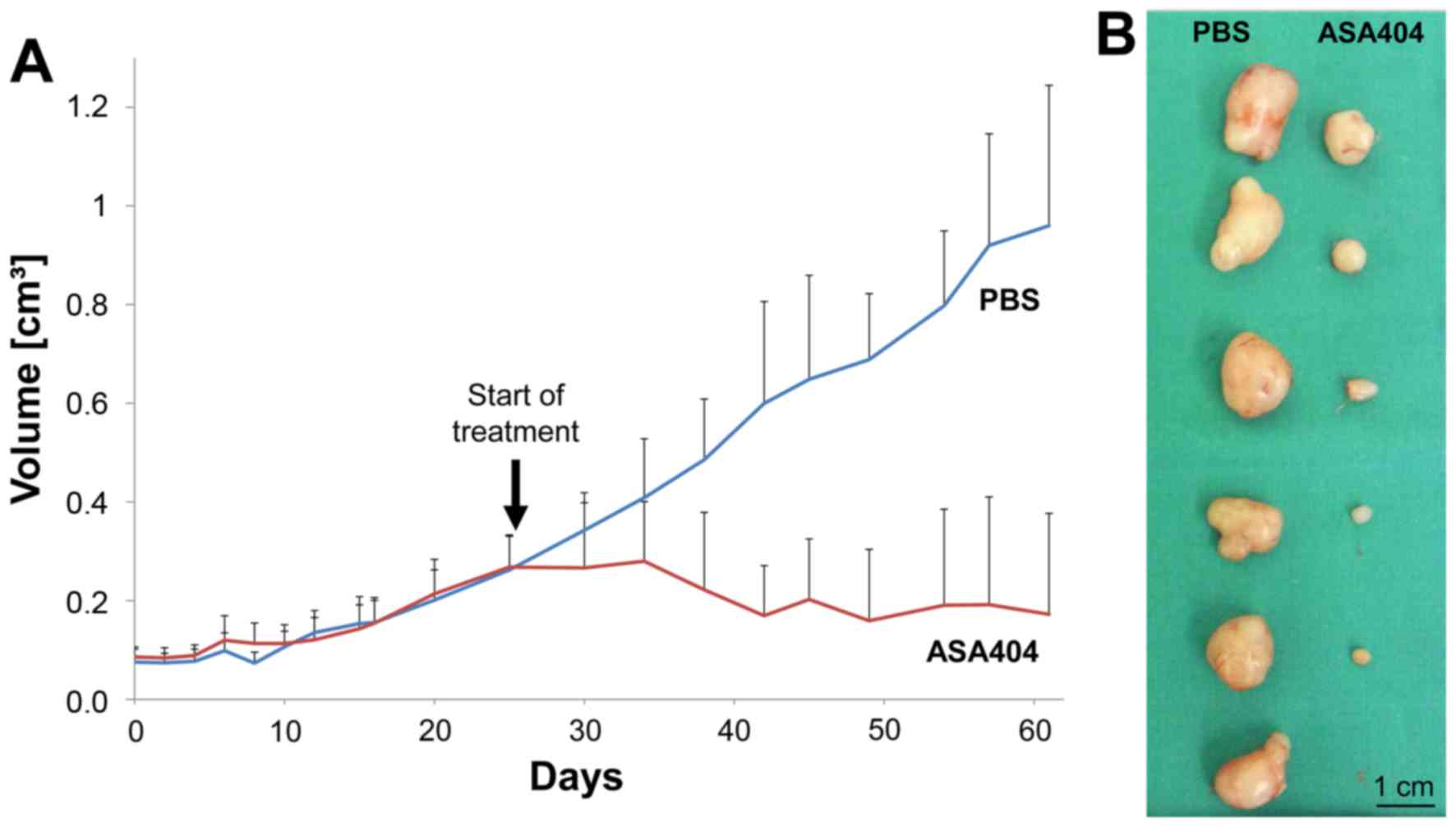
ASA404 dramatically reduces tumor
growth in a subcutaneous glioma model
To evaluate long-term effects of ASA404 on tumor
growth we continuously treated mice with subcutaneous LN-229
tumors. After all tumors reached a sufficient size (diameter 5 to
10 mm) animals were randomized to receive PBS or ASA404 (25 mg/kg)
twice per week. Treatment started on day 25. Animals treated with
PBS showed the expected increase in tumor volume (Fig. 5A, blue line), while in animals treated
with ASA404 tumor size decreased (Fig.
5A, red line). After 60 days the study was stopped and tumors
excised. The mean tumor volume of PBS treated mice reached 0.96
cm3 (Fig. 5A and B, left
column), while tumor volume in ASA404 treated mice reached 0.17
cm3 (Fig. 5A and B, right
column).
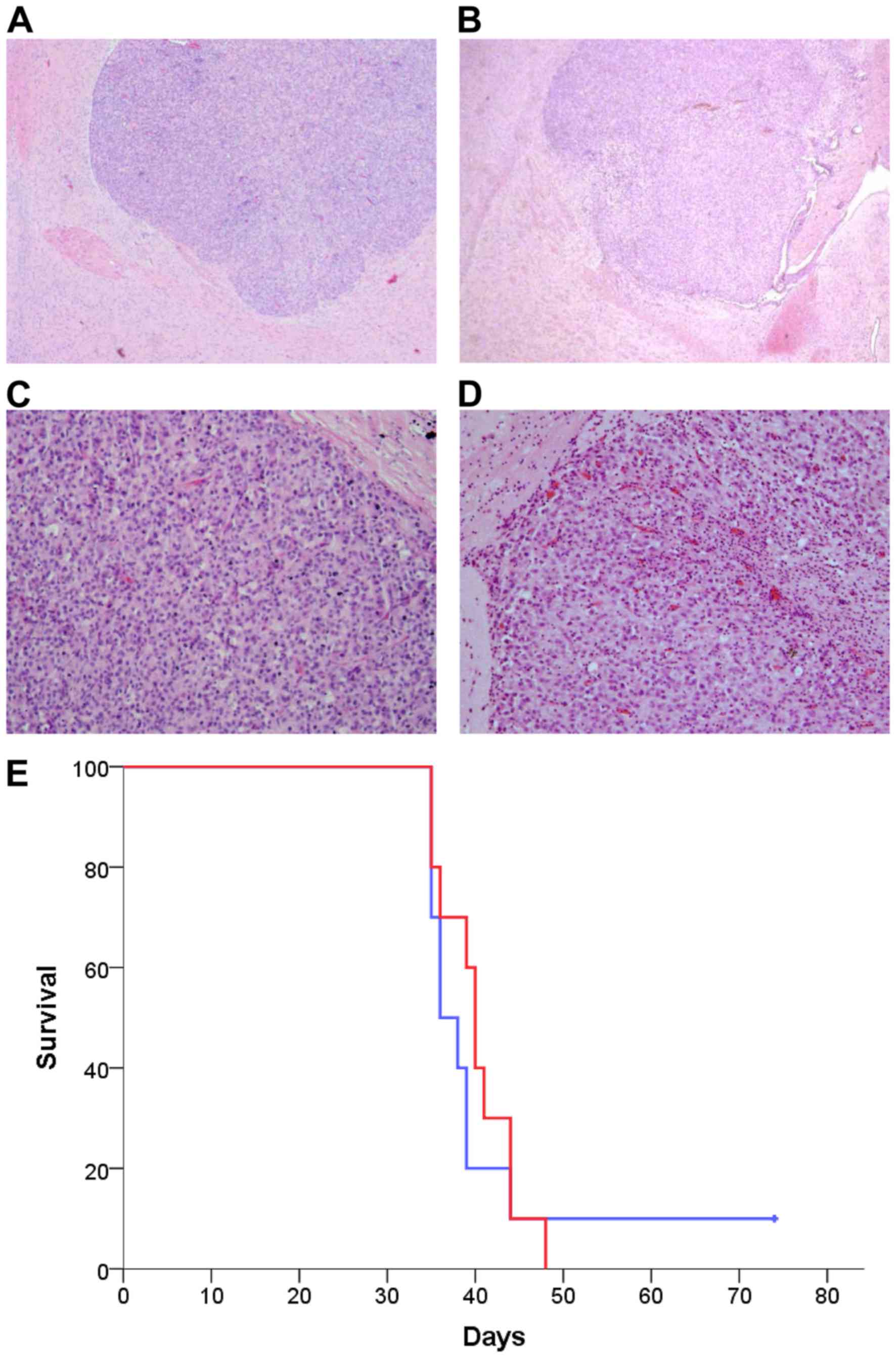
ASA404 has no activity in orthotopic
glioma models
U-87 cells were implanted stereotactically in
athymic nude mice. When the first mouse showed symptoms all animals
were randomized to receive either PBS (Fig. 6A and C) or ASA404 (25 mg/kg, Fig. 6B and D) intraperitoneally. 24 h after
treatment mice were sacrificed and brains excised. We did not find
relevant differences on H&E slides between PBS and ASA404
treated tumors (Fig. 6A-D).
Especially, we did not find necrotic areas or an increase in
intratumoral hemorrhage. Identical results were found for U-251 and
Tu-2449 tumors.
Further, we evaluated the influence of ASA404 on
symptom-free survival. Athymic nude mice with U-87 tumors were
continuously treated with PBS or ASA404 twice a week starting at
day 7 after tumor implantation. Median survival for PBS was 36 days
(Fig. 6E, blue line) and for ASA404
40 days (Fig. 6E, red line).
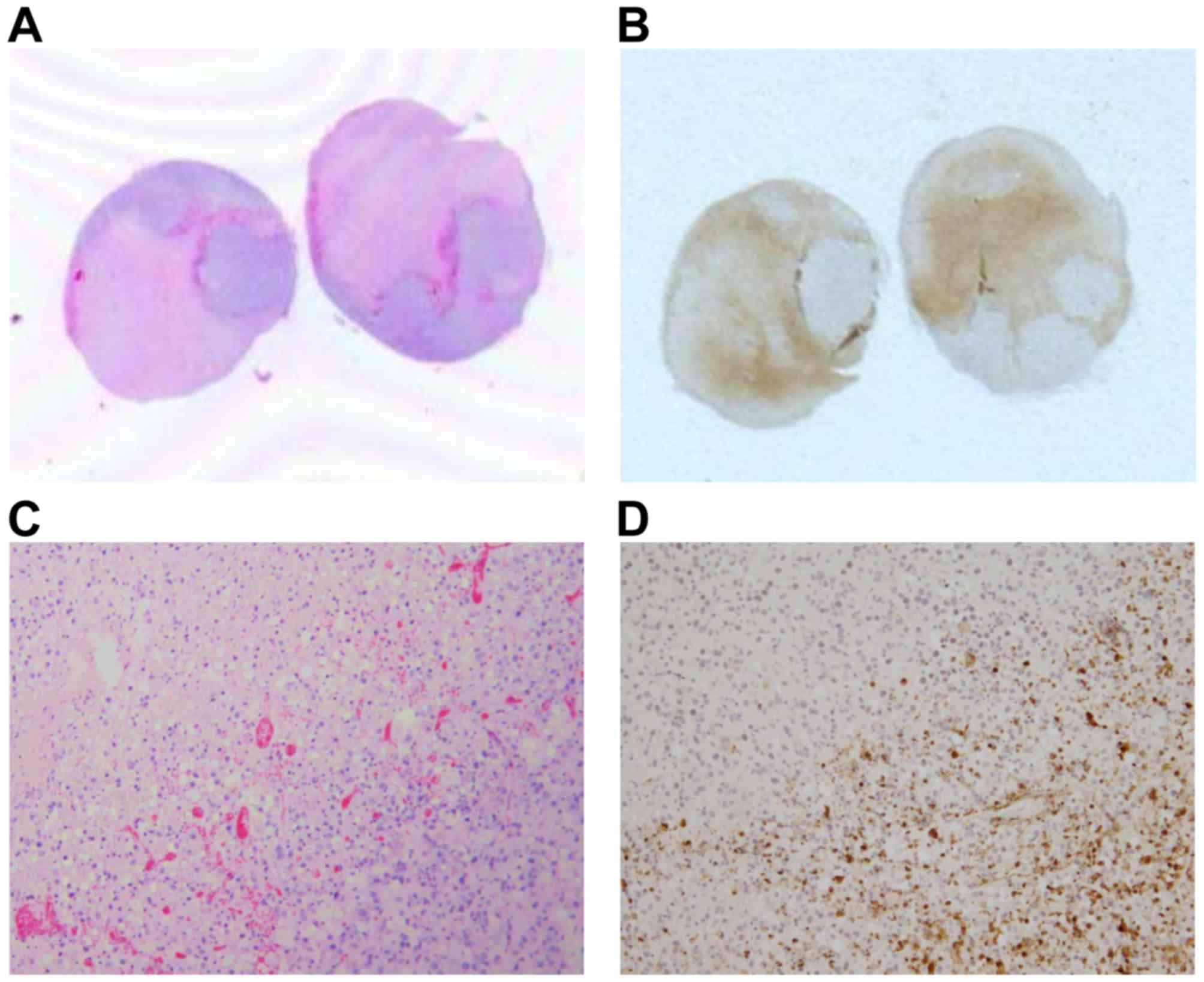
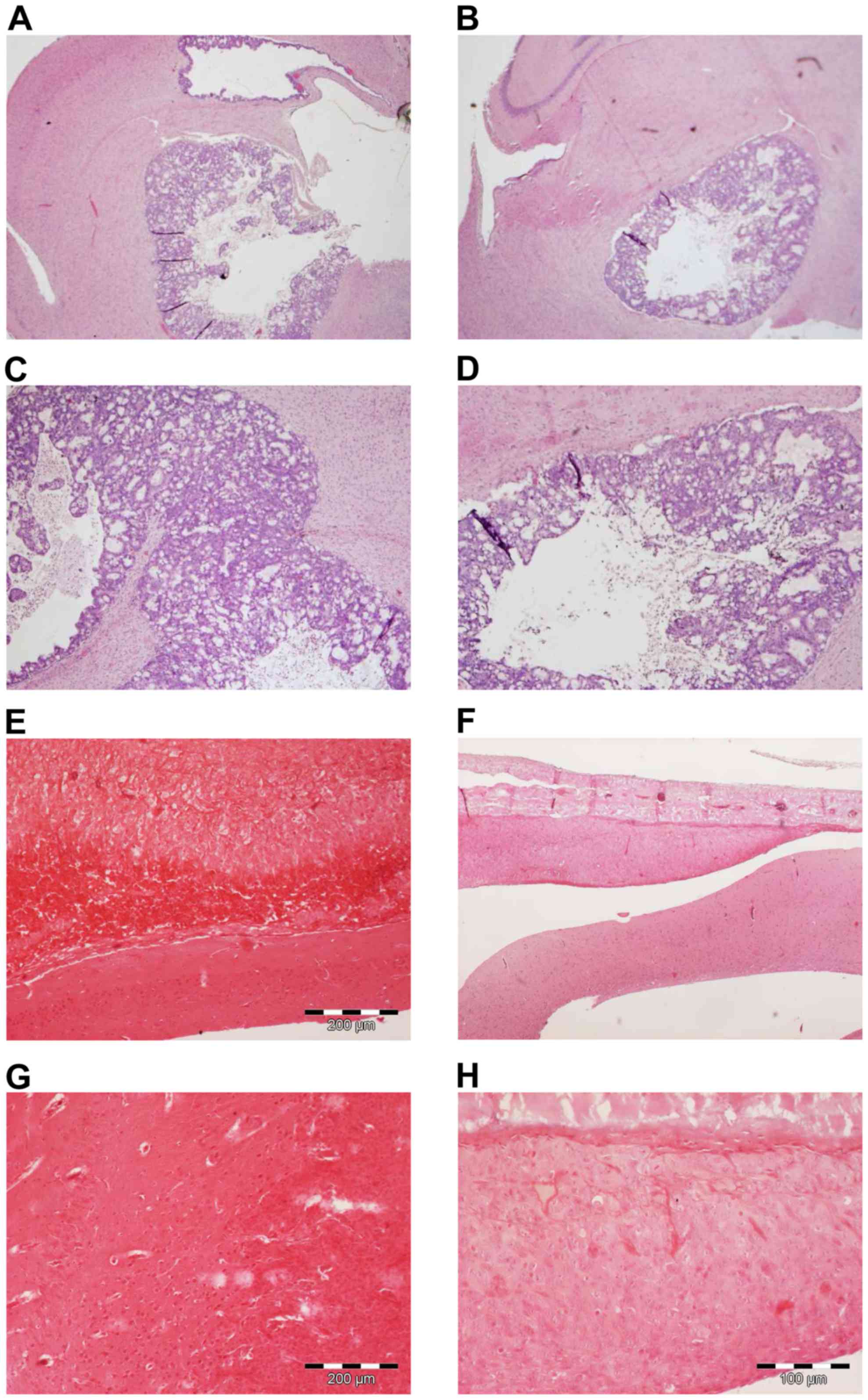
ASA404 has no activity in models for
CNS metastasis and malignant meningioma
To further evaluate the brain activity of ASA404 we
applied an animal model for CNS metastasis using the colon
carcinoma cell line HT-29 injected to the right striatum. As a
meningioma model the malignant meningioma cell line IOMM-Lee was
inoculated into the subarachnoid space. Mice were sacrificed 24 h
after treatment with PBS or ASA404 (25 mg/kg) and brains/tumors
excised for histological analyses (Fig.
7). As shown in Fig. 7A and C
(PBS treated mice) HT-29 cells formed large and necrotic tumors.
After treatment with ASA404 (Fig. 7B and
D) viability was not reduced and we did not see additional
necrotic areas, suggesting that ASA404 had no activity in this
model. No obvious activity of ASA404 has been detected in the
malignant meningioma model (Fig. 7E and
G shows PBS treatment, Fig. 7F and
H shows ASA404 treatment).
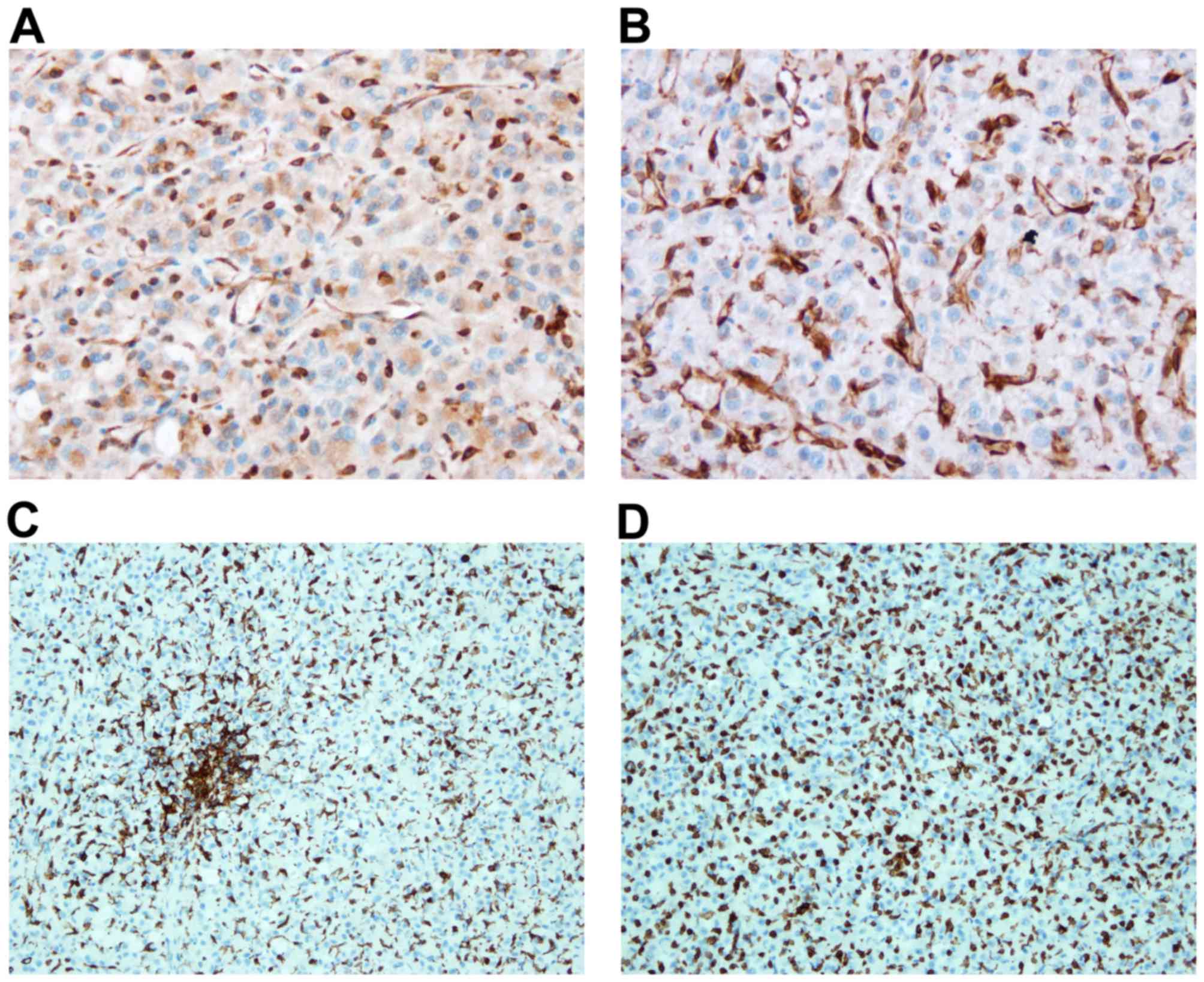
STING expression and iba1 positive
cells in subcutaneous and intracranial U-87 tumors
As STING is the molecular target of ASA404 in a
murine setting we examined tumor tissue for STING expression using
a monoclonal antibody recognizing both human and murine STING. As
shown in Fig. 8 STING expression in a
subcutaneous (Fig. 8A) and an
intracranial (Fig. 8B) U-87 tumor. We
did not find a clear difference in STING expression explaining the
differential activity of ASA404. Notably, STING expression was more
focused in the intracranial tumor, while most cells in the
subcutaneous tumor showed a more widespread, cytosolic STING
expression.
As macrophages in the tumor microenvironment are
recognized as the mediator cells of STING activity we used iba1
immunohistochemistry to stain for macrophages and microglia. Again
no relevant differences were observed between subcutaneous
(Fig. 8C) and intracranial (Fig. 8D) tumors. We found a high number of
iba1 positive cells in both, subcutaneous and intracranial
tumors.
Discussion
More efficacious therapies are desperately needed
for patients with malignant brain tumors, in particular for
glioblastoma. One new approach is using VDA. ASA404, or
DMXAA/vadimezan, is a vascular disrupting agent that has shown
preclinical activity in several different tumor models (17–20).
Earlier phase clinical trials showed promising results, but a phase
III trial in non-small cell lung cancer was negative (31,32).
Preclinical studies on the efficacy of ASA404 in brain tumors are
rare, clinical trials do not exist (20,44).
In the current study we can show that ASA404 has
moderate to low in vitro activity in human glioma cell lines
(U-87, LN-229), a colon carcinoma cell line (HT-29) and a brain
endothelial cell line (bEnd.3). In contrast, ASA404 has dramatic
effects in several subcutaneous glioma models (U-87, LN-229, U-251
and LN-308) and a colon carcinoma model (HT-29). Treatment with
ASA404 resulted in extensive necrosis and hemorrhages within a few
hours. When applied continuously ASA404 lead to a marked reduction
of tumor growth. Unfortunately, these impressive results did not
translate to the orthotopic, intracerebral glioma models (U-87,
U-251). To evaluate whether this could be a cell type specific
issue, we evaluated ASA404 in a murine, syngeneic glioma model
(Tu-2449), a model for CNS metastasis (HT-29) and a model for
malignant meningioma (IOMM-Lee). Again, ASA404 was not able to
induce the strong effects seen in the subcutaneous models. In
summary, we report on diverging effects in subcutaneous and
intracranial tumor models.
In general, this once again illustrates that
subcutaneous animal models are usually futile for brain tumor
research and should be limited to specific mechanistic issues.
Several explanations are conceivable for our
diverging effects. First, it has been shown that ASA404 does not
cross the blood-brain-barrier to a relevant degree in a syngeneic
glioma mouse model using the GL261 glioma cell line and C57BL/6
mice (44). At a dose of 25 mg/kg
ASA404 reached a Cmax of 474 µmol/l in the plasma of
tumor-bearing mice. In contrast, Cmax in the brain of
tumor-bearing mice only reached 6.9 µmol/l. For this
pharmacokinetic analysis whole brain lysates but not brain tumor
lysates were used (44). Further, the
GL261 tumors only show minor contrast enhancement on MRI scans
suggesting only a mild disruption of the blood-brain-barrier
(45). In other tumor models and the
clinical situation the blood-brain-barrier in malignant brain
tumors is rather markedly dysfunctional and ASA404 might reach
higher concentrations in tumor tissue. However, this issue of
limited brain penetration still might be the explanation why ASA404
did not show intracranial activity in our models. In contrast to
our results, ASA404 increased survival in a previous study using
orthotopic the GL261 and the U-87 mouse model (20). In both models only minimal contrast
enhancement was detected before treatment. Notably, in both models
marked extravasation of the contrast agent was detected 24 h after
treatment with ASA404. A more recent study did not find increased
survival with ASA404 in an orthotopic GL261 model corroborating our
results (44).
Second, there might be a difference in molecular
and/or cellular effectors. In 2005 it has been shown that tumor
associated macrophages are the main cellular target of ASA404
(22,33). Moreover, the STING has been identified
as the molecular target of ASA404 recently (23,24,26,27,29,30).
Interestingly, only murine STING binds to ASA404 and leads to
TANK-binding kinase 1 and IFN regulatory factor 3 signaling
(25,29). Human STING failed to bind to or signal
in response to ASA404, which might be the explanation why ASA404
failed in clinical trials for human cancer (29). These findings would make it essential
that a relevant number of murine tumor associated macrophages are
present in a tumor to make it susceptible to ASA404. Therefore, we
screened tumor tissue for STING expression and tumor associated
macrophages. As shown in Fig. 8 we
did not detect fundamental differences in STING expression between
subcutaneous (Fig. 8A) and
intracranial tumors (Fig. 8B).
Whether the more diffuse and more cytosolic expression of STING in
subcutaneous tumors has relevance, is beyond the scope of this
study. STING expression in intracranial tumors (Fig. 8B) seemed to be more focused and
restricted to fewer cells. Moreover, we were not able to
specifically detect murine STING as available antibodies either
recognize human and murine STING or just human STING. In the light
of the brain penetration issues further evaluations on this topic
might be of limited interest. Regarding tumor associated
macrophages we used an antibody against iba1 to detect macrophages
in the examined tumor tissue. Obviously, iba1 can be found on
macrophages and microglia, impeding differentiation between
macrophages and microglia particularly in the intracranial tumors.
Again we did not find considerable differences in the number of
iba1 positive cells in the examined tumor tissue from subcutaneous
(Fig. 8C) and intracranial (Fig. 8D) tumor tissue.
In summary, we can show that ASA404 (DMXAA,
vadimezan) leads to strong responses in the subcutaneous setting
when using several human glioma cell lines and one colon carcinoma
cell line in athymic mice. This is of particular importance,
regarding STING, the molecular target of ASA404. It has been shown
that ASA404 only binds to the murine form of STING and not to human
STING. Therefore, effects of ASA404 cannot directly be conveyed via
human tumor cells. On the one hand, this might be an explanation
for failure of ASA404 in later stage clinical trials. On the other
hand, this emphasizes the importance of tumor infiltrating host
cells in our setting with human cancer cells growing in a rodent
host. This renders host macrophages as a promising and
untransformed therapeutic target. Current knowledge on vascular
disrupting properties of ASA404 suggests that tumor infiltrating
macrophages sense ASA404 via STING and then initiate the release of
cytokines leading to disrupted blood vessels and hemorrhagic
necrosis. Unfortunately, activity of ASA404 was limited to the
subcutaneous setting and we did not detect changes in several
orthotopic brain tumor models. This might be due to a different
cellular composition of the tumor microenvironment, but most likely
is the result of the missing ability of ASA404 to cross the
blood-brain-barrier (46).
Besides vascular disruption, STING agonists also
lead to an activation of the innate immunity (24). STING activation results in type I
interferon production and thereby activating dendritic cells in the
tumor and augmenting the priming of tumor-specific T cells.
Further, STING activation enhances recruitment of effector T cells
into the tumor microenvironment (22–24,27,29,30,33,47).
Several synthetic agonists of human STING have been developed as
immunotherapeutics and at least one is currently tested in a phase
1 trial (NCT02675439) (23,24,26).
Whether these new STING agonists cross the blood-brain-barrier has
not been demonstrated so far. Nonetheless, agonists for human STING
are highly promising new drugs as VDA and more importantly as
immunotherapeutics.
Acknowledgements
This work has been partially supported by an
unrestricted Grant from Novartis. The Dr. Senckenberg Institute of
Neurooncology is supported by the Dr. Senckenberg Foundation. M.M.
would like to thank the Luxembourg National Research Fond (FNR) for
the support (FNR PEARL P16/BM/11192868 grant). O.B. has served as a
consultant for Roche, BMS and NOXXON and has received travel grants
from Roche, BMS and medac. J.P.S. has received a grant from Merck
as well as honoraria for lectures, travel or advisory board
participation from Roche, Boehringer, Bristol-Myers Squibb, Medac
and Mundipharma.
References
|
1
|
Carmeliet P and Jain RK: Molecular
mechanisms and clinical applications of angiogenesis. Nature.
473:298–307. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Louis DN, Perry A, Reifenberger G, von
Deimling A, Figarella-Branger D, Cavenee WK, Ohgaki H, Wiestler OD,
Kleihues P and Ellison DW: The 2016 world health organization
classification of tumors of the central nervous system: A summary.
Acta Neuropathol. 131:803–820. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Preusser M, Hassler M, Birner P, Rudas M,
Acker T, Plate KH, Widhalm G, Knosp E, Breitschopf H, Berger J and
Marosi C: Microvascularization and expression of VEGF and its
receptors in recurring meningiomas: Pathobiological data in favor
of anti-angiogenic therapy approaches. Clin Neuropathol.
31:352–360. 2012. View
Article : Google Scholar : PubMed/NCBI
|
|
4
|
Takano S: Glioblastoma angiogenesis: VEGF
resistance solutions and new strategies based on molecular
mechanisms of tumor vessel formation. Brain Tumor Pathol. 29:73–86.
2012. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Batchelor TT, Reardon DA, de Groot JF,
Wick W and Weller M: Antiangiogenic therapy for glioblastoma:
Current status and future prospects. Clin Cancer Res. 20:5612–5619.
2014. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Friedman HS, Prados MD, Wen PY, Mikkelsen
T, Schiff D, Abrey LE, Yung WK, Paleologos N, Nicholas MK, Jensen
R, et al: Bevacizumab alone and in combination with irinotecan in
recurrent glioblastoma. J Clin Oncol. 27:4733–4740. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Chinot OL, Wick W, Mason W, Henriksson R,
Saran F, Nishikawa R, Carpentier AF, Hoang-Xuan K, Kavan P, Cernea
D, et al: Bevacizumab plus radiotherapy-temozolomide for newly
diagnosed glioblastoma. N Engl J Med. 370:709–722. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Gilbert MR, Dignam JJ, Armstrong TS, Wefel
JS, Blumenthal DT, Vogelbaum MA, Colman H, Chakravarti A, Pugh S,
Won M, et al: A randomized trial of bevacizumab for newly diagnosed
glioblastoma. N Engl J Med. 370:699–708. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Batchelor TT, Mulholland P, Neyns B,
Nabors LB, Campone M, Wick A, Mason W, Mikkelsen T, Phuphanich S,
Ashby LS, et al: Phase III randomized trial comparing the efficacy
of cediranib as monotherapy and in combination with lomustine,
versus lomustine alone in patients with recurrent glioblastoma. J
Clin Oncol. 31:3212–3218. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Wick W, Puduvalli VK, Chamberlain MC, van
den Bent MJ, Carpentier AF, Cher LM, Mason W, Weller M, Hong S,
Musib L, et al: Phase III study of enzastaurin compared with
lomustine in the treatment of recurrent intracranial glioblastoma.
J Clin Oncol. 28:1168–1174. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Wick W, Steinbach JP, Platten M, Hartmann
C, Wenz F, von Deimling A, Shei P, Moreau-Donnet V, Stoffregen C
and Combs SE: Enzastaurin before and concomitant with radiation
therapy, followed by enzastaurin maintenance therapy, in patients
with newly diagnosed glioblastoma without MGMT promoter
hypermethylation. Neuro Oncol. 15:1405–1412. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Stupp R, Hegi ME, Gorlia T, Erridge SC,
Perry J, Hong YK, Aldape KD, Lhermitte B, Pietsch T, Grujicic D, et
al: Cilengitide combined with standard treatment for patients with
newly diagnosed glioblastoma with methylated MGMT promoter (CENTRIC
EORTC 26071–22072 study): A multicentre, randomised, open-label,
phase 3 trial. Lancet Oncol. 15:1100–1108. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Baguley BC: Antivascular therapy of
cancer: DMXAA. Lancet Oncol. 4:141–148. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Hinnen P and Eskens FA: Vascular
disrupting agents in clinical development. Br J Cancer.
96:1159–1165. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Thorpe PE: Vascular targeting agents as
cancer therapeutics. Clin Cancer Res. 10:415–427. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Tozer GM, Kanthou C and Baguley BC:
Disrupting tumour blood vessels. Nat Rev Cancer. 5:423–435. 2005.
View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Folaron M, Kalmuk J, Lockwood J, Frangou
C, Vokes J, Turowski SG, Merzianu M, Rigual NR, Sullivan-Nasca M,
Kuriakose MA, et al: Vascular priming enhances chemotherapeutic
efficacy against head and neck cancer. Oral Oncol. 49:893–902.
2013. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Kanwar JR, Kanwar RK, Pandey S, Ching LM
and Krissansen GW: Vascular attack by
5,6-dimethylxanthenone-4-acetic acid combined with B7.1
(CD80)-mediated immunotherapy overcomes immune resistance and leads
to the eradication of large tumors and multiple tumor foci. Cancer
Res. 61:1948–1956. 2001.PubMed/NCBI
|
|
19
|
Matthews KE, Hermans IF, Roberts JM, Ching
LM and Ronchese F: 5,6-Dimethylxanthenone-4-acetic acid treatment
of a non-immunogenic tumour does not synergize with active or
passive CD8+ T-cell immunotherapy. Immunol Cell Biol.
84:383–389. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Seshadri M and Ciesielski MJ: MRI-based
characterization of vascular disruption by
5,6-dimethylxanthenone-acetic acid in gliomas. J Cereb Blood Flow
Metab. 29:1373–1382. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Ching LM, Zwain S and Baguley BC:
Relationship between tumour endothelial cell apoptosis and tumour
blood flow shutdown following treatment with the antivascular agent
DMXAA in mice. Br J Cancer. 90:906–910. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Jassar AS, Suzuki E, Kapoor V, Sun J,
Silverberg MB, Cheung L, Burdick MD, Strieter RM, Ching LM, Kaiser
LR, et al: Activation of tumor-associated macrophages by the
vascular disrupting agent 5,6-dimethylxanthenone-4-acetic acid
induces an effective CD8+ T-cell-mediated antitumor
immune response in murine models of lung cancer and mesothelioma.
Cancer Res. 65:11752–11761. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Corrales L, Glickman LH, McWhirter SM,
Kanne DB, Sivick KE, Katibah GE, Woo SR, Lemmens E, Banda T, Leong
JJ, et al: Direct activation of STING in the tumor microenvironment
leads to potent and systemic tumor regression and immunity. Cell
Rep. 11:1018–1030. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Corrales L, McWhirter SM, Dubensky TW Jr
and Gajewski TF: The host STING pathway at the interface of cancer
and immunity. J Clin Invest. 126:2404–2411. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Roberts ZJ, Goutagny N, Perera PY, Kato H,
Kumar H, Kawai T, Akira S, Savan R, van Echo D, Fitzgerald KA, et
al: The chemotherapeutic agent DMXAA potently and specifically
activates the TBK1-IRF-3 signaling axis. J Exp Med. 204:1559–1569.
2007. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Curran E, Chen X, Corrales L, Kline DE,
Dubensky TW Jr, Duttagupta P, Kortylewski M and Kline J: STING
pathway activation stimulates potent immunity against acute myeloid
leukemia. Cell Rep. 15:2357–2366. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Gao P, Ascano M, Zillinger T, Wang W, Dai
P, Serganov AA, Gaffney BL, Shuman S, Jones RA, Deng L, et al:
Structure-function analysis of STING activation by c[G (2′,5′)pA
(3′,5′)p] and targeting by antiviral DMXAA. Cell. 154:748–762.
2013. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Ishikawa H and Barber GN: STING is an
endoplasmic reticulum adaptor that facilitates innate immune
signalling. Nature. 455:674–678. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Conlon J, Burdette DL, Sharma S, Bhat N,
Thompson M, Jiang Z, Rathinam VA, Monks B, Jin T, Xiao TS, et al:
Mouse, but not human STING, binds and signals in response to the
vascular disrupting agent 5,6-dimethylxanthenone-4-acetic acid. J
Immunol. 190:5216–5225. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Prantner D, Perkins DJ, Lai W, Williams
MS, Sharma S, Fitzgerald KA and Vogel SN:
5,6-Dimethylxanthenone-4-acetic acid (DMXAA) activates stimulator
of interferon gene (STING)-dependent innate immune pathways and is
regulated by mitochondrial membrane potential. J Biol Chem.
287:39776–39788. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
McKeage MJ, Von Pawel J, Reck M, Jameson
MB, Rosenthal MA, Sullivan R, Gibbs D, Mainwaring PN, Serke M,
Lafitte JJ, et al: Randomised phase II study of ASA404 combined
with carboplatin and paclitaxel in previously untreated advanced
non-small cell lung cancer. Br J Cancer. 99:2006–2012. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Lara PN Jr, Douillard JY, Nakagawa K, von
Pawel J, McKeage MJ, Albert I, Losonczy G, Reck M, Heo DS, Fan X,
et al: Randomized phase III placebo-controlled trial of carboplatin
and paclitaxel with or without the vascular disrupting agent
vadimezan (ASA404) in advanced non-small-cell lung cancer. J Clin
Oncol. 29:2965–2971. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Wallace A, LaRosa DF, Kapoor V, Sun J,
Cheng G, Jassar A, Blouin A, Ching LM and Albelda SM: The vascular
disrupting agent, DMXAA, directly activates dendritic cells through
a MyD88-independent mechanism and generates antitumor cytotoxic T
lymphocytes. Cancer Res. 67:7011–7019. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Smilowitz HM, Weissenberger J, Weis J,
Brown JD, O'Neill RJ and Laissue JA: Orthotopic transplantation of
v-src-expressing glioma cell lines into immunocompetent mice:
Establishment of a new transplantable in vivo model for malignant
glioma. J Neurosurg. 106:652–659. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Tuchen M, Wilisch-Neumann A, Daniel EA,
Baldauf L, Pachow D, Scholz J, Angenstein F, Stork O, Kirches E and
Mawrin C: Receptor tyrosine kinase inhibition by
regorafenib/sorafenib inhibits growth and invasion of meningioma
cells. Eur J Cancer. 73:9–21. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Pachow D, Andrae N, Kliese N, Angenstein
F, Stork O, Wilisch-Neumann A, Kirches E and Mawrin C: mTORC1
inhibitors suppress meningioma growth in mouse models. Clin Cancer
Res. 19:1180–1189. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
McCutcheon IE, Friend KE, Gerdes TM, Zhang
BM, Wildrick DM and Fuller GN: Intracranial injection of human
meningioma cells in athymic mice: An orthotopic model for
meningioma growth. J Neurosurg. 92:306–314. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Lee WH: Characterization of a newly
established malignant meningioma cell line of the human brain:
IOMM-Lee. Neurosurgery. 27:389–396. 1990. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Kliese N, Gobrecht P, Pachow D, Andrae N,
Wilisch-Neumann A, Kirches E, Riek-Burchardt M, Angenstein F,
Reifenberger G, Riemenschneider M, et al: miRNA-145 is
downregulated in atypical and anaplastic meningiomas and negatively
regulates motility and proliferation of meningioma cells. Oncogene.
32:4712–4720. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
RayChaudhury A, Frazier WA and D'Amore PA:
Comparison of normal and tumorigenic endothelial cells: Differences
in thrombospondin production and responses to transforming growth
factor-beta. J Cell Sci. 107:39–46. 1994.PubMed/NCBI
|
|
41
|
Muhlner U, Mohle-Steinlein U,
Wizigmann-Voos S, Christofori G, Risau W and Wagner EF: Formation
of transformed endothelial cells in the absence of VEGFR-2/Flk-1 by
Polyoma middle T oncogene. Oncogene. 18:4200–4210. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Montesano R, Pepper MS, Möhle-Steinlein U,
Risau W, Wagner EF and Orci L: Increased proteolytic activity is
responsible for the aberrant morphogenetic behavior of endothelial
cells expressing the middle T oncogene. Cell. 62:435–445. 1990.
View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Weller M, Rieger J, Grimmel C, Van Meir
EG, Tribolet N, De Krajewski S, Reed JC, von Deimling A and
Dichgans J: Predicting chemoresistance in human malignant glioma
cells: The role of molecular genetic analyses. Int J Cancer.
79:640–644. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Yung R, Seyfoddin V, Guise C, Tijono S,
McGregor A, Connor B and Ching LM: Efficacy against subcutaneous or
intracranial murine GL261 gliomas in relation to the concentration
of the vascular-disrupting agent, 5,6-dimethylxanthenone-4-acetic
acid (DMXAA), in the brain and plasma. Cancer Chemother Pharmacol.
73:639–649. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Renner DN, Jin F, Litterman AJ, Balgeman
AJ, Hanson LM, Gamez JD, Chae M, Carlson BL, Sarkaria JN, Parney
IF, et al: Effective treatment of established GL261 murine gliomas
through picornavirus vaccination-enhanced tumor antigen-specific
CD8+ T cell responses. PLoS One. 10:e01255652015.
View Article : Google Scholar : PubMed/NCBI
|
|
46
|
Zhao L, Ching LM, Kestell P and Baguley
BC: The antitumour activity of 5,6-dimethylxanthenone-4-acetic acid
(DMXAA) in TNF receptor-1 knockout mice. Br J Cancer. 87:465–470.
2002. View Article : Google Scholar : PubMed/NCBI
|
|
47
|
Conlon J, Burdette DL, Sharma S, Bhat N,
Thompson M, Jiang Z, Rathinam VA, Monks B, Jin T, Xiao TS, et al:
Mouse, but not human STING, binds and signals in response to the
vascular disrupting agent 5,6-dimethylxanthenone-4-acetic acid. J
Immunol. 190:5216–5225. 2013. View Article : Google Scholar : PubMed/NCBI
|